

Abstract
Three-dimensional super-resolution imaging in thick, semi-transparent biological specimens is hindered by light scattering, which increases background and degrades both contrast and optical sectioning. We describe a simple method that mitigates these issues, improving image quality in our recently developed two-photon instant structured illumination microscope without requiring any hardware modifications to the instrument. By exciting the specimen with three laterally-structured, phase-shifted illumination patterns and post-processing the resulting images, we digitally remove both scattered and out-of-focus emissions that would otherwise contaminate our raw data. We demonstrate the improved performance of our approach in biological samples, including pollen grains, primary mouse aortic endothelial cells cultured in a three–dimensional collagen matrix and live tumor-like cell spheroids.
OCIS codes: (110.0180) Microscopy, (100.6640) Superresolution, (170.2520) Fluorescence microscopy, (180.0180) Microscopy
1. Introduction
The ability to acquire high-resolution, three-dimensional (3D) images is vital in biology [1]. This task is particularly difficult in thick, densely-labeled samples where scattering of both emission and excitation light can dramatically reduce image quality. Our recently developed two-photon (2P) instant structured illumination microscope (2P ISIM) [2] combines the depth penetration of a multiphoton microscope (reducing excitation-side scattering and providing superior optical sectioning) [3] with the resolving power of point-based structured illumination (enabling super-resolution imaging) [4–6]. Nevertheless, 2P ISIM still suffers from image quality degradation in thick, densely labeled samples due to emission-side light scattering, as we use widefield detection (a camera) to collect the fluorescence.
We present a method to reduce the effects of emission-side scattering in 2P ISIM by illuminating the sample with a series of phase-shifted, laterally-structured incoherent illumination patterns and digitally post-processing the resulting images. This approach is particularly attractive because it requires no physical modification of the existing instrument and can be easily optimized for any biological sample.
Incoherent patterned illumination coupled with digital post-processing has been successfully implemented in single photon excitation (1P) applications, including conventional widefield microscopy [7] and light-sheet microscopy [8,9], where it resulted in improved optical sectioning and enhanced image contrast on a variety of samples, including pollen grains and live Drosophila embryos. It has also been shown to improve axial sectioning in widefield 2P temporal focusing microscopy [10], but has not yet been implemented in conjunction with 2P superresolution imaging. Here we demonstrate that combining incoherent structured illumination with 2P ISIM maintains the super-resolving power of 2P ISIM while simultaneously improving both optical sectioning and contrast in thick, scattering biological specimens.
2. Results
By sequentially projecting three fine, phase-shifted incoherent horizontal illumination patterns ( , , ) onto the sample at axial position z, collecting the resulting images and digitally processing the resulting data according to:
| (1) |
unstructured background emissions (including both scattered and out-of-focus emissions) are largely separated and eliminated from the desired signal (non-scattered, structured emission). The incoherent structured illumination imaging process on our 2P ISIM system is illustrated in Fig. 1 . Individual raw images of single z-slices from an autofluorescent ragweed pollen grain for each phase of the sinusoidal illumination pattern [Fig. 1(a)] are displayed with reconstructions of the normal image and the processed image (showing enhanced optical sectioning and contrast). Maximum intensity projections of the entire imaging volume [Fig. 1(b) and Fig. 1(c)] as well as line profiles derived from single slices [Fig. 1(d)] demonstratethe capacity of patterned illumination and digital post processing to remove haze from scattering and unstructured out-of-focus emissions.
Fig. 1.

Incoherent structured illumination improves contrast in 2P ISIM. (a) Raw images of a 20 μm diameter ragweed pollen grain when using patterned excitation in 2P ISIM. The relative phase shift (0°, 120°, or 240°) is indicated above each image. Raw images may be subsequently summed (bottom left) to recreate the normal image or processed according to Eq. (1) to yield an image with improved contrast (bottom right). Images were acquired 4 μm from the coverslip surface. (b) Comparative maximum intensity projections after summing, or (c) otherwise processing each slice in the imaging volume, and (d) line profiles derived from single slices in (a) confirm the superior contrast afforded by combining incoherent structured illumination with 2P ISIM. Pattern spacing: 1.6 μm. All scalebars: 4 μm. These images have not been deconvolved.
