

Abstract
Although the mechanisms of action (MoA) of nonstructural protein 3 inhibitors (NS3i) and NS5B inhibitors (NS5Bi) are well understood, the MoA of cyclophilin inhibitors (CypI) and NS5A inhibitors (NS5Ai) are not fully defined. In this study, we examined whether CypI and NS5Ai interfere with hepatitis C virus (HCV) RNA synthesis of replication complexes (RCs) or with an earlier step of HCV RNA replication, the creation of double-membrane vesicles (DMVs) essential for HCV RNA replication. In contrast to NS5Bi, both CypI and NS5Ai do not block HCV RNA synthesis by way of RCs, suggesting that they exert their antiviral activity prior to the establishment of enzymatically active RCs. We found that viral replication is not a precondition for DMV formation, since the NS3-NS5B polyprotein or NS5A suffices to create DMVs. Importantly, only CypI and NS5Ai, but not NS5Bi, mir-122, or phosphatidylinositol-4 kinase IIIα (PI4KIIIα) inhibitors, prevent NS3-NS5B-mediated DMV formation. NS3-NS5B was unable to create DMVs in cyclophilin A (CypA) knockdown (KD) cells. We also found that the isomerase activity of CypA is absolutely required for DMV formation. This not only suggests that NS5A and CypA act in concert to build membranous viral factories but that CypI and NS5Ai mediate their early anti-HCV effects by preventing the formation of organelles, where HCV replication is normally initiated. This is the first investigation to examine the effect of a large panel of anti-HCV agents on DMV formation, and the results reveal that CypI and NS5Ai act at the same membranous web biogenesis step of HCV RNA replication, thus indicating a new therapeutic target of chronic hepatitis C.
INTRODUCTION
Chronic hepatitis C infection affects approximately 200 million people worldwide and is a leading cause of acute and chronic liver diseases (1), and 4 million new hepatitis C virus (HCV) infections occur each year (2, 3). HCV accounts for 2/3 of liver cancer and transplant cases in the developed world (4). Until 2011, the combination of pegylated alpha interferon (IFN-α) and ribavirin (RBV) had a success rate of ∼80% in patients with genotypes 2 and 3 but only ∼50% in patients with genotype 1; most importantly, it causes severe side effects (5–9). There was thus an imperative demand for the identification and development of new anti-HCV agents with diversified mechanisms of action (MoA) in order to deliver interchangeable IFN/RBV-free therapies. Remarkably, new classes of safe and efficacious inhibitors, including direct-acting antiviral (DAAs), such as nonstructural protein 3 inhibitors (NS3i), NS5Ai, and NS5Bi, as well as host-targeting antivirals (HTAs), such as cyclophilin inhibitors (CypI), mir-122 inhibitors (mir-122i), and phosphatidylinositol-4 kinase IIIα inhibitors (PI4KIIIαi), have emerged (10). A number of these compounds have reached IFN- or IFN/RBV-free clinical trials or have been approved. Interestingly, although the mechanisms of action (MoA) of NS3i and NS5Bi are well understood, the MoA of CypI and NS5Ai are not fully defined. In this study, we conducted a set of experiments aimed at gaining an understanding of how these two distinct classes of inhibitors mediate their antiviral effects.
Three CypI, alisporivir, NIM811, and SCY-635, have been tested in clinical studies (11–30). Alisporivir is the most advanced in this class, as it has been exposed to the largest number of patients, and it is currently is in clinical development in IFN-free combination regimens (31). We and others have showed that the isomerase pocket of cyclophilin A (CypA) is essential for HCV replication (32–35). CypI blocks HCV replication by neutralizing the enzymatic activity of CypA. We and others showed that CypA interacts directly with NS5A of multiple genotypes and that this interaction is abrogated by CypI (36–45). Thus, there is a direct correlation between preventing NS5A-CypA interactions and blocking HCV replication. However, it remains unknown why the contact between CypA and NS5A is vital for HCV. Since CypI represent an attractive class of host-targeting antivirals due to their pangenotypic activities and high barrier to resistance, they may represent useful drug partners for DAAs in IFN/RBV-free regimens.
Several NS5Ai are already key components of safe and efficacious IFN/RBV-free regimens. Among them are the original daclatasvir (DCV) and the structurally related ledipasvir and ombitasvir. Elbasvir (MK-8742), ACH-3102, and GS-5816 are newer NS5Ai that are in earlier stages of clinical development (46–57). These NS5Ai have a high barrier to resistance, since multiple mutations should emerge in NS5A to decrease HCV susceptibility to these compounds (46–57). Because NS5A has no recognized enzymatic activity, the MoA of NS5Ai and NS5A function in HCV RNA replication remain obscure. We and others showed that NS5Ai failed to prevent NS5A dimerization (58) and contact between NS5A and various ligands, including NS5B (58, 59), core (60), and CypA (58). On the other hand, recent studies provided evidence that NS5Ai decrease the interaction between NS5A and phosphatidylinositol-4 kinase IIIα (PI4KIIIα) (61–63). Importantly, PI4KIIIα is vital for HCV replication. Indeed, the suppression of either the expression of PI4KIIIα or its enzymatic activity blocks HCV replication (64–68). However, as for NS5A-CypA interactions, it remains totally unknown how NS5A-PI4KIIIα interactions control HCV RNA replication. Since NS5A is an RNA binding protein (40, 69, 70), one cannot exclude the possibility that NS5Ai block HCV by diminishing the affinity of NS5A to the viral RNA, eventually modulating the degree of hyperphosphorylation of NS5A, leading to abortive replicase activity, as recently suggested (62, 63).
In this study, we designed and conducted a set of experiments aimed at gaining an understanding of how these two highly efficacious classes of inhibitors mediate their antiviral activities. Specifically, we examined the steps of HCV RNA replication at which these inhibitors act. These experiments are also aimed at gaining an understanding of the precise functions of CypA and NS5A in the HCV RNA synthesis machinery.
MATERIALS AND METHODS
Compounds.
The HCV NS5Ai daclatasvir (Daklinza; Bristol-Myers Squibb) and ledipasvir (Gilead), the NS5Bi sofosbuvir (Sovaldi; Gilead) and mericitabine (Roche), and the NS3i boceprevir (Victrelis; Merck) and telaprevir (Incivek; Vertex) were all obtained from MedchemExpress (Princeton, NJ, USA). The CypI alisporivir (ALV) and NIM811 were provided by Novartis Pharma, Basel, Switzerland. The mir-122i SPC3649 (71) was obtained from Exiqon, the NS5Bi 3′-deoxycytidine-5′-triphosphate (3′-dCTP) was obtained from TriLink Biotechnologies, and the PI4KIIIαi C23 was provided by R. De Francesco (72).
Plasmids and cells.
The full-genomic luciferase reporter replicon Luc-Neo-JFH-1 was created previously (60), as follows. The plasmid pFK-Luc-JFH1 was obtained from T. Wakita and T. Pietschmann (73–75), and the XbaI site in the firefly luciferase gene and the NotI site in the encephalomyocarditis virus (EMCV) internal ribosome entry site (IRES) were utilized to clone the luciferase/ubiquitin-neomycin phosphotransferase II (NPT II) fusion cassette out of pFK389ILuc-Neo (wild-type replicon from genotype 1b [GT1b]) (74, 75) and placed into the pFK-Luc-JFH1 plasmid, creating the full-length Luc-Neo-JFH-1 construct. The JFH1 sequence from NS3 through NS5B was subcloned from the pSGR-JFH1 subgenomic replicon construct into the pTM1 vector, as described previously (76). The pTM1 vector was designed for T7 RNA polymerase-driven protein expression and contains a T7 promoter and EMCV IRES upstream of a multiple cloning site and T7 transcriptional terminator. T7 RNA polymerase-expressing Huh7.5.1 cells (T7/Huh7.5.1 cells) were described previously (76). The T7 RNA polymerase-expressing pSMPUW-IRES-Bsd vector and pTM1(NS3-NS5B) plasmid were used for cotransfections of Huh7.5.1 cells. CypA knockdown (KD) Huh7.5.1 cells and CypA KD cells stably expressing either short hairpin RNA (shRNA)-resistant wild-type CypA or the shRNA-resistant isomerase-deficient H126Q CypA were described previously (33).
In vitro replicase assay.
Replication complexes (RCs) were isolated as described previously (77). An HCV in vitro replicase activity assay was carried out in a reaction mixture containing 20 mM Tris-HCl (pH 8), 10 mM MgCl2, 5 mM dithiothreitol, 5 mM KCl, 40 μg/ml actinomycin D, 16 mM creatine phosphate, 40 ng/μl creatine kinase, 1 U of RNasin, and a 10- or 30-μl RC sample fraction in a total volume of 50 μl at 35°C for 60 min. The nucleotide concentrations were as follows for different experimental setups: 1 mM (each) ATP, CTP, and UTP and 5 mM GTP for total HCV RNA quantification; 20 μCi of [α-32P]CTP, 10 μM CTP, 1 mM (each) ATP and UTP, and 5 mM GTP for the in vitro replicase activity assays and measurement by autoradiography. Radioactively labeled RNA reaction products were purified by phenol-chloroform extraction and isopropanol precipitation and then analyzed by denaturing glyoxal agarose gel electrophoresis prior to autoradiography, as described previously (77, 78). Drugs (1 or 10 μM) were added to the reaction mixture prior to the addition of the RCs. HCV RNA synthesis was quantified by autoradiography. Total synthesized HCV RNA was quantified by autoradiography before and after the replicase assay, and the data were represented as the fold increase, as previously reported (78).
