

Abstract
Enterococcus faecalis and Enterococcus faecium are frequently resistant to vancomycin and β-lactams. In enterococcal infections with reduced glycopeptide susceptibility, combination therapy is often administered. Our objective was to conduct pharmacokinetic/pharmacodynamic (PK/PD) models to evaluate β-lactam synergy with daptomycin (DAP) against resistant enterococci. One E. faecalis strain (R6981) and two E. faecium strains (R6370 and 8019) were evaluated. DAP MICs were obtained. All strains were evaluated for response to LL37, an antimicrobial peptide, in the presence and absence of ceftaroline (CPT), ertapenem (ERT), and ampicillin (AMP). After 96 h, in vitro models were run simulating 10 mg DAP/kg body weight/day, 600 mg CPT every 8 h (q8h), 2 g AMP q4h, and 1 g ERT q24h, both alone and in combination against all strains. DAP MICs were 2, 4, and 4 μg/ml for strains R6981, R6370, and 8019, respectively. PK/PD models demonstrated bactericidal activity with DAP-CPT, DAP-AMP, and DAP-ERT combinations against strain 8019 (P < 0.001 and log10 CFU/ml reduction of >2 compared to any single agent). Against strains R6981 and R6370, the DAP-AMP combination demonstrated enhancement against R6370 but not R6981, while the combinations of DAP-CPT and DAP-ERT were bactericidal, demonstrated enhancement, and were statistically superior to all other regimens at 96 h (P < 0.001) against both strains. CPT, ERT, and AMP similarly augmented LL37 killing against strain 8019. In strains R6981 and R6370, CPT and ERT aided LL37 more than AMP (P < 0.001). Compared to DAP alone, combination regimens provide better killing and prevent resistance. Clinical research involving DAP combinations is warranted.
INTRODUCTION
Enterococcus faecalis and Enterococcus faecium together account for 12% of hospital-acquired infections in the United States (1). Often, enterococcal infections are caused by multidrug-resistant strains. For example, 0.4 to 5.2% and 70 to 92.6% of E. faecalis and E. faecium strains are resistant to ampicillin, respectively, and vancomycin resistance is present in up to 12.5% of E. faecalis and 79.7% of E. faecium (2–4). Vancomycin-resistant enterococci (VRE) are associated with increased mortality and complicated infections, such as infective endocarditis (5). Treatment of VRE infections can prove problematic, as available treatment options are potentially limited by static activity and/or platelet suppression with long-term use (6–8). Daptomycin (DAP) is a bactericidal lipopeptide often used against resistant enterococci (9). Mechanistically, it binds with calcium to form a cationic moiety that disrupts membrane potential to confer its antimicrobial effects, similar to endogenous, cationic antimicrobial peptides (10, 11). DAP is frequently dosed at 6 mg/kg body weight daily, although recent clinical and in vitro data suggest improved efficacy at higher doses (7, 12–14). DAP retains excellent in vitro activity against E. faecalis and E. faecium, with MIC50/90 values of 1/2 and 2/4 μg/ml, respectively (15). Reports of DAP-nonsusceptible enterococcal strains, defined as an MIC of >4 μg/ml, are becoming more prevalent, however (16). Recent data from a single center demonstrate that up to 0.5% of E. faecalis and 4.7% of E. faecium strains possess DAP MICs of 4 μg/ml (17). At these MICs, mutations that confer DAP inactivity are more prevalent than at lower MIC values, necessitating caution when treating these isolates (18). The nonsusceptibility among these isolates appears to be mediated at least partially through charge repulsion, with resistant strains possessing a more positive surface charge, repelling DAP (19). Indeed, in vitro data suggest that there is a strong correlation between DAP nonsusceptibility and resistance to other positively charged, endogenous antimicrobial proteins (20–22).There is a need to find appropriate measures to prevent DAP nonsusceptibility and treat such organisms.