In order to deliver a sinusoidal illumination pattern to the sample, we modulate the intensity of the excitation beam during scanning with a voltage-controlled Pockel’s cell, an approach that affords significantly more flexibility than previous implementations that translated a patterned grid in a conjugate imaging plane [7,8]. Figure 2(a) demonstrates our capacity to project patterns of variable spacing onto a thick fluorescent lake by altering the frequency of the waveform input to the Pockel’s cell (a square root sine function, because signal is proportional to the square of the illumination intensity in a 2P microscope). Regardless of the pattern spacing, summing the total emission from the sample does not improve the sectioning capability of the microscope [Fig. 2(b)]. However, processing the images according to Eq. (1) does improve sectioning: use of finer grid patterns results in better rejection of out-of-focus emissions [Fig. 2(c)], with the relative improvement diminishing as the pattern spacing decreases (i.e. improvement in optical sectioning after processing approaches a limit as a function of pattern spacing). Imaging subdiffractive fluorescent beads embedded in a 3D scattering matrix also established that the use of patterned illumination and digital post-processing did not alter the inherent lateral and axial resolution of our microscope [Fig. 2(d) and Fig. 2(e)].
Fig. 2.

Using incoherent structured illumination to improve sectioning and contrast in thick samples. (a) Exemplary patterns at different spatial frequencies, as viewed in a thick fluorescent lake. Scalebar: 10 μm. ‘NA' corresponds to the case without a pattern, i.e. scanning conventional point-illumination through the sample. Patterns are shown at 0° phase offset. (b) and (c) Sectioning response in a thick fluorescent lake. In (b), images corresponding to different phases were summed, normalized according to their intensity at the thick lake interface (defined as the axial position at which the derivative of the change in intensity as a function of axial position is maximized) and compared. (c) The same data shown in (b) were processed according to Eq. (1), and normalized as before. Using higher frequency patterns results in better optical sectioning (less fluorescence) when imaging deeper into the lake (positive values of axial position in the graph). (d) XZ slice through a single 100 nm diameter fluorescent bead in a scattering matrix, as imaged conventionally (upper) and with 1.6 μm spaced pattern (lower). Note the reduction in background after applying patterned illumination. Scalebar: 500 nm. Bead data are deconvolved. (e) The FWHM of 100 nm subdiffractive fluorescent beads placed in the same scattering matrix as in (d), without (no pattern) and with incoherent structured illumination (1.6 μm spacing). Mean and standard deviations from 10 beads, after deconvolution, are shown. Beads appear the same size regardless of imaging condition. (f) Improvement in contrast, comparing different patterns at different depths in the scattering matrix used in (d) and (e).
We also investigated the relative increase in contrast as a function of pattern spacing in this sample [Fig. 2(f)]. Here ‘contrast’ is defined as the ratio of the standard deviation (s.d.) of the energy-normalized image histogram with patterned illumination to the s.d. without patterned illumination, which provides an objective measure for comparing images produced with and without incoherent structured illumination [9]. The s.d. of the energy-normalized image histogram is defined as:
| (2) |
where ci is the pixel count for intensity level i in an image, the set of pairs (i, ci) defines the intensity histogram of the image, the image intensity sum is:
| (3) |
the total pixel count is:
| (4) |
and the average intensity of the image . For all patterns and depths analyzed, the contrast of the processed image is improved compared to the normal image. Near the surface of the sample and at shallow depths, tightly spaced patterns offer the best performance, while deeper into the sample coarser patterns perform best [Fig. 2(f)]. These results can be explained by noting that the ability to deliver finely spaced patterns diminishes more rapidly as a function of depth than does the ability to deliver coarsely spaced patterns. The optimalchoice of pattern thus depends on the depth and sample under investigation.
We next applied our method to a series of particularly challenging (i.e. thick, densely labeled and highly scattering) biological samples. Figure 3(a) shows phalloidin-stained actin structures in primary mouse aortic endothelial cells, cultured within a 3D collagen gel. In a 1P imaging system, scattering due to the collagen matrix inhibits high-resolution imaging deep into this sample [11], but 2P ISIM permits observation of fine details at depths of ~100 μm [Fig. 3(c)] and incoherent patterned illumination further improves images by removing additional out-of-focus haze [Fig. 3(b)]. Far from the coverslip, our method resolves parallel actin bundles <200 nm apart [Fig. 3(d) and Fig. 3(e)], and many individual bundles at apparent widths of <150 nm. Improved optical sectioning is also visible in axial reslices of these data, where small features and voids (blue arrows in Fig. 3(f) and Fig. 3(g)) are much more apparent than in conventional 2P ISIM.
Fig. 3.