DMV analyses.
After electroporation, Huh7.5.1 cells were plated in Falcon 35-mm dishes at 350,000/plate in triplicate and allowed to attach overnight. Anti-HCV drugs were applied for 24 h and the medium aspirated and replaced with fresh prewarmed medium to wash away any detached cells and cell debris. The cells were fixed with glutaraldehyde in cacodylate buffer. For the transfections, parental Huh7.5.1 cells or those expressing T7 RNA polymerase (T7/Huh7.5.1 cells) (76) were seeded at 1.2 × 106/10-cm dish for 24 h and transfected with 20 μg of pTM1(NS3-NS5B) (76) or NS5A mammalian expression construct (58) using Fugene, as per the manufacturer's (Promega Corp., Madison, WI) recommendations. After 24 h, the cells were split, counted, and seeded in triplicate to 35-mm dishes, followed by DAA or HTA treatment, as described above. Following drug treatments, hepatoma cells were fixed in 2.5% glutaraldehyde in 0.1 M cacodylate buffer and, after a brief buffer wash, were postfixed in cacodylate-buffered 1% osmium tetroxide with 1% potassium ferricyanide. Following a further buffer wash, the cells were dehydrated in ethanol series, transitioned in 2-hydroxypropyl methacrylate (HPMA), and embedded in LX112 (Ladd Research, Williston, VT). Pieces of the flat embedded resin-containing cells were glued to a blank block face, and 60-nm thin sections were cut, mounted on copper slot grids coated with Parlodion, and stained with uranyl acetate and lead citrate for examination on a Philips CM100 electron microscope (FEI, Hillsborough, OR) at 80 kV and images collected using a MegaView III CCD camera (Olympus Soft Imaging Solutions GmbH, Münster, Germany). Thin sections were cut in the direction parallel to the substrate, and the slice producing the largest nuclear diameter was analyzed, since this plane was generally found to contain the largest number of DMVs. Electron micrographs (between 20 and 100) covering the entire cross-section of the cell were recorded, and to facilitate counting, these were digitally merged to produce a single image representing a 60-nm-thick plane through the center of the infected cell. The merged images were analyzed using ImageJ software (http://rsb.info.nih.gov/ij/). For the detection of NS5A within the DMVs, immunogold labeling was conducted as described previously (79).
RESULTS
In contrast to NS5Bi, CypI and NS5Ai failed to block the replicase activity of isolated HCV replication complexes.
In order to determine at which steps CypI and NS5Ai and CypA and NS5A act during HCV RNA replication, we asked whether CypI and NS5Ai mediate their antiviral activities prior to forming enzymatically active RCs. Using a protocol developed by the Bartenschlager lab (77), we demonstrate significant replicase activity of the RCs isolated from HCV-infected cells (Fig. 1). The removal of ribonucleotide triphosphates (rNTPs) from the replicase reaction mixture decreases RNA replication in RCs (Fig. 1). The addition of NS5Bi, such as sofosbuvir and mericitabine, to RCs inhibits HCV RNA synthesis when used at a high concentration (10 μM; Fig. 1, top) but not at a low concentration (1 μM; Fig. 1, bottom). This suggests that high concentrations of NS5Bi are required for conversion to sufficient amounts of active triphosphates to HCV RNA synthesis. Supporting this notion, the active triphosphate 3′-dCTP efficiently blocks the replicase activity of isolated RCs, even at a low concentration (1 μM) (Fig. 1). The degree of NS5Bi-mediated inhibition correlates with the mass of the RCs used for the replicase reaction. Specifically, replicase inhibition is more profound when fewer RCs are used (Fig. 1). In contrast, CypI, including ALV and NIM811, exhibit no inhibitory effect, even at a high concentration (10 μM) (Fig. 1). Similarly, the NS5Ai daclatasvir and ledipasvir failed to block the replicase activity of RCs (Fig. 1). Shown at the top of Fig. 1 is a representative autoradiograph of HCV RNA synthesis by isolated RCs in the presence of a control (dimethyl sulfoxide [DMSO]), a CypI (ALV), an NS5Ai (daclatasvir), and an NS5Bi (3′-dCTP). Note that we used a drug concentration that completely prevents JFH-1 replication in vitro (10 μM) (80). These data suggest that CypI and NS5Ai exhibit no inhibitory activities on HCV RNA replication after the establishment of enzymatically active RCs. They also suggest that CypA and/or NS5A plays a major role in HCV RNA synthesis prior to RC formation.
FIG 1.
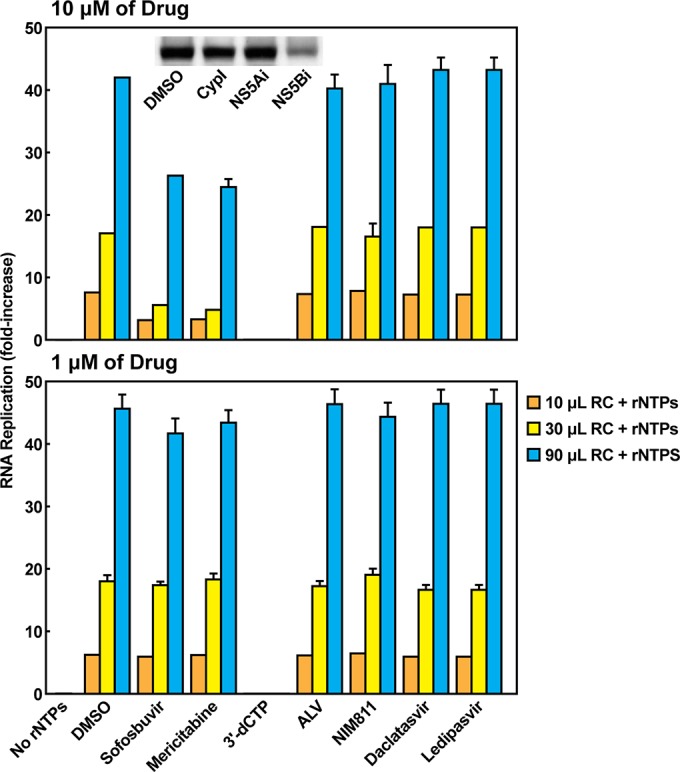
CypI and NS5Ai do not block HCV RNA synthesis of isolated replication complexes (RCs). RCs were isolated from JFH-1-infected Huh7.5.1 cells, as described previously (77, 89). HCV replicase activity was determined in a reaction mixture containing 20 mM Tris-HCl (pH 8), 10 mM MgCl2, 5 mM dithiothreitol (DTT), 5 mM KCl, 40 μg/ml actinomycin D, 20 μCi of [α-32P]CTP, 10 μM CTP, 1 mM (each) ATP and UTP, 5 mM GTP, 2.5 mM phosphoenolpyruvate, 1 U of pyruvate kinase, 1 U of RNasin, and 10, 30, or 90 μl of RC fraction in a total volume of 120 μl at 35°C for 60 min, as previously reported (77, 78). Drugs (10 or 1 μM) were added to the reaction mixture prior the addition of the RCs. HCV RNA synthesis was quantified by autoradiography, as illustrated in top panel, in which DMSO, the CypI ALV, the NS5Ai daclatasvir, and the NS5Bi 3′-dCTP were used with 90 μl of RCs. Total HCV RNA was quantified before and after the replicase assay and is represented as the fold increase, as previously reported (78). The error bars represent the standard errors of duplicates. The data are the mean values of five replicates from 3 independent experiments.
Requirements for HCV-mediated DMV formation.