Several in vitro studies have demonstrated successful synergism between DAP and other antimicrobials. Ampicillin (AMP), ceftriaxone (CRO), and ceftaroline (CPT) specifically have demonstrated synergy, and CPT has demonstrated the ability to restore DAP susceptibility to DAP-nonsusceptible strains (23–25). Mechanistically, it appears that these β-lactams decrease the surface charge of enterococcal cells and increase DAP binding to confer their synergistic effects (23–25). Two of these studies also demonstrated that AMP and CPT restore not only DAP activity but also endogenous, cationic antimicrobial peptide activity, demonstrating that the effect β-lactams have on membrane charge may be imperative for the synergistic effect (24, 25). Case reports have also demonstrated the clinical efficacy of the DAP-AMP and DAP-CPT combinations against E. faecalis and E. faecium (24, 26, 27).
Previous data suggest that several β-lactams possess synergy with DAP against enterococci, although the data are currently limited to static, time-kill experiments (28). To the best of our knowledge, there are no available data comparing several β-lactam agents based on their abilities to provide enhancement with DAP against E. faecalis and E. faecium in pharmacokinetic/pharmacodynamic (PK/PD) models. Our study was designed to compare the synergistic activities of CPT, ertapenem (ERT), and AMP with DAP in a one-compartment, PK/PD model experiment. We also sought to further explore the mechanism of synergy by performing antimicrobial peptide (LL37) time-kill assays to examine the effect of β-lactams on this endogenous cationic mimic of DAP activity.
MATERIALS AND METHODS
Bacterial strains.
One clinical strain of ampicillin-resistant E. faecalis and two clinical strains of E. faecium were selected for this study. The E. faecalis strain (R6981) and one E. faecium strain (R6370) were chosen randomly from our library at the Anti-Infective Research Laboratory (ARL). The other E. faecium strain (8019) has been previously described and was donated by George Sakoulas and his laboratory (19).
Antimicrobials.
DAP was purchased commercially from Cubist Pharmaceuticals (Lexington, MA). AMP and ERT were purchased commercially from Sigma Chemical Co. (St. Louis, MO). CPT was obtained from Actavis (formerly Forest Pharmaceuticals; Parsippany, NJ).
Susceptibility testing.
MIC values of studied antimicrobials were determined in duplicate by broth microdilution at approximately 106 CFU/ml in Mueller-Hinton broth (MHB; Difco, Detroit, MI) supplemented with 50 μg/ml calcium per CLSI guidelines (16). DAP MICs were also determined in the presence of the biologic-free peaks of CPT (17 μg/ml), ERT (15.5 μg/ml), and AMP (70 μg/ml) to determine the ability of the respective β-lactams to reduce the DAP MIC.
In vitro PK/PD model.
An in vitro, one-compartment PK/PD model with a 250-ml capacity and input and outflow ports was used. The target starting inoculum was ∼107 CFU/ml. Fresh medium (MHB) was continuously supplied and removed from the compartment along with the drug via a peristaltic pump (Masterflex; Cole-Parmer Instrument Company, Chicago, IL) at an appropriate rate to simulate the average human clearance and half-lives (t1/2) of the antimicrobials. The antimicrobial regimens evaluated were simulations of 10 mg/kg DAP every 24 h (q24h) (targeted maximum free-drug concentration in serum [fCmax], 11.3 μg/ml; t1/2, 8 h; protein binding, 92%; area under the concentration-time curve for the free, unbound fraction of a drug over 24 h [fAUC0–24], 114.8 μg · h/ml), 600 mg CPT every 8 h (fCmax, 17.04 μg/ml; t1/2, 2.66 h; protein binding, 20%), 1 g ERT every 24 h (fCmax, 15.5 μg/ml; t1/2, 4 h; protein binding, 90%), 2 g AMP every 4 h (fCmax,70 μg/ml; t1/2, 1.9 h; protein binding, 20%), 10 mg/kg DAP q24h plus 600 mg CPT q8h, 10 mg/kg DAP q24h plus 1 g ERT q24h, and 10 mg/kg DAP q24h plus 2 g AMP q4h (29–34). The models were performed in duplicate to ensure reproducibility. Supplemental DAP was added at an appropriate rate to CPT, ERT, and AMP to compensate for the higher flow rate required to simulate CPT, ERT, and AMP clearance (35).