Contrast improvement in densely tagged, thick biological specimens. (a) Partial maximum intensity projection (0-12μm) of a 30-μm thick volume, ~100 μm from the coverslip surface, highlighting phalloidin-labeled actin in a fixed primary culture mouse aortic endothelial cell in a 3D collagen matrix. Scalebar: 10 μm. (b) Digitally processed and (c) conventional 2P ISIM single slices (8 μm into volume), showing higher magnification views of yellow rectangle in (a). Note improved contrast in (b) compared to (c). Scale Bars: 5 μm. (d) Higher magnification view of yellow rectangle in (b), showing two actin bundles just resolved and (e) line profile (peaks are spaced 180 nm apart). Scalebar: 500 nm. Axial cuts through volume, corresponding to magenta dotted line in (a) are shown in digitally processed (f) and conventional (g) 2P ISIM. Blue arrows highlight void areas within cell that are visible in (f), due to improved sectioning, but masked in (g). Pattern spacing: 1.6 μm. Scalebar: 5 μm. These images have been deconvolved.
We were also able to improve multicolor 2P ISIM imaging of peroxisomes and tightly packed mitochondria in live tumor-like spheroid cell co-cultures using incoherent structured illumination [Fig. 4(a) ]. Such samples are particularly challenging due to their dense labeling and scattering nature. In both lateral and axial views of the spheroid, individual mitochondria are more readily identifiable when utilizing structured illumination as compared to normal illumination, allowing for improved reconstruction of the 3D spheroid volume (compare Fig. 4(b) and Fig. 4(d) to Fig. 4(c) and Fig. 4(e)).
Fig. 4.

Incoherent structured illumination improves contrast and axial sectioning in thick, scattering biological specimens. (a) 3D rendering of a 35-μm thick volume showing MitoTracker Red-labeled mitochondria and GFP-tagged peroxisomes in a live tumor-like spheroid. The spheroid is a co-culture of transformed keratinocytes (HACAT) growing on human lung fibroblasts (MRL-TR). MitoTracker Red labels mitochondria in both cell types while peroxisomal targeted GFP is only expressed in the stromal layer fibroblasts. (b) Digitally processed and (c) conventional 2P ISIM single slices at indicated axial depth from the coverslip. Yellow arrows highlight specific structures displaying improved contrast in (b) compared to (c). (d) Digitally processed and (e) conventional XZ cuts through the spheroid volume at indicated Y position. Yellow arrows highlight specific structures displaying improved contrast in (d) compared to (e). Pattern spacing: 1.6 μm. All scalebars: 10 μm. These images have been deconvolved.
3. Discussion
Our approach provides a straightforward method to improve contrast and optical sectioning of 2P ISIM, facilitating observation of structures in thick, densely labeled samples that would be otherwise obscured by scattered light in a 1P super-resolution microscope or diffractively blurred in a conventional 2P microscope. Limitations of the method include: (i). a reduction in imaging bit depth compared to conventional (no pattern) imaging; (ii). a slight striping pattern in the data after digital processing (which may be corrected by applying a simple de-striping algorithm as shown in Fig. 5 ); (iii) increased light exposure and a reduction in imaging speed relative to conventional 2P ISIM, due to acquisition of three images instead of one. Nevertheless, our acquisition rate was sufficient for imaging the live mitochondria and peroxisomes in Fig. 4 without motion blur.
Fig. 5.
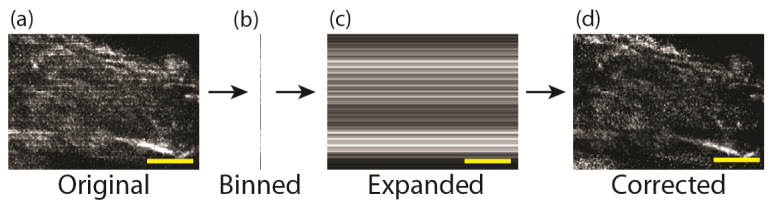
Stripe-correction algorithm. (a) Variable striping artifacts as seen in Alexa488-phalloidin stained primary mouse aortic endothelial cells are corrected on a slice-by-slice basis by (b) horizontally binning the image data, (c) expanding the binned image to match the dimensions of (a) and (d) dividing the original image (a) by (c) to obtain the corrected image. Scale bars: 4 μm. These images have not been deconvolved.
An alternative strategy to improve sectioning would be to introduce a physical pinhole in a conjugate image plane [12]. This option requires no post-processing and provides a signal proportional to fluorophore density. However, introducing a physical pinhole would requiresignificant modification of our existing microscope, including additional optics and optical surfaces for descanning and rescanning, with an accompanying reduction in signal.