With this goal in mind, we examined the impact of HCV on the formation of new endoplasmic reticulum (ER)-derived structures necessary to ensure efficient viral replication in a protected membranous compartment. To address this issue, Huh7.5.1 cells were transfected with full-genomic JFH-1 RNA and examined by electron microscopy (EM) analysis 3 days postelectroporation. We first confirmed that HCV mediates tremendous endoplasmic reticulum (ER) membrane reorganization close to the nucleus, the so-called membranous web (MW), as previously described (78, 81–84) (Fig. 2A, middle and bottom). No ER membrane rearrangement was observed in the cells electroporated without RNA (Fig. 2A, top). In addition to creating the MW, HCV also creates a large number of DMVs, which represent the main components of the MW (61, 76, 78, 81, 82). Figure 2B shows that HCV-formed DMVs are heterogeneous in terms of shape and size. We measured a diameter of 200 to 550 nm for typical spherical DMVs (Fig. 2C). We do not know at this stage whether or not this heterogeneity in DMV size influences HCV infection and RNA replication. We observed DMVs without (Fig. 2D, top) and with cytosol. At this stage, we do not know which of these structures more or less efficiently support HCV RNA replication. The viral composition of DMVs was recently reported (78). Specifically, the Bartenschlager lab generated a functional replicon containing a hemagglutinin (HA) affinity tag in NS4B, the supposed scaffold protein of the viral replication complex. Using HA-specific affinity purification, they were able to isolate DMVs, as demonstrated by EM. Moreover, they showed that affinity-purified DMVs contain NS5A, NS4B, NS3, and replicase activity (78). Using a DMV counting assay based on EM analysis, followed by ImageJ and the ITEM software for DMV quantification, we estimated that there are 200 to 400 DMVs per cell (Fig. 3A). Representative EM images are presented in Fig. 3A. We asked whether HCV replication is a precondition for DMV formation. To test this hypothesis, we took advantage of a T7 promoter-driven JFH-1 NS3-NS5B DNA plasmid [pTM1(NS3-5B)] developed by the Tai lab (76) that permits the expression of NS3, NS4A, NS4B, NS5A, and NS5B upon HCV polyprotein processing, as well as viral RNA synthesis when transfected in T7 RNA polymerase-expressing Huh7.5.1 cells (T7/Huh7.5.1 cells) (76). Importantly, the expression of the HCV NS3-NS5B polyprotein suffices to create both the MV and DMVs (76) (Fig. 3A). Our observation that both full-genomic JFH-1 and NS3-NS5B plasmid trigger the formation of similar numbers of DMVs (Fig. 3A) suggests not only that the NS3-NS5B polyprotein contains all sufficient HCV proteins necessary for DMV formation but also that viral RNA replication is not a precondition for the formation of this specific ER-derived organelle. Since both CypI and NS5Ai indirectly and directly target NS5A, we asked whether NS5A alone is capable of creating DMVs. We found that NS5A mediates the formation of DMVs upon Huh7.5.1 cell transfection, although at lower levels (∼6-fold) than full-genomic or NS3-NS5B JFH-1 (Fig. 3A), suggesting that NS5A is sufficient for DMV formation, albeit less efficiently in the absence of other nonstructural proteins. By reverse transcription-PCR (RT-PCR) (Fig. 3A, left) and Western blot (Fig. 3A, right) analyses, we found that pTM1(NS3-NS5B)-expressing cells contain higher levels of HCV RNA and NS5A protein than do cells electroporated with JFH-1 RNA (Fig. 3A). However, the numbers of DMVs induced by pTM1(NS3-NS5B) and JFH-1 are similar (Fig. 3A). Importantly, NS3-NS5B is unable to create DMVs in CypA knockdown (KD) Huh7.5.1 cells (Fig. 3B), which we previously created (33). Together, these findings strongly suggest that both NS5A and CypA are key viral and host factors for the establishment of this specific ER-derived compartment, DMVs. Moreover, we found that the reintroduction of shRNA-resistant wild-type CypA into the CypA KD cells restores NS3-NS5B-mediated DMV formation (Fig. 3B), further suggesting that CypA is a critical host factor for HCV-mediated DMV creation. As we previously reported (33), the reintroduction of the shRNA-resistant wild-type CypA into the CypA KD cells rescues both CypA expression and HCV RNA replication (Fig. 3B, lower images). Importantly, the reintroduction of shRNA-resistant isomerase-deficient H126Q CypA into the CypA KD cells fails to restore DMV formation (Fig. 3B), suggesting that the isomerase activity of CypA is vital for HCV-mediated DMV creation. This is in accordance with our previous work showing that the reintroduction of shRNA-resistant wild-type, but not isomerase-deficient H126Q CypA, into the CypA KD cells restores HCV replication (33).
FIG 2.

HCV mediates formation of MW and DMVs. (A) Huh7.5.1 cells were electroporated with (middle and bottom) or without (top) full-genomic JFH-1 replicon and analyzed by EM for MW formation. (B) DMVs are shown at higher magnification to illustrate shape heterogeneity. LD, lipid droplet; MMV, multiple-membrane vesicles; N, nucleus. (C) Illustration of the diameter of a typical spherical HCV (two diameter measurements in green and red). (D) Examples of closed (top panel) and opened (bottom panel) DMVs are presented. The DMVs are representative of >20 independent experiments. (E) NS5A is labeled with 10-nm gold particles within DMVs.
FIG 3.

Both NS5A and the isomerase activity of CypA govern DMV formation. (A) Parental or T7 polymerase-expressing Huh7.5.1 cells were transfected in triplicate with either full-genomic JFH-1 RNA, NS3-NS5B, or NS5A JFH-1 DNA plasmids and analyzed by EM as well as by ImageJ and the ITEM software for DMV quantification. Illustrated EM images of spherical or more tubular DMVs are presented. The two lower images display RNA and protein levels of NS5A in cells expressing JFH-1, NS3-NS5B, or NS5A. A quantitative RT-PCR using primers targeting the NS5A region was conducted, as we described previously (33). RNA levels of HCV and NS5A were normalized to GAPDH RNA levels. The mean normalized values from triplicate samples are plotted; the error bars indicate the standard deviation. Western blot analysis of CypA was conducted, as we described previously (33). (B) Parental Huh7.5.1 cells, CypA-KD cells, CypA-KD cells expressing shRNA for CypA-resistant wild-type CypA, or CypA-KD cells expressing shRNA for CypA-resistant isomerase-deficient H126Q CypA were cotransfected in triplicate with NS3-NS5B JFH-1 [pTM1(NS3-NS5B)] and T7 polymerase-expressing DNA plasmids and analyzed by EM as well as by ImageJ and the ITEM software for DMV quantification. Illustrated EM images are presented. The two lower images display a rescue analysis of CypA expression and HCV RNA replication in CypA-KD cells transfected with shRNA-resistant wild-type or isomerase-deficient H126Q CypA plasmids, as we described previously (33). The data are representative of two independent experiments.
CypI and NS5Ai, but not NS5Bi, Mir-122i, or PI4KIIIαi, prevent HCV-mediated DMV formation.
Our data above suggest that both NS5A and CypA are necessary for the formation of DMVs. We then asked whether other classes of anti-HCV agents are capable of preventing the formation of these ER-derived organelles. Specifically, we tested a panel of DAAs and HTAs for their effects on NS3-NS5B-mediated DMV creation. We used the NS3-NS5B DNA plasmid rather than full-genomic JFH-1 in order to examine the specific and direct impact of DAAs and HTAs on the creation of DMVs that occurs independent of viral replication and polyprotein synthesis. Importantly, we found that all tested CypI (cyclosporine [CsA], ALV, and NIM811) profoundly decrease the NS3-NS5B-mediated formation of DMVs compared to that with the DMSO control (Fig. 4A). In contrast, two other HTAs, the mir-122i SPC3649 and the PI4KIIIαi C23, exhibit no significant inhibitory effect on DMV formation (Fig. 4A). In terms of DAAs, the two tested NS5Ai (daclatasvir and ledipasvir), like CypI, greatly diminish the numbers of de novo-formed DMVs, whereas the two tested NS5Bi (sofosbuvir and mericitabine) have no effect (Fig. 4A). These data indicate that only specific classes of anti-HCV agents prevent the NS3-NS5B-mediated formation of DMVs and further suggest that both CypA and NS5A control DMV formation by HCV. The CypI- and NS5Ai-mediated inhibition of NS3-NS5B- or NS5A-mediated DMV formation is dose dependent (Fig. 4B). This also further supports that our DMV counting assay is both reliable and sensitive. We found that the CypI ALV and the NS5Ai daclatasvir do not inhibit the formation of DMVs created by the CypI-resistant or the NS5Ai-resistant NS5A protein, respectively (Fig. 4C), further demonstrating that DMVs block specificity by these two distinct classes of HCV inhibitors.
FIG 4.

CypI and NS5Ai, but not other anti-HCV agents, prevent HCV-mediated DMV formation in a dose-dependent manner. (A) T7 polymerase-expressing Huh7.5.1 cells were transfected in triplicate with NS3-NS5B JFH-1 DNA plasmid in the presence or absence of selected anti-HCV agents (2 μM) and analyzed by EM, as well as by ImageJ and the ITEM software for DMV quantification. Illustrated EM images are presented. (B) Decreasing concentrations of the CypI ALV (in μM) or the NS5Ai daclatasvir (in nM) were used on NS3-NS5B- or NS5A-created DMVs. Graph curves of the data are presented. (C) ALV or daclatasvir was used on wild-type (WT), NS5Ai-resistant (Y93H), or CypI-resistant (D316F/Y317N) NS5A-created DMVs. The data are representative from three independent experiments.