Pharmacodynamic analysis.
Samples from each model were collected at 0, 4, 8, 24, 32, 48, 72, and 96 h in duplicate. Colony counts were determined by spiral plating appropriate dilutions using an automatic spiral plater (WASP; DW Scientific, West Yorkshire, England). Colonies were counted using a laser colony counter (ProtoCOL; Synoptics Limited, Frederick, MD). Bacteria were plated on brain heart infusion agar (BHIA; Difco, Detroit, MI). These methods have a low limit of reliable detection of 2 log10 CFU/ml. The total reduction in log10 CFU/ml over 96 h was determined by plotting model time-kill curves based on the number of remaining organisms over the 96-h time period. Bactericidal activity (99.9% kill) was defined as a ≥3-log10 CFU/ml decrease in colony count from the initial inoculum. Bacteriostatic activity was defined as a <3-log10 CFU/ml reduction in colony count from the initial inoculum. Enhancement of combinations was defined as a ≥2-log10 CFU/ml reduction over the most active single agent.
Pharmacokinetic analysis.
CPT, ERT, and AMP concentrations were determined by bioassay using Kocuria rhizophila (formerly Micrococcus luteus) ATCC 9341. Each sample was tested in duplicate by placing antibiotic-permeated disks of known, standard concentrations as well as disks containing unknown time point concentrations on agar plates (Antibiotic Medium Number 11; Difco, Detroit, MI) inoculated with a 0.5 McFarland suspension of the test organisms. DAP concentrations were determined using standard high-performance liquid chromatography (HPLC) (36). The half-lives, areas under the curve (AUC), and peak concentrations of the antibiotics were determined by the trapezoidal method using PK Analyst software (version 1.10; MicroMath Scientific Software, Salt Lake City, UT).
Resistance.
The emergence of resistance was evaluated at 96 h by plating 100-μl samples from the model on BHIA plates supplemented with DAP at a concentration 3 times the DAP MIC of the tested organism (e.g., a concentration of 1.5 μg/ml for an organism with an MIC of 0.5 μg/ml). Resistant colonies growing on screening plates were evaluated by the broth microdilution method to determine the DAP MIC.
Cathelicidin LL37 microbicidal assay.
The R6981, R6370, and 8019 strains were grown to stationary phase (16 to 20 h) in lysogeny broth (LB) in either the presence or absence of 17 μg/ml CPT, 15.5 μg/ml ERT, or 70 μg/ml AMP, pelleted, washed with phosphate-buffered saline (PBS), and exposed at an inoculum of 105 CFU/ml to 32 μM, 16 μM, and 16 μM LL37, respectively, in RPMI-5% LB. The percentage of surviving bacteria (±standard deviation [SD]) after 2 h of incubation at 35°C was calculated by plating on BHIA plates.
Statistical analysis.
Changes in CFU per milliliter at 96 h were compared by one-way analysis of variance (ANOVA) for in vitro models. A P value of ≤0.05 was considered significant. All statistical analyses were performed using SPSS Statistical Software (release 21; SPSS, Inc., Chicago, IL).
RESULTS
Susceptibility testing.
MIC values of all organisms against all antimicrobials are listed in Table 1. All isolates were resistant to vancomycin. DAP MIC values in the presence of β-lactams are listed in Table 2.
TABLE 1.
MICs of antibiotics to tested organisms
| Strain | MIC (μg/ml) |
|||
|---|---|---|---|---|
| DAP | CPT | ERT | AMP | |
| R6981 | 2 | >32 | >64 | >128 |
| R6370 | 4 | >32 | >64 | >128 |
| 8019 | 4 | >32 | >64 | >128 |
TABLE 2.
MICs of DAP alone or in the presence of CPT (17 μg/ml), ERT (15.5 μg/ml), and AMP (70 μg/ml)
| Strain | MIC (μg/ml) |
|||
|---|---|---|---|---|
| DAP | DAP-CPT | DAP-ERT | DAP-AMP | |
| R6981 | 2 | 0.25 | 0.5 | 0.5 |
| R6370 | 4 | 0.25 | 1 | 1 |
| 8019 | 4 | 0.13 | 1 | 0.5 |
In vitro PK/PD models.