4. Methods
4.1 Image acquisition
For a detailed description of instrumentation and the image acquisition process in 2P ISIM please see [2]. Deconvolution: Richardson-Lucy algorithm for 40 iterations, assuming a Gaussian PSF with lateral FWHM of 220 nm and axial FWHM of 660 nm. Other image acquisition parameters include:
Figure 1. Autofluorescent ragweed pollen grains were imaged with an exposure time of 500 ms, EM gain setting of 200, Z-step size of 250 nm, 6x frame averaging, 1.6 μm pattern spacing at an excitation wavelength of 900 nm.
Figure 2. The thick fluorescent lake (autofluorescent plastic slide) was imaged with exposure time of 500 ms, EM gain setting of 20, Z-step size of 500 nm, 1x frame averaging, variable pattern spacing at an excitation wavelength of 900 nm. The fluorescent beads in a 3D scattering gel were imaged with an exposure time of 500 ms, EM gain setting of 200, Z-step size of 250 nm, 8x frame averaging at an excitation wavelength of 900 nm.
Figure 3 and Fig. 5. The phalloidin-labeled primary mouse endothelial cells in a 3D collagen matrix were imaged with an exposure time of 500 ms, EM gain setting of 200, Z-step size of 250 nm, 4x frame averaging, 1.6 μm pattern spacing at an excitation wavelength of 900 nm.
Figure 4. Peroxisomal-targeted GFP in the fibroblast cell layers of the tumor-like spheroid co-cultures was imaged with an exposure time of 500 ms, EM gain setting of 200, Z-step size of 250 nm, 4x frame averaging, 1.6 μm pattern spacing at an excitation wavelength of 900 nm. A 525/50 nm emission filter (Cat # FF01-525/50-25 Semrock, NY) was included in the emission path to exclude MitoTracker Red emission. Following this, the MitoTracker Red-labeled mitochondria in both cell types were imaged with an exposure time of 500 ms, EM gain setting of 200, Z-step size of 250 nm, 4x frame averaging, 1.6 μm pattern spacing at an excitation wavelength of 900 nm. For MitoTracker Red imaging a 617/73 nm emission filter (Cat # FF01-617/73-25 Semrock, NY) was included in the emission path to exclude GFP emission.
4.2 Sample preparation
Figure 1. Individual ragweed pollen grains (Cat #07673, Polysciences, PA) were deposited on #1.5 glass bottomed 35 mm dishes (Cat # P35-1.5-20-C, MatTek, MA) and immersed in deionized water for imaging.
Figure 2. The yellow autofluorescent plastic slide (Cat # 92001, Chroma, VT) was imaged without modification. 100 nm diameter yellow/green fluorescent beads (Cat # F8803, Invitrogen, OR) were embedded at 1:250 v/v dilution inside a 3D gel made from 5% w/w agarose (Cat # A9414-5G, Sigma, MO) containing 0.5% v/v nonfluorescent scattering 62 nm diameter polystyrene beads (Cat # L031030C, Bangs Laboratories, IN) in deionized water. The contents were vortexed, sonicated and microwaved, the liquefied gel deposited on a #1.5 glass bottomed 35 mm dish, immersed in additional deionized water, and imaged.
Figure 3 and Fig. 5. Endothelial cells derived from primary mouse aortic explants were embedded in 1.6 mg/ml collagen gels as previously described [11]. Cells in collagen gels were fixed with 3% paraformaldehyde in cytoskeleton buffer (CB; 10mM MES pH 6.1, 138mM KCl, 3mM MgCl, 2mM EGTA) for 30 minutes at 37 °C, rinsed twice with CB, then extracted in CB with 0.4% triton for 60 minutes at room temperature. Cells were labeled in CB with Alexa 488 phalloidin (1:200 dilution; Life Sciences Technologies catalog #A12379) for 4-12 hours at 4 °C and rinsed with CB for 10 minutes at room temperature prior to imaging.
Figure 4. Cells were maintained in DMEM (Cat # 10569-010, Life Technologies) supplemented with 10% FBS (Cat # SH30070.03, Hyclone) in a CO2 supplied incubator (5%) at 37 °C. The stromal layer consisted of human transformed lung fibroblasts (MRL-TR) originating from MRC-5 primary lung fibroblasts (ATCC) that were stably transfected with peroxisomal-targeted GFP [6]. For live imaging, the cells were plated on 25 mm round #1.5 coverslips (Cat # 64-0715, Warner Instruments) inserted into 35 mm tissue culture dishes (Thermo Scientific), and prior to reaching complete confluency the culture was seeded with HaCaT transformed keratinocytes at a density of 105 cells per 35 mm well. After 3-5 days, niches of HaCaT spheroids began to grow over the fibroblastic layer. Prior to live imaging, the cells were incubated with 200 nM MitoTracker Red CM-H2XRos (Cat# M-7512, Life Technologies) for 30 minutes and the coverslips rinsed with fresh medium and mounted on a single depression glass slide (Cat # 260241, Ted Pella) containing imaging medium (phenol red free-DMEM with 10% FBS supplemented with 5 mM Hepes) (Cat # 15630160, Life Technologies).