DISCUSSION
CypI and NS5Ai are two classes of anti-HCV agents that we previously showed in vitro to demonstrate strong synergy for anti-HCV activity (80). However, the precise MoA remains poorly understood. In this study, we conducted a set of experiments aimed at gaining an understanding of their MoA during the early steps of HCV replication. Using an in vitro replicase assay, we obtained indirect evidence that both CypA and NS5A play critical roles in HCV RNA replication prior to the establishment of enzymatically active RCs. Specifically, we showed that in contrast to NS5Bi, CypI and NS5Ai do not block the replicase activity of isolated RCs. These observations do not exclude the possibility that either CypA or NS5A plays a role in the later stages of HCV replication, such as particle assembly and release, as suggested by others (85–88). We previously showed that CypI, such as CsA and ALV, deplete isolated RCs of CypA (89). Thus, our data suggest that CypA has no main function in HCV RNA replication after the establishment of active RCs. Note our somehow surprising finding that NS5Bi used at a high concentration (10 μM) inhibit the replicase activity of isolated RCs; this suggests that some nucleo(t)sides are converted by kinases present in the “crude” RC mixture (e.g., contaminating mitochondria known to convert nucleosides into nucleoside triphosphates). We were able to detect by liquid chromatography-tandem mass spectrometry (LC-MS/MS) the active form of sofosbuvir, GS-461203, in RCs (50 μl) 2 h after addition of 10 μM sofosbuvir (data not shown). No GS-461203 was detected in the absence of RCs. The GS-461203 levels in RCs were ∼1,000-fold less abundant than those in the cell extracts (37 femtomoles [fmol]/106 cells versus 51 pmol/106 cells), suggesting that the conversion of sofosbuvir into its active form is poor in crude RCs but sufficient enough to provide a very small amount of GS-461203 capable of neutralizing the replicase activity of crude RCs.
Since HCV modulates the structure of the ER to facilitate its RNA replication, we examined the impact of CypI and NS5Ai on the HCV-mediated formation of new membranous organelles necessary for viral replication. In accordance with recent reports (61, 76, 78, 81, 82), we found that HCV creates a large number of organelles in the MW called DMVs. We obtained evidence that viral replication is not a precondition for DMV formation. Indeed, the expression of the NS3-NS5B polyprotein creates DMVs of similar numbers, shapes, and sizes to those created by full-length replicating HCV. The important roles of NS5A and CypA in DMV formation are demonstrated by the induction of DMVs in cells expressing NS5A alone and the inhibition of DMV formation by the suppression of CypA expression. We also obtained evidence that the isomerase activity of CypA is required for DMV formation. This is in accordance with data from our lab and others that suggest the importance of the isomerase pocket of CypA in affecting HCV replication (32–35). In accordance with previous work from the Bartenschlager lab (61, 78, 81, 82), we observed that the number and size of the DMVs created by NS5A are lower than those created by HCV or NS3-NS5B.
The formation of DMVs is a novel function for NS5A, since although NS5A was recognized as a membrane-associated protein, its ability to alter the ER-derived structure was unrecognized. Previous studies identified an N-terminal amphipathic helix in NS5A necessary for membrane localization and being conserved across isolates (90). Genetically disrupting the amphipathic helix impairs HCV replication (90). In collaboration with the Chisari lab, we reported that an 18-amino-acid peptide (C5A) derived from the amphipathic α-helical N-terminal membrane anchor domain of NS5A ruptures the membrane integrity of HCV, HIV-1, and herpes simplex virus 1 (HSV) (91–93). Thus, NS5A may possess the ability to modify the structure and/or shape of its ER membranes. It will be interesting to determine whether NS5A generates DMVs in nonhepatoma cells, such as 293T cells, or whether this phenotype is unique to hepatocyte-like cells. Our finding that NS3-NS5B is unable to form DMVs in CypA KD cells or in isomerase-deficient CypA cells suggests that CypA via its isomerase acts in concert with NS5A to govern DMV formation. Since CypA via its isomerase pocket binds to the domain II of NS5A and CypI prevent these contacts, it is likely that CypA-NS5A interactions are critical for DMV formation.
We found that both CypI and NS5Ai prevent NS3-NS5B-mediated DMV formation, whereas NS5Bi, mir-122i, and PI4KIIIαI do not exhibit an inhibitory effect. This is important because it suggests that although CypI and NS5Ai are structurally unrelated and target distinct proteins (CypA and NS5A) they exert a similar antiviral MoA. CypI might indirectly target NS5A, since CypA binds NS5A and might influence its activity in HCV RNA replication. This would be in accordance with our hypothesis that NS5A-CypA interactions govern DMV formation. The inhibition of DMV formation by NS5Ai is more obscure, since we showed that NS5Ai do not block NS5A-CypA interactions (58). Although recently challenged (94), one study has reported that NS5Ai inhibit the association between NS5A and PI4KIIIα (62). It has been proposed that the binding of NS5Ai to NS5A induces a conformational change in NS5A that decreases the association between the viral protein and PI4KIIIα (62, 63). Thus, it will be interesting to determine whether NS5A-PI4KIIIα interactions, like NS5A-CypA interactions, also control DMV formation. It is important to emphasize that PI4KIIIαi (i.e., AL-9) do not block NS5A-PI4KIIIα interactions, whereas NS5Ai do (62). Note that in contrast to a previous study, which used the PI4KIIIαi AL-9 (62), we found here that the PI4KIIIαI C23 does not block DMV formation at a drug concentration that totally blocks both HCV replication and HCV protein expression (100 nM). This apparent discrepancy may arise from drug solubility issues, as suggested previously (62), or from distinct experimental methodologies. However, we found that C23 reduces the size/diameter and cellular distribution of DMVs, as previously reported for AL-9 (62, 95, 96) (data not shown). Since PI4KIIIαi block HCV replication, the latest observation may suggest that DMV size is a critical parameter in HCV replication.
During the course of this study, the Bartenschlager lab (61, 82) reported that the CypI cyclosporine D and the NS5Ai daclatasvir block HCV-created DMVs in hepatoma cells, which is consistent with our own findings. After a detailed analysis of the three-dimensional (3D) morphology and biogenesis of intracellular membrane structures induced by HCV, the Bartenschlager lab identified lasso-like structures of DMVs, with a pore-like opening that connects the interior of the DMV with the cytosol (81), which is very similar to those shown in Fig. 2D (bottom). Moreover, they obtained evidence that the biogenesis and morphology of MWs created by HCV are unexpectedly similar to those of distantly related coronaviruses and arteriviruses (81). Ultimately, they proposed that this similarity may reflect a common use of host cell pathways for biogenesis and functionality of the membranous structures induced by these viruses (81). Our findings suggest that two distinct classes of anti-HCV agents, CypI and NS5Ai, inhibit the same step of HCV replication but have distinct targets, CypA and NS5A, respectively. How can these findings be reconciled? One possibility is that CypA, by binding to and mediating the proper folding of NS5A, enhances the ability of the ER membrane-associated viral protein to drive the creation of DMVs (Fig. 5, middle model). In this model, CypI, by preventing CypA-NS5A contact, prevent NS5A-mediated DMV creation and therefore HCV RNA synthesis (Fig. 5, left model). In a similar model, NS5Ai directly bind to NS5A and thus interfere with the ability of NS5A to create DMVs (Fig. 5, right panel). We and others previously demonstrated that NS5A dimerizes and that NS5A dimerization remains resistant to both CypI and NS5Ai (58). Thus, the prevention of NS5A dimerization or NS5A-CypA contacts cannot explain the block in DMV formation by NS5Ai. However, NS5Ai, by binding to the domain I of NS5A that contains the ER membrane anchor, may interfere with the shaping/bending of the ER membrane for DMV formation either by interfering with the intrinsic capacity of NS5A to shape the ER membrane or by interfering with the association of NS5A with host ligands having ER membrane-bending properties. Since PI4KIIIαi might be one of those host NS5A ligands, it will be interesting to examine its impact on HCV-mediated DMV formation.
FIG 5.

Models for the MoA of CypI and NS5Ai on HCV-mediated DMV formation and viral RNA replication. In the middle model, in the absence of inhibitors, HCV components accumulate at the ER via their membrane anchor domains and initiate the formation of enzymatically active replication complexes (RCs) within newly formed protective DMVs. In the left model, due to the binding of CypI to the isomerase pocket of CypA, the host isomerase protein is incapable of interacting with the domain II of NS5A during RC formation. Without a proper folding by CypA, NS5A is unable to drive DMV formation, where HCV RNA synthesis and replication normally occur in a shielded compartment, leading to an abortive replication. In the right model, due to the binding of NS5Ai to the domain I of NS5A, the viral protein for a still undefined reason is unable to drive DMV formation. In the absence of a protective cellular compartment, RCs, especially viral RNA, are completely exposed to cellular sensors of foreign RNA, as well as to cellular-degrading factors of viral components.
In conclusion, our results demonstrate that CypA and NS5A are both required for DMV formation and suggest that CypI and NS5Ai mediate their early anti-HCV effects by preventing the formation of HCV replication organelles. This study provides the first demonstration that the isomerase activity of CypA is vital for HCV-mediated DMV formation. This is the first comprehensive investigation examining the effect of a large panel of anti-HCV agents on DMV formation, and the results reveal that CypI and NS5Ai act at the same MW biogenesis step of HCV RNA replication, and they indicate a new therapeutic target for chronic hepatitis C infection.