The average observed fCmax for DAP was 11.8 μg/ml (target, 11.3 μg/ml), the average fAUC0–24 was 121.5 μg · h/ml (target, 114.8 μg · h/ml), and the average t1/2 was 7.9 h (target, 8 h) (29). For CPT, the average observed fCmax was 16.5 μg/ml (target, 17.04 μg/ml), and the average t1/2 was 2.5 h (target, 2.66 h). For ERT, the average observed fCmax was 15 μg/ml (target, 15.5 μg/ml) with an average t1/2 of 4 h (target, 4 h). The average observed fCmax for AMP was 73 μg/ml (target, 70 μg/ml), and the average t1/2 was 2.1 h (target, 1.9 h).
Against E. faecalis R6981 (Table 3, Fig. 1A), DAP alone was initially bactericidal but exhibited regrowth at 32 h and demonstrated little reduction at 96 h compared to the drug-free growth control. At 96 h, the DAP MIC had increased from the initial 2 μg/ml to 4 μg/ml, although this MIC increase is within the 1-fold margin of error allotted by CLSI. The fAUC0–24/MIC of DAP for this organism was 60.8 based on the initial isolate DAP MIC of 2 μg/ml (Table 4). CPT, ERT, and AMP demonstrated little to no activity when used as single agents. Combination regimens of DAP-CPT and DAP-ERT demonstrated similar, sustained bactericidal activity and were significantly more effective than all other regimens (P < 0.001). The DAP-AMP combination, however, demonstrated sustained regrowth and significantly greater bacterial growth at 96 h than the other combination regimens (P < 0.001). The fAUC0–24/MIC values for DAP in the presence of the β-lactams, based on the reduced combination DAP MICs, are listed in Table 4.
TABLE 3.
In vitro activities of regimens against R6981, R6370, and 8019 at 96 h
| Regimen | Log10 CFU/ml ± SD at 96 h |
||
|---|---|---|---|
| R6981 | R6370 | 8019 | |
| DAP | 6.43 ± 0.26 | 6.65 ± 0.21 | 7.75 ± 0.52 |
| CPT | 8.58 ± 0.05 | 8.02 ± 0.05 | 7.38 ± 0.28 |
| ERT | 9.42 ± 0.47 | 8.15 ± 0.07 | 8.18 ± 0.14 |
| AMP | 7.53 ± 0.11 | 7.60 ± 0.14 | 7.63 ± 0.15 |
| DAP-CPT | 2.00 ± 0.00a,b | 2.25 ± 0.35a,b | 2.00 ± 0.00a,b |
| DAP-ERT | 2.34 ± 0.26a,b | 2.00 ± 0.00a,b | 2.14 ± 0.08a,b |
| DAP-AMP | 4.75 ± 0.21a | 4.63 ± 0.01a,b | 2.09 ± 0.10a,b |
| Growth control | 8.94 ± 0.13 | 8.50 ± 0.14 | 8.42 ± 0.08 |
Significantly different from any single-agent regimen.
Enhancement from any single-agent regimen.
FIG 1.