Acknowledgments
This work was supported by the Intramural Research Programs of the National Institute of Biomedical Imaging and Bioengineering (P.W.W., P.C., Y.W., and H.S.), and the National Heart, Lung, and Blood Institute (R.S.F. and C.M.W.) of the National Institutes of Health. The NIH does not endorse or recommend any commercial products, processes, or services. The views and opinions of authors expressed here do not necessarily state or reflect those of the U.S. government and they may not be used for advertising or product endorsement purposes. We thank Christian Combs and Daniela Malide for help in rendering 3D data.
References and links
- 1.Fischer R. S., Wu Y., Kanchanawong P., Shroff H., Waterman C. M., “Microscopy in 3D: a biologist’s toolbox,” Trends Cell Biol. 21(12), 682–691 (2011). 10.1016/j.tcb.2011.09.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Winter P. W., York A. G., Nogare D. D., Ingaramo M., Christensen R., Chitnis A., Patterson G. H., Shroff H., “Two-photon instant structured illumination microscopy improves the depth penetration of super-resolution imaging in thick scattering samples,” Optica 1(3), 181–191 (2014). 10.1364/OPTICA.1.000181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Helmchen F., Denk W., “Deep tissue two-photon microscopy,” Nat. Methods 2(12), 932–940 (2005). 10.1038/nmeth818 [DOI] [PubMed] [Google Scholar]
- 4.Müller C. B., Enderlein J., “Image scanning microscopy,” Phys. Rev. Lett. 104(19), 198101 (2010). 10.1103/PhysRevLett.104.198101 [DOI] [PubMed] [Google Scholar]
- 5.De Luca G. M. R., Breedijk R. M. P., Brandt R. A. J., Zeelenberg C. H. C., de Jong B. E., Timmermans W., Azar L. N., Hoebe R. A., Stallinga S., Manders E. M. M., “Re-scan confocal microscopy: scanning twice for better resolution,” Biomed. Opt. Express 4(11), 2644–2656 (2013). 10.1364/BOE.4.002644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.York A. G., Chandris P., Nogare D. D., Head J., Wawrzusin P., Fischer R. S., Chitnis A., Shroff H., “Instant super-resolution imaging in live cells and embryos via analog image processing,” Nat. Methods 10(11), 1122–1126 (2013). 10.1038/nmeth.2687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Neil M. A. A., Juskaitis R., Wilson T., “Method of obtaining optical sectioning by using structured light in a conventional microscope,” Opt. Lett. 22(24), 1905–1907 (1997). 10.1364/OL.22.001905 [DOI] [PubMed] [Google Scholar]
- 8.Breuninger T., Greger K., Stelzer E. H. K., “Lateral modulation boosts image quality in single plane illumination fluorescence microscopy,” Opt. Lett. 32(13), 1938–1940 (2007). 10.1364/OL.32.001938 [DOI] [PubMed] [Google Scholar]
- 9.Keller P. J., Schmidt A. D., Santella A., Khairy K., Bao Z., Wittbrodt J., Stelzer E. H. K., “Fast, high-contrast imaging of animal development with scanned light sheet-based structured-illumination microscopy,” Nat. Methods 7(8), 637–642 (2010). 10.1038/nmeth.1476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Choi H., Yew E. Y. S., Hallacoglu B., Fantini S., Sheppard C. J. R., So P. T. C., “Improvement of axial resolution and contrast in temporally focused widefield two-photon microscopy with structured light illumination,” Biomed. Opt. Express 4(7), 995–1005 (2013). 10.1364/BOE.4.000995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fischer R. S., Gardel M. L., Ma X., Adelstein R. S., Waterman C. M., “Local cortical tension by myosin II guides 3D endothelial cell branching,” Curr. Biol. 19(3), 260–265 (2009). 10.1016/j.cub.2008.12.045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gauderon R., Lukins P. B., Sheppard C. J. R., “Effect of a confocal pinhole in two-photon microscopy,” Microsc. Res. Tech. 47(3), 210–214 (1999). [DOI] [PubMed] [Google Scholar]