ACKNOWLEDGMENTS
We thank F. Chisari for the Huh7.5.1 cells, T. Pietschmann, T. Wakita, and R. Bartenschlager for the Luc-JFH-1 plasmid, Novartis for alisporivir and NIM811, C. Rice for the anti-NS5A 9E10 antibody, and R. De Francesco for C23.
This work was supported by the U.S. Public Health Service grant AI087746 from the National Institute of Allergy and Infectious Diseases (NIAID) and by a research grant from Novartis Pharma, Basel, Switzerland (SFP-1599).
This is publication no. 29009 from the Department of Immunology & Microbial Science, The Scripps Research Institute, La Jolla, CA.
REFERENCES
- 1.Dienstag JL, McHutchison JG. 2006. American Gastroenterological Association technical review on the management of hepatitis C. Gastroenterology 130:231–264. doi: 10.1053/j.gastro.2005.11.010. [DOI] [PubMed] [Google Scholar]
- 2.Alter MJ. 2007. Epidemiology of hepatitis C virus infection. World J Gastroenterol 13:2436–2441. doi: 10.3748/wjg.v13.i17.2436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Soriano V, Madejon A, Vispo E, Labarga P, Garcia-Samaniego J, Martin-Carbonero L, Sheldon J, Bottecchia M, Tuma P, Barreiro P. 2008. Emerging drugs for hepatitis C. Expert Opin Emerg Drugs 13:1–19. doi: 10.1517/14728214.13.1.1. [DOI] [PubMed] [Google Scholar]
- 4.Shepard CW, Finelli L, Alter MJ. 2005. Global epidemiology of hepatitis C virus infection. Lancet Infect Dis 5:558–567. doi: 10.1016/S1473-3099(05)70216-4. [DOI] [PubMed] [Google Scholar]
- 5.Armstrong GL, Wasley A, Simard EP, McQuillan GM, Kuhnert WL, Alter MJ. 2006. The prevalence of hepatitis C virus infection in the United States, 1999 through 2002. Ann Intern Med 144:705–714. doi: 10.7326/0003-4819-144-10-200605160-00004. [DOI] [PubMed] [Google Scholar]
- 6.Cross TJ, Antoniades CG, Harrison PM. 2008. Current and future management of chronic hepatitis C infection. Postgrad Med J 84:172–176. doi: 10.1136/pgmj.2008.068205. [DOI] [PubMed] [Google Scholar]
- 7.Manns MP, Wedemeyer H, Cornberg M. 2006. Treating viral hepatitis C: efficacy, side effects, and complications. Gut 55:1350–1359. doi: 10.1136/gut.2005.076646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sy T, Jamal MM. 2006. Epidemiology of hepatitis C virus (HCV) infection. Int J Med Sci 3:41–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tong MJ, Reddy KR, Lee WM, Pockros PJ, Hoefs JC, Keeffe EB, Hollinger FB, Hathcote EJ, White H, Foust RT, Jensen DM, Krawitt EL, Fromm H, Black M, Blatt LM, Klein M, Lubina J. 1997. Treatment of chronic hepatitis C with consensus interferon: a multicenter, randomized, controlled trial. Consensus Interferon Study Group. Hepatology 26:747–754. [DOI] [PubMed] [Google Scholar]
- 10.Pawlotsky JM. 2014. New hepatitis C virus (HCV) drugs and the hope for a cure: concepts in anti-HCV drug development. Semin Liver Dis 34:22–29. doi: 10.1055/s-0034-1371007. [DOI] [PubMed] [Google Scholar]
- 11.Fischer G, Gallay P, Hopkins S. 2010. Cyclophilin inhibitors for the treatment of HCV infection. Curr Opin Investig Drugs 11:911–918. [PubMed] [Google Scholar]
- 12.Gallay PA. 2012. Cyclophilin inhibitors: a novel class of promising host-targeting anti-HCV agents. Immunol Res 52:200–210. doi: 10.1007/s12026-011-8263-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lin K. 2010. Development of novel antiviral therapies for hepatitis C virus. Virol Sin 25:246–266. doi: 10.1007/s12250-010-3140-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pawlotsky JM. 2012. New antiviral agents for hepatitis C. F1000 Biol Rep 4:5. doi: 10.3410/B4-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pockros PJ. 2010. New direct-acting antivirals in the development for hepatitis C virus infection. Ther Adv Gastroenterol 3:191–202. doi: 10.1177/1756283X10363055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Vermehren J, Sarrazin C. 2011. New hepatitis C therapies in clinical development. Eur J Med Res 16:303–314. doi: 10.1186/2047-783X-16-7-303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.von Hahn T, Ciesek S, Manns MP. 2011. Arrest all accessories–inhibition of hepatitis C virus by compounds that target host factors. Discov Med 12:237–244. [PubMed] [Google Scholar]
- 18.Lin K, Gallay P. 2013. Curing a viral infection by targeting the host: the example of cyclophilin inhibitors. Antiviral Res 99:68–77. doi: 10.1016/j.antiviral.2013.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gallay PA, Lin K. 2013. Profile of alisporivir and its potential in the treatment of hepatitis C. Drug Des Dev Ther 7:105–115. doi: 10.2147/DDDT.S30946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Flisiak R, Horban A, Gallay P, Bobardt M, Selvarajah S, Wiercinska-Drapalo A, Siwak E, Cielniak I, Higersberger J, Kierkus J, Aeschlimann C, Grosgurin P, Nicolas-Métral V, Dumont JM, Porchet H, Crabbé R, Scalfaro P. 2008. The cyclophilin inhibitor Debio-025 shows potent anti-hepatitis C effect in patients coinfected with hepatitis C and human immunodeficiency virus. Hepatology 47:817–826. doi: 10.1002/hep.22131. [DOI] [PubMed] [Google Scholar]
- 21.Hopkins S, DiMassimo B, Rusnak P, Heuman D, Lalezari J, Sluder A, Scorneaux B, Mosier S, Kowalczyk P, Ribeill Y, Baugh J, Gallay P. 2012. The cyclophilin inhibitor SCY-635 suppresses viral replication and induces endogenous interferons in patients with chronic HCV genotype 1 infection. J Hepatol 57:47–54. doi: 10.1016/j.jhep.2012.02.024. [DOI] [PubMed] [Google Scholar]
- 22.Lawitz E, Godofsky E, Rouzier R, Marbury T, Nguyen T, Ke J, Huang M, Praestgaard J, Serra D, Evans TG. 2011. Safety, pharmacokinetics, and antiviral activity of the cyclophilin inhibitor NIM811 alone or in combination with pegylated interferon in HCV-infected patients receiving 14 days of therapy. Antiviral Res 89:238–245. doi: 10.1016/j.antiviral.2011.01.003. [DOI] [PubMed] [Google Scholar]
- 23.Herrmann E, Kafer A, Flisiak R, Nicolas-Metral V, Zeuzem S, Crabbe R. 2009. PK-PD modeling of viral kinetics during treatment with Debio-025 plus pegylated interferon α-2a in treatment-naive HCV patients. J Hepatol 50(Suppl 1):S344. doi: 10.1016/S0168-8278(09)60948-X. [DOI] [Google Scholar]
- 24.Flisiak R, Feinman SV, Jablkowski M, Horban A, Kryczka W, Pawlowska M, Heathcote JE, Mazzella G, Vandelli C, Nicolas-Métral V, Grosgurin P, Liz JS, Scalfaro P, Porchet H, Crabbé R. 2009. The cyclophilin inhibitor Debio 025 combined with Peg-IFN2a significantly reduces viral load in treatment naïve hepatitis C patients. Hepatology 49:1460–1468. doi: 10.1002/hep.22835. [DOI] [PubMed] [Google Scholar]
- 25.Flisiak R, Pawlotsky JM, Crabbe R, Calistru PI, Kryczka W, Haussinger D, Mazzella G, Romero-Gomez M, Purcea D, Vuagniaux G, Bao W, Avila C, Zeuzem S. 2011. Once daily alisporivir (DEB025) plus PEG-IFN-alfa-2A/ribavirin results in superior sustained virologic response (SVR24) in chronic hepatitis C genotype 1 treatment-naive patients—the ESSENTIAL study, abstr 190 EASL 46th Annu Meet, 30 March to 3 April 2011, Berlin, Germany. [Google Scholar]
- 26.Pawlotsky JM, Sarin SK, Foster GR, Peng CY, Rasenack J, Flisiak R, Piratvisuth T, Wedemeyer H, Chuang WL, Zhang WM, Naoumov NV. 2012. Alisporivir plus ribavirin is highly effective as interferon-free or interferon-add-on regimen in previously untreated HCV-G2 or G3 patients: SVR12 results from VITAL-1 phase 2b study, abstr 1405 J Hepatol 56(Suppl 2):S553. doi: 10.1016/S0168-8278(12)61416-0. [DOI] [Google Scholar]
- 27.Pawlotsky JM, Sarin SK, Foster GR, Peng CY, Rasenack J, Flisiak R, Piratvisuth T, Wedemeyer H, Chuang WL, Zhang WM, Naoumov NV. 2012. Alisporivir plus ribavirin achieves high rates of sustained HCV clearance (SVR24) as interferon (IFN)-free or IFN-add-on regimen in treatment-naive patients with HCV GT2 or GT3: final results from VITAL-1 study, abstr 233 63rd Annu Meet Am Assoc Study Liver Dis, 9 to 12 November 2012, Boston, MA. [Google Scholar]