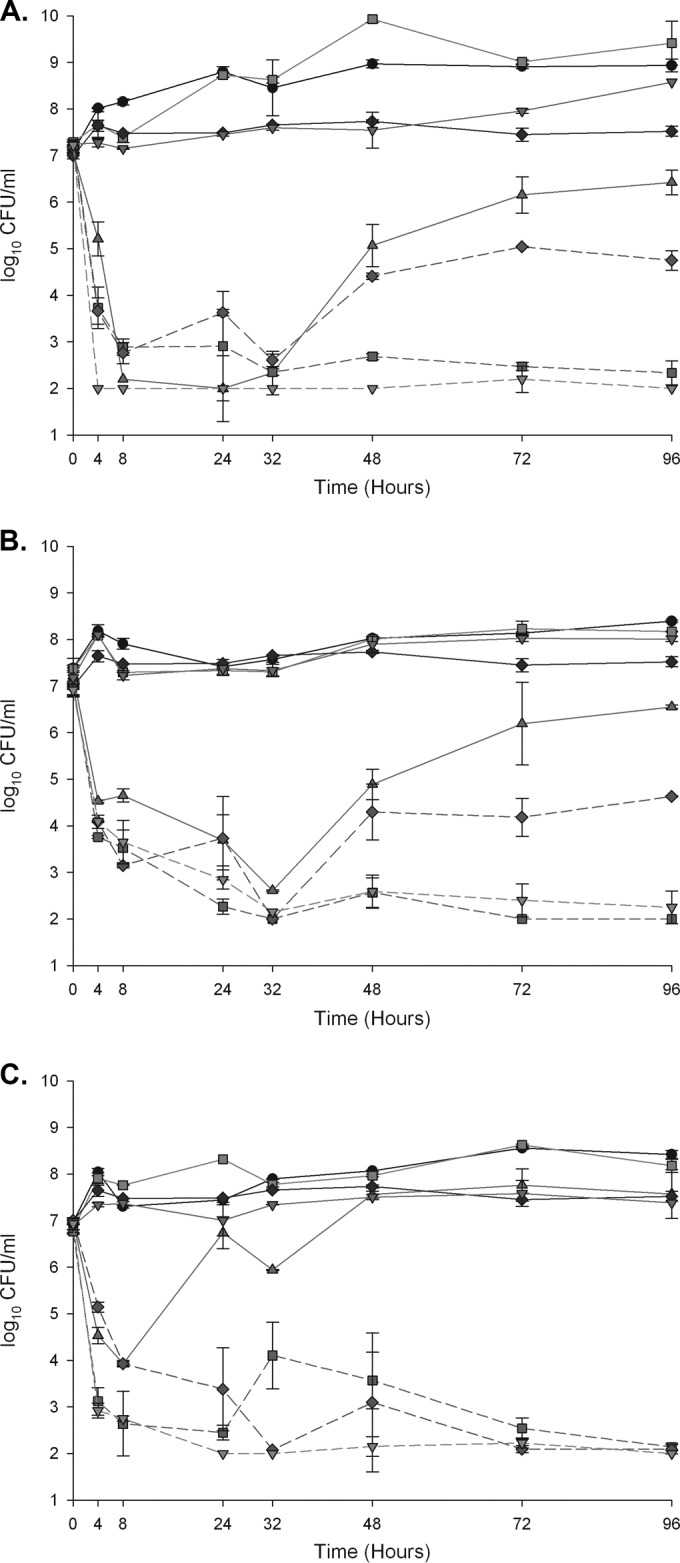
A 96-hour, one-compartment PK/PD model. Solid line with circles, growth control; solid line with upward triangles, DAP at 10 mg/kg/day; solid line with downward triangles, CPT at 600 mg q12h; solid line with squares, ERT at 1 g q24h; solid line with diamonds, AMP at 2 g q4h; dashed line with downward triangles, DAP-CPT; dashed line with squares, DAP-ERT; dashed line with diamonds, DAP-AMP. (A) E. faecalis R6981; (B) E. faecium R6370; (C) E. faecium 8019.
TABLE 4.
DAP fAUC0–24/MIC ratios observed in PK/PD regimens based on reduced DAP MICs in the presence of β-lactams
| Regimen | DAP fAUC0–24/MIC ratioa |
||
|---|---|---|---|
| R6981 | R6370 | 8019 | |
| DAP | 60.8 | 30.4 | 30.4 |
| DAP-CPT | 486 | 486 | 973 |
| DAP-ERT | 243 | 121.5 | 121.5 |
| DAP-AMP | 243 | 121.5 | 243 |
The fAUC0–24/MIC ratio corresponds to the initial isolate DAP MIC for DAP, DAP MIC in the presence of CPT for DAP-CPT, DAP MIC in the presence of ERT for DAP-ERT, and DAP MIC in the presence of AMP for DAP-AMP.