- 28.Alberti M, Chuang WL, Flisiak R, Mazella G, Horban A, Goeser T, Calistru Buti PM, Davis G, Gong Y. 2012. Alisporivir (ALV) plus PEG-interferon/ribavirin (PR) in HCV G1 treatment-experienced patients achieves primary endpoint with superior efficacy at treatment week 12 compared to retreatment with PR, abstr 1406. J Hepatol 56(Suppl 2):S553–S554. doi: 10.1016/S0168-8278(12)61417-2. [DOI] [Google Scholar]
- 29.Buti M, Flisiak R, Kao JH, Chuang WL, Streinu-Cercel A, Tabak F, Calistru P, Goeser T, Rasenack J, Horban A, Davis GL, Alberti A, Mazzella G, Pol S, Orsenigo R, Brass C. 2014. Alisporivir with peginterferon/ribavirin in patients with chronic hepatitis C genotype 1 infection who failed to respond to or relapsed after prior interferon-based therapy: FUNDAMENTAL, a phase II trial. J Viral Hepat, in press. doi: 10.1111/jvh.12360. [DOI] [PubMed] [Google Scholar]
- 30.Griffel L, Bao W, Orsenigo R, Guo V, Wu M, Loeffler C, Brass C, Avila C, Naoumov NV. 2013. Interferon (IFN)-free alisporivir has a better overall safety profile compared to IFN-containing treatment: a pooled analysis of the ALV development program. J Hepatol 58(Suppl 1):S336–S337. doi: 10.1016/S0168-8278(13)60823-5. [DOI] [Google Scholar]
- 31.Naoumov NV. 2014. Cyclophilin inhibition as potential therapy for liver diseases. J Hepatol 61:1166–1174. doi: 10.1016/j.jhep.2014.07.008. [DOI] [PubMed] [Google Scholar]
- 32.Yang F, Robotham JM, Nelson HB, Irsigler A, Kenworthy R, Tang H. 2008. Cyclophilin A is an essential cofactor for hepatitis C virus infection and the principal mediator of cyclosporine resistance in vitro. J Virol 82:5269–5278. doi: 10.1128/JVI.02614-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chatterji U, Bobardt M, Selvarajah S, Yang F, Tang H, Sakamoto N, Vuagniaux G, Parkinson T, Gallay P. 2009. The isomerase active site of cyclophilin A is critical for hepatitis C virus replication. J Biol Chem 284:16998–17005. doi: 10.1074/jbc.M109.007625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kaul A, Stauffer S, Berger C, Pertel T, Schmitt J, Kallis S, Zayas M, Lohmann V, Luban J, Bartenschlager R. 2009. Essential role of cyclophilin A for hepatitis C virus replication and virus production and possible link to polyprotein cleavage kinetics. PLoS Pathog 5:e1000546. doi: 10.1371/journal.ppat.1000546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Liu Z, Yang F, Robotham JM, Tang H. 2009. Critical role of cyclophilin A and its prolyl-peptidyl isomerase activity in the structure and function of the hepatitis C virus replication complex. J Virol 83:6554–6565. doi: 10.1128/JVI.02550-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hanoulle X, Badillo A, Wieruszeski JM, Verdegem D, Landrieu I, Bartenschlager R, Penin F, Lippens G. 2009. Hepatitis C virus NS5A protein is a substrate for the peptidyl-prolyl cis/trans isomerase activity of cyclophilins A and B. J Biol Chem 284:13589–13601. doi: 10.1074/jbc.M809244200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chatterji U, Lim P, Bobardt MD, Wieland S, Cordek DG, Vuagniaux G, Chisari F, Cameron CE, Targett-Adams P, Parkinson T, Gallay PA. 2010. HCV resistance to cyclosporin A does not correlate with a resistance of the NS5A-cyclophilin A interaction to cyclophilin inhibitors. J Hepatol 53:50–56. doi: 10.1016/j.jhep.2010.01.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Coelmont L, Hanoulle X, Chatterji U, Berger C, Snoeck J, Bobardt M, Lim P, Vliegen I, Paeshuyse J, Vuagniaux G, Vandamme AM, Bartenschlager R, Gallay P, Lippens G, Neyts J. 2010. DEB025 (alisporivir) inhibits hepatitis C virus replication by preventing a cyclophilin A induced cis-trans isomerisation in domain II of NS5A. PLoS One 5:e13687. doi: 10.1371/journal.pone.0013687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fernandes F, Ansari IU, Striker R. 2010. Cyclosporine inhibits a direct interaction between cyclophilins and hepatitis C NS5A. PLoS One 5:e9815. doi: 10.1371/journal.pone.0009815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Foster TL, Gallay P, Stonehouse NJ, Harris M. 2011. Cyclophilin A interacts with domain II of hepatitis C virus NS5A and stimulates RNA binding in an isomerase-dependent manner. J Virol 85:7460–7464. doi: 10.1128/JVI.00393-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gregory MA, Bobardt M, Obeid S, Chatterji U, Coates NJ, Foster T, Gallay P, Leyssen P, Moss SJ, Neyts J, Nur-e-Alam M, Paeshuyse J, Piraee M, Suthar D, Warneck T, Zhang MQ, Wilkinson B. 2011. Preclinical characterization of naturally occurring polyketide cyclophilin inhibitors from the sanglifehrin family. Antimicrob Agents Chemother 55:1975–1981. doi: 10.1128/AAC.01627-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Verdegem D, Badillo A, Wieruszeski JM, Landrieu I, Leroy A, Bartenschlager R, Penin F, Lippens G, Hanoulle X. 2011. Domain 3 of NS5A protein from the hepatitis C virus has intrinsic alpha-helical propensity and is a substrate of cyclophilin A. J Biol Chem 286:20441–20454. doi: 10.1074/jbc.M110.182436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yang F, Robotham JM, Grise H, Frausto S, Madan V, Zayas M, Bartenschlager R, Robinson M, Greenstein AE, Nag A, Logan TM, Bienkiewicz E, Tang H. 2010. A major determinant of cyclophilin dependence and cyclosporine susceptibility of hepatitis C virus identified by a genetic approach. PLoS Pathog 6:e1001118. doi: 10.1371/journal.ppat.1001118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Moss SJ, Bobardt M, Leyssen P, Coates N, Chatterji U, Dejian X, Foster T, Liu J, Nur-e-Alam M, Suthar D, Yongsheng C, Warneck T, Neyts J, Gallay P, Wilkinson B, Gregory MA. 2012. Sangamides, a new class of cyclophilin inhibiting host targeted antivirals for treatment of HCV infection. MedChemComm 3:944–949. doi: 10.1039/c1md00227a. [DOI] [Google Scholar]
- 45.Hopkins S, Bobardt M, Chatterji U, Garcia-Rivera JA, Lim P, Gallay PA. 2012. The cyclophilin inhibitor SCY-635 disrupts hepatitis C virus NS5A-cyclophilin A complexes. Antimicrob Agents Chemother 56:3888–3897. doi: 10.1128/AAC.00693-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Feeney ER, Chung RT. 2014. Antiviral treatment of hepatitis C. BMJ 348:g3308. doi: 10.1136/bmj.g3308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Izumi N. 2014. Efficacy of daclatasvir in hepatitis C virus. Expert Rev Anti Infect Ther 12:1025–1031. doi: 10.1586/14787210.2014.942282. [DOI] [PubMed] [Google Scholar]
- 48.Kim do Y, Ahn SH, Han KH. 2014. Emerging therapies for hepatitis C. Gut Liver 8:471–479. doi: 10.5009/gnl14083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Manns M, Pol S, Jacobson IM, Marcellin P, Gordon SC, Peng CY, Chang TT, Everson GT, Heo J, Gerken G, Yoffe B, Towner WJ, Bourliere M, Metivier S, Chu CJ, Sievert W, Bronowicki JP, Thabut D, Lee YJ, Kao JH, McPhee F, Kopit J, Mendez P, Linaberry M, Hughes E, Noviello S, HALLMARK-DUAL Study Team. 2014. All-oral daclatasvir plus asunaprevir for hepatitis C virus genotype 1b: a multinational, phase 3, multicohort study. Lancet 384:1–7. doi: 10.1016/S0140-6736(14)61059-X. [DOI] [PubMed] [Google Scholar]
- 50.Kapoor R, Kohli A, Sidharthan S, Sims Z, Petersen TL, Osinusi A, Nelson AK, Silk R, Kotb C, Sugarman K, Lam BP, Pang PS, Subramanian M, McHutchison JG, Masur H, Kottilil S, Rustgi VK. 2014. Treatment of hepatitis C genotype 4 with ledipasvir and sofosbuvir for 12 weeks: results of the SYNERGY trial, abstr 240 65th Annu Meet Am Assoc Study Liver Dis (AASLD), 7 to 12 November 2014, Boston, MA. [Google Scholar]
- 51.Pol S, Reddy R, Hezode C, Hassanein T, Marcellin P, Berenguer M, Fleischer-Stepniewska K, Mobashery N, Hall C, Collins C, Vilchez R. 2014. Interferon-free regimens of smbitasvir and ABT-450/r with or without ribavirin in patients with HCV genotype 4 infection: PEARL-I study results, abstr 1928 65th Annu Meet Am Assoc Study Liver Dis (AASLD), 7 to 12 November 2014, Boston, MA. [Google Scholar]
- 52.Lawitz E, Gane EJ, Pearlman B, Tam E, Ghesquiere W, Guyader D, Alric L, Bronowicki JP, Lester L, Sievert W, Ghalib RH, Balart LA, Sund F, Lagging M, Dutko F, Howe A, Shaughnessy M, Hwang P, Wahl J, Robertson M, Barr E, Haber B. 2014. Efficacy and safety of MK-5172 and MK-8742 ± ribavirin in hepatitis C genotype 1 infected patients with cirrhosis or previous null response: final results of the C-WORTHY study (parts A and B), abstr 196 65th Annu Meet Am Assoc Study Liver Dis (AASLD), 7 to 12 November 2014, Boston, MA. [Google Scholar]
- 53.Sulkowski MS, Hezode C, Gerstoft J, Vierling JM, Mallolas J, Pol S, Kugelmas M, Murillo A, Weis N, Nahass R, Shibolet O, Serfaty L, Bourlière M, DeJesus E, Zuckerman E, Dutko F, Howe A, Shaughnessy M, Hwang P, Wahl J, Robertson M, Barr E, Haber B. 2014. Efficacy and safety of MK-5172 + MK-8742 ± ribavirin in HCV mono-infected and HIV/HCV co-infected treatment-naive, non-cirrhotic patients with HCV GT1 infection: the C-WORTHY study (final results, parts A and B), abstr 236 65th Annu Meet Am Assoc Study Liver Dis (AASLD), 7 to 12 November 2014, Boston, MA. [Google Scholar]
- 54.Lawitz E, Poordad F, Gutierrez JA, Evans B, Hwang P, Howe A, Feng HP, Robertson M, Wahl J, Barr E, Haber B. 2014. C-SWIFT: MK-5172 + MK-8742 + sofosbuvir in treatment-naive patients with hepatitis C virus genotype 1 infection, with and without cirrhosis, for durations of 4, 6, or 8 weeks. 65th Annu Meet Am Assoc Study Liver Dis (AASLD), 7 to 12 November 2014, Boston, MA. [Google Scholar]
- 55.Lawitz E, Gane E, Pearlman B, Tam E, Ghesquiere W, Guyader D, Laurent Alric Bronowicki J-P, Lester L, Sievert W, Ghalib R, Balart L, Sund F, Lagging M, Dutko F, Shaughnessy M, Hwang P, Anit Howe AYM, Wahl J, Robertson M, Barr E, Haber B. 2014. Efficacy and safety of 12 weeks versus 18 weeks of treatment with grazoprevir (MK-5172) and elbasvir (MK-8742) with or without ribavirin for hepatitis C virus genotype 1 infection in previously untreated patients with cirrhosis and patients with previous null response with or without cirrhosis (C-WORTHY): a randomized, open-label phase 2 trial. Lancet, in press. doi: 10.1016/S0140-6736(14)61795-5. [DOI] [PubMed] [Google Scholar]
- 56.Gane EJ, Kocinsky H, Schwabe C, Mader M, Suri V, Donohue MK, Huang M, Hui J, Yang J, Apelian D. 2014. Interim sustained virologic response (SVR), safety and tolerability results of 8-week treatment with ACH-3102 and sofosbuvir in chronic hepatitis C (HCV), genotype-1 (GT-1), treatment-naïve patients: a phase 2 “proxy” study, abstr LB-23 65th Annu Meet Am Assoc Study Liver Dis (AASLD), 7 to 12 November 2014, Boston, MA. [Google Scholar]
- 57.Lawitz E, Poordad F. 2014. C-SWIFT: MK-5172 + MK-8742 + sofosbuvir in treatment-naive patients with hepatitis C virus genotype 1 infection, with and without cirrhosis, for durations of 4, 6, or 8 weeks, abstr LB-33 65th Annu Meet Am Assoc Study Liver Dis (AASLD), 7 to 12 November 2014, Boston, MA. [Google Scholar]
- 58.Lim PJ, Chatterji U, Cordek D, Sharma SD, Garcia-Rivera JA, Cameron CE, Lin K, Targett-Adams P, Gallay PA. 2012. Correlation between NS5A dimerization and hepatitis C virus replication. J Biol Chem 287:30861–30873. doi: 10.1074/jbc.M112.376822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Garcia-Rivera JA, Bobardt M, Chatterji U, Hopkins S, Gregory MA, Wilkinson B, Lin K, Gallay PA. 2012. Multiple mutations in hepatitis C virus NS5A domain II are required to confer a significant level of resistance to alisporivir. Antimicrob Agents Chemother 56:5113–5121. doi: 10.1128/AAC.00919-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Gawlik K, Baugh J, Chatterji U, Lim PJ, Bobardt MD, Gallay PA. 2014. HCV core residues critical for infectivity are also involved in core-NS5A complex formation. PLoS One 9:e88866. doi: 10.1371/journal.pone.0088866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Berger C, Romero-Brey I, Radujkovic D, Terreux R, Zayas M, Paul D, Harak C, Hoppe S, Gao M, Penin F, Lohmann V, Bartenschlager R. 2014. Daclatasvir-like inhibitors of NS5A block early biogenesis of hepatitis C virus-induced membranous replication factories, independent of RNA replication. Gastroenterology 147:1094–1105.e25. doi: 10.1053/j.gastro.2014.07.019. [DOI] [PubMed] [Google Scholar]
- 62.Reghellin V, Donnici L, Fenu S, Berno V, Calabrese V, Pagani M, Abrignani S, Peri F, De Francesco R, Neddermann P. 2014. NS5A inhibitors impair NS5A-phosphatidylinositol 4-kinase IIIα complex formation and cause a decrease of phosphatidylinositol 4-phosphate and cholesterol levels in hepatitis C virus-associated membranes. Antimicrob Agents Chemother 58:7128–7140. doi: 10.1128/AAC.03293-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Eyre NS, Beard MR. 2014. HCV NS5A inhibitors disrupt replication factory formation: a novel mechanism of antiviral action. Gastroenterology 147:959–962. doi: 10.1053/j.gastro.2014.09.024. [DOI] [PubMed] [Google Scholar]
- 64.Berger KL, Cooper JD, Heaton NS, Yoon R, Oakland TE, Jordan TX, Mateu G, Grakoui A, Randall G. 2009. Roles for endocytic trafficking and phosphatidylinositol 4-kinase III alpha in hepatitis C virus replication. Proc Natl Acad Sci U S A 106:7577–7582. doi: 10.1073/pnas.0902693106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Berger KL, Kelly SM, Jordan TX, Tartell MA, Randall G. 2011. Hepatitis C virus stimulates the phosphatidylinositol 4-kinase III alpha-dependent phosphatidylinositol 4-phosphate production that is essential for its replication. J Virol 85:8870–8883. doi: 10.1128/JVI.00059-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lim YS, Hwang SB. 2011. Hepatitis C virus NS5A protein interacts with phosphatidylinositol 4-kinase type IIIalpha and regulates viral propagation. J Biol Chem 286:11290–11298. doi: 10.1074/jbc.M110.194472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Bianco A, Reghellin V, Donnici L, Fenu S, Alvarez R, Baruffa C, Peri F, Pagani M, Abrignani S, Neddermann P, De Francesco R. 2012. Metabolism of phosphatidylinositol 4-kinase IIIα-dependent PI4P is subverted by HCV and is targeted by a 4-anilino quinazoline with antiviral activity. PLoS Pathog 8:e1002576. doi: 10.1371/journal.ppat.1002576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Vaillancourt FH, Brault M, Pilote L, Uyttersprot N, Gaillard ET, Stoltz JH, Knight BL, Pantages L, McFarland M, Breitfelder S, Chiu TT, Mahrouche L, Faucher AM, Cartier M, Cordingley MG, Bethell RC, Jiang H, White PW, Kukolj G. 2012. Evaluation of phosphatidylinositol-4-kinase IIIα as a hepatitis C virus drug target. J Virol 86:11595–11607. doi: 10.1128/JVI.01320-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Huang L, Hwang J, Sharma SD, Hargittai MR, Chen Y, Arnold JJ, Raney KD, Cameron CE. 2005. Hepatitis C virus nonstructural protein 5A (NS5A) is an RNA-binding protein. J Biol Chem 280:36417–36428. doi: 10.1074/jbc.M508175200. [DOI] [PubMed] [Google Scholar]