Against E. faecium R6370 (Table 3, Fig. 1B), DAP alone was initially bactericidal but exhibited regrowth at 48 h and failed to maintain bactericidal activity at 96 h. The fAUC0–24/MIC of DAP for this organism was 30.4 based on the initial isolate DAP MIC of 4 μg/ml. At 96 h, the recovered isolate from the DAP single regimen exhibited a DAP MIC of 8 μg/ml. The DAP-ERT and DAP-CPT combinations demonstrated enhancement and bactericidal activity, and they were statistically superior to all other regimens (P < 0.001). Although the DAP-AMP combination was initially bactericidal and demonstrated enhancement, it was inferior to the other combination regimens and did not sustain bactericidal activity at 96 h (P < 0.001). The fAUC0–24/MIC values for DAP in the presence of the β-lactams are listed in Table 4.
Against E. faecium 8019 (Table 3, Fig. 1C), DAP alone produced initial bactericidal activity but sustained substantial regrowth at 96 h, and resistance plating revealed a DAP-nonsusceptible mutant with a DAP MIC of 32 μg/ml. Based on the initial isolate DAP MIC of 4 μg/ml, the fAUC0–24/MIC of DAP for this organism was 30.4, as well. CPT, ERT, and AMP provided no activity as single agents. However, combination regimens of DAP with CPT, ERT, and AMP produced rapid, sustained bactericidal activity. Each combination produced enhancement, and each combination was significantly more active than any single agent at 96 h (P < 0.001). All combination regimens also prevented the emergence of DAP nonsusceptibility. The fAUC0–24/MIC values for DAP in the presence of the β-lactams are listed in Table 4.
Cathelicidin LL37 microbicidal assay.
LL37 killing of E. faecalis R6981 at 2 h was significantly enhanced with exposure to subinhibitory concentrations of CPT (17 μg/ml) and ERT (15.5 μg/ml), while AMP (70 μg/ml) provided no benefit compared to no exposure (P < 0.001) (Fig. 2A). Against E. faecium R6370, CPT and ERT demonstrated significant benefit compared to AMP (P < 0.001) (Fig. 2B). AMP was superior to no antimicrobial exposure but did not achieve the same level of killing as CPT or ERT (P < 0.001), which is consistent with the model experiments. Against E. faecium 8019, CPT, ERT, and AMP all provided significantly enhanced LL37 killing compared to that of no antimicrobial exposure at 2 h (P < 0.001) (Fig. 2C).
FIG 2.
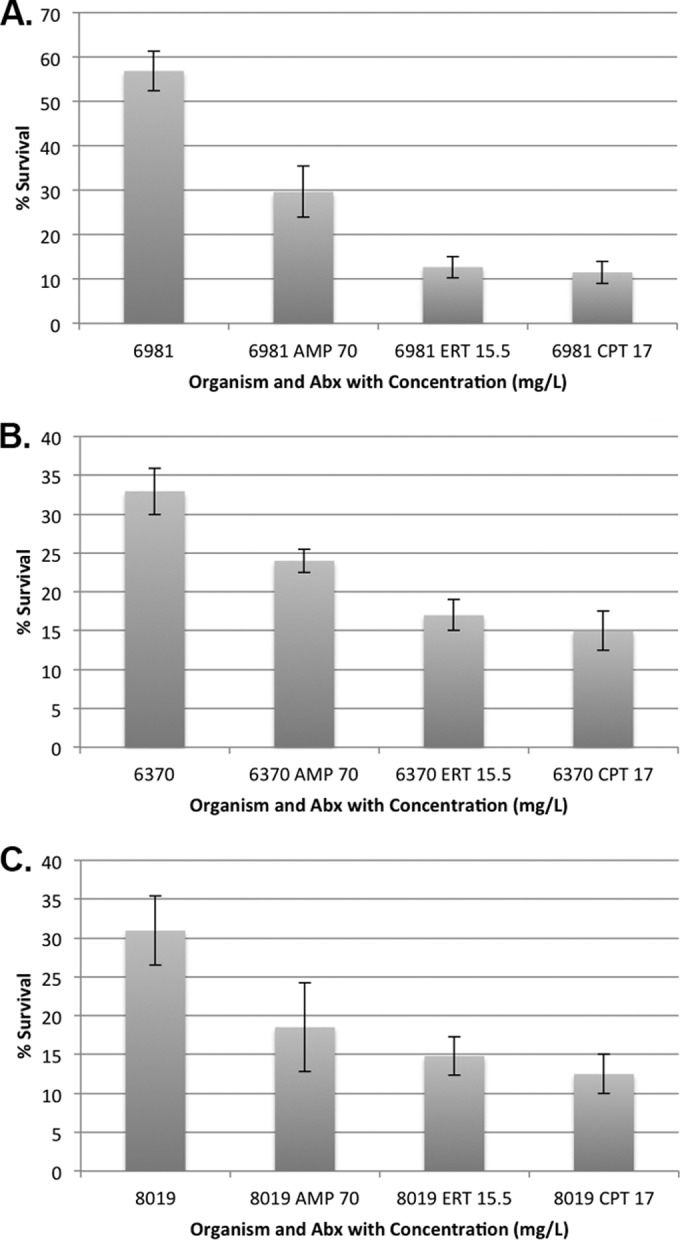
Percent survival of E. faecalis R6981 against 32 μM LL37 (A), E. faecium R6370 against 16 μM LL37 (B), and E. faecium 8019 against 16 μM LL37 (C) compared to the untreated growth control at 2 h. Whiskers indicate standard deviations.
DISCUSSION
Our study compared several β-lactam agents in combination with DAP in an in vitro PK/PD model experiment. Here, we have demonstrated that β-lactams provide enhancement to DAP and prevent DAP nonsusceptibility, as AMP was able to provide this effect against one strain, and CPT and ERT were able to demonstrate this effect against all strains tested.
Against E. faecalis R6981 and E. faecium R6370, DAP in combination with CPT or ERT was superior to DAP-AMP, somewhat unexpectedly. Nitrocefin testing revealed no β-lactamase present within the strain, so other factors may be responsible for the lack of enhancement by AMP. Against DAP-susceptible E. faecium 8019, we were able to demonstrate resistance prevention and bactericidal activity with DAP in combination with CPT, ERT, or AMP. Alone, DAP was unable to provide sustained bactericidal activity, and the DAP MIC value at 96 h elevated to 32 μg/ml, even though DAP was dosed at 10 mg/kg/day. Notably, the starting daptomycin MIC value of strain 8019 was 4 μg/ml, which has been previously demonstrated to be associated with diminished DAP efficacy (18, 37). In time-kill studies, the authors who demonstrated this effect were able to restore the activity of DAP against several of these strains with the addition of AMP. It should be noted, however, that these strains possessed mutations in the liaFSR three-component regulatory system, and genetic analysis has revealed no such mutation in strain 8019 (19). When we evaluated DAP MICs in the presence of biologic free peak concentrations of CPT, ERT, and AMP, we observed impressive reductions. Our data suggest that the addition of not only AMP but also a number of β-lactams may be important as the DAP MIC reaches the breakpoint, even without the presence of selected mutations. Further research is warranted to investigate the ability of β-lactam agents to provide synergistic activity against strains harboring different resistance mutations important for daptomycin susceptibility.
A factor that may have contributed to the lack of efficacy of DAP alone against any of these strains is the fAUC0–24/MIC ratio we obtained in our PK/PD models. In an in vitro model, Werth and colleagues recently demonstrated that the prevention of DAP resistance is accomplished with a DAP AUC0–24/MIC ratio of 1,562 for E. faecalis and 781 for E. faecium (38). In our study, DAP alone was able to achieve fAUC0–24/MIC ratios of 60.8 and 30.4 for E. faecalis and E. faecium, respectively. Although our experiments were performed in protein-free medium, based on the 92% protein binding of DAP, these fAUC0–24/MIC values translate to AUC0–24/MIC ratios of 770 and 385 if total DAP doses were given, as is the case in clinical practice. Since these values are well below the demonstrated resistance prevention cutoffs, it is not surprising that DAP resistance developed within our models, even at regimens based on 10 mg/kg/day. The addition of β-lactams and their respective MIC