- 70.Foster TL, Belyaeva T, Stonehouse NJ, Pearson AR, Harris M. 2010. All three domains of the hepatitis C virus nonstructural NS5A protein contribute to RNA binding. J Virol 84:9267–9277. doi: 10.1128/JVI.00616-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Lanford RE, Hildebrandt-Eriksen ES, Petri A, Persson R, Lindow M, Munk ME, Kauppinen S, Ørum H. 2010. Therapeutic silencing of microRNA-122 in primates with chronic hepatitis C virus infection. Science 327:198–201. doi: 10.1126/science.1178178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Leivers AL, Tallant M, Shotwell JB, Dickerson S, Leivers MR, McDonald OB, Gobel J, Creech KL, Strum SL, Mathis A, Rogers S, Moore CB, Botyanszki J. 2014. Discovery of selective small molecule type III phosphatidylinositol 4-kinase alpha (PI4KIIIα) inhibitors as anti hepatitis C (HCV) agents. J Med Chem 57:2091–2106. doi: 10.1021/jm400781h. [DOI] [PubMed] [Google Scholar]
- 73.Wakita T, Pietschmann T, Kato T, Date T, Miyamoto M, Zhao Z, Murthy K, Habermann A, Kräusslich HG, Mizokami M, Bartenschlager R, Liang TJ. 2005. Production of infectious hepatitis C virus in tissue culture from a cloned viral genome. Nat Med 11:791–796. doi: 10.1038/nm1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Vrolijk JM, Kaul A, Hansen BE, Lohmann V, Haagmans BL, Schalm SW, Bartenschlager R. 2003. A replicon-based bioassay for the measurement of interferons in patients with chronic hepatitis C. J Virol Methods 110:201–209. doi: 10.1016/S0166-0934(03)00134-4. [DOI] [PubMed] [Google Scholar]
- 75.Koutsoudakis G, Kaul A, Steinmann E, Kallis S, Lohmann V, Pietschmann T, Bartenschlager R. 2006. Characterization of the early steps of hepatitis C virus infection by using luciferase reporter viruses. J Virol 80:5308–5320. doi: 10.1128/JVI.02460-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Tai AW, Salloum S. 2011. The role of the phosphatidylinositol 4-kinase PI4KA in hepatitis C virus-induced host membrane rearrangement. PLoS One 6:e26300. doi: 10.1371/journal.pone.0026300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Quinkert D, Bartenschlager R, Lohmann V. 2005. Quantitative analysis of the hepatitis C virus replication complex. J Virol 79:13594–13605. doi: 10.1128/JVI.79.21.13594-13605.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Paul D, Hoppe S, Saher G, Krijnse-Locker J, Bartenschlager R. 2013. Morphological and biochemical characterization of the membranous hepatitis C virus replication compartment. J Virol 87:10612–10627. doi: 10.1128/JVI.01370-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Salloum S, Wang H, Ferguson C, Parton RG, Tai AW. 2013. Rab18 binds to hepatitis C virus NS5A and promotes interaction between sites of viral replication and lipid droplets. PLoS Pathog 9:e1003513. doi: 10.1371/journal.ppat.1003513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Chatterji U, Garcia-Rivera JA, Baugh J, Gawlik K, Wong KA, Zhong W, Brass CA, Naoumov NV, Gallay PA. 2014. Alisporivir plus NS5A inhibitor combinations provide additive to synergistic anti-HCV activity with no cross-resistance. Antimicrob Agents Chemother 58:3327–3334. doi: 10.1128/AAC.00016-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Romero-Brey I, Merz A, Chiramel A, Lee JY, Chlanda P, Haselman U, Santarella-Mellwig R, Habermann A, Hoppe S, Kallis S, Walther P, Antony C, Krijnse-Locker J, Bartenschlager R. 2012. Three-dimensional architecture and biogenesis of membrane structures associated with hepatitis C virus replication. PLoS Pathog 8:e1003056. doi: 10.1371/journal.ppat.1003056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Madan V, Paul D, Lohmann V, Bartenschlager R. 2014. Inhibition of HCV replication by cyclophilin antagonists is linked to replication fitness and occurs by inhibition of membranous web formation. Gastroenterology 146:1361–1372.e1-9. doi: 10.1053/j.gastro.2014.01.055. [DOI] [PubMed] [Google Scholar]
- 83.Gosert R, Egger D, Lohmann V, Bartenschlager R, Blum HE, Bienz K, Moradpour D. 2003. Identification of the hepatitis C virus RNA replication complex in Huh-7 cells harboring subgenomic replicons. J Virol 77:5487–5492. doi: 10.1128/JVI.77.9.5487-5492.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Wölk B, Büchele B, Moradpour D, Rice CM. 2008. A dynamic view of hepatitis C virus replication complexes. J Virol 82:10519–10531. doi: 10.1128/JVI.00640-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Nag A, Robotham JM, Tang H. 2012. Suppression of viral RNA binding and the assembly of infectious hepatitis C virus particles in vitro by cyclophilin inhibitors. J Virol 86:12616–12624. doi: 10.1128/JVI.01351-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.McGivern DR, Masaki T, Williford S, Ingravallo P, Feng Z, Lahser F, Asante-Appiah E, Neddermann P, De Francesco R, Howe AY, Lemon SM. 2014. Kinetic analyses reveal potent and early blockade of hepatitis C virus assembly by NS5A inhibitors. Gastroenterology 147:453–462.e7. doi: 10.1053/j.gastro.2014.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Tellinghuisen TL, Foss KL, Treadaway J. 2008. Regulation of hepatitis C virion production via phosphorylation of the NS5A protein. PLoS Pathog 4:e1000032. doi: 10.1371/journal.ppat.1000032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Appel N, Zayas M, Miller S, Krijnse-Locker J, Schaller T, Friebe P, Kallis S, Engel U, Bartenschlager R. 2008. Essential role of domain III of nonstructural protein 5A for hepatitis C virus infectious particle assembly. PLoS Pathog 4:e1000035. doi: 10.1371/journal.ppat.1000035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Chatterji U, Bobardt MD, Lim P, Gallay PA. 2010. Cyclophilin A-independent recruitment of NS5A and NS5B into hepatitis C virus replication complexes. J Gen Virol 91:1189–1193. doi: 10.1099/vir.0.018531-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Penin F, Brass V, Appel N, Ramboarina S, Montserret R, Ficheux D, Blum HE, Bartenschlager R, Moradpour D. 2004. Structure and function of the membrane anchor domain of hepatitis C virus nonstructural protein 5A. J Biol Chem 279:40835–40843. doi: 10.1074/jbc.M404761200. [DOI] [PubMed] [Google Scholar]
- 91.Cheng G, Montero A, Gastaminza P, Whitten-Bauer C, Wieland SF, Isogawa M, Fredericksen B, Selvarajah S, Gallay PA, Ghadiri MR, Chisari FV. 2008. A virocidal amphipathic α-helical peptide that inhibits hepatitis C virus infection in vitro. Proc Natl Acad Sci U S A 105:3088–3093. doi: 10.1073/pnas.0712380105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Bobardt MD, Cheng G, de Witte L, Selvarajah S, Chatterji U, Sanders-Beer BE, Geijtenbeek TB, Chisari FV, Gallay PA. 2008. Hepatitis C virus NS5A anchor peptide disrupts human immunodeficiency virus. Proc Natl Acad Sci U S A 105:5525–5530. doi: 10.1073/pnas.0801388105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.de Witte L, Bobardt MD, Chatterji U, van Loenen FB, Verjans GM, Geijtenbeek TB, Gallay PA. 2011. HSV neutralization by the microbicidal candidate C5A. PLoS One 6:e18917. doi: 10.1371/journal.pone.0018917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Chukkapalli V, Berger KL, Kelly SM, Thomas M, Deiters A, Randall G. 2014. Daclatasvir inhibits hepatitis C virus NS5A motility and hyper-accumulation of phosphoinositides. Virology 476C:168–179. doi: 10.1016/j.virol.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Reiss S, Harak C, Romero-Brey I, Radujkovic D, Klein R, Ruggieri A, Rebhan I, Bartenschlager R, Lohmann V. 2013. The lipid kinase phosphatidylinositol-4 kinase III alpha regulates the phosphorylation status of hepatitis C virus NS5A. PLoS Pathog 9:e1003359. doi: 10.1371/journal.ppat.1003359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Wang H, Perry JW, Lauring AS, Neddermann P, De Francesco R, Tai AW. 2014. Oxysterol-binding protein is a phosphatidylinositol 4-kinase effector required for HCV replication membrane integrity and cholesterol trafficking. Gastroenterology 146:1373–1385.e1-11. doi: 10.1053/j.gastro.2014.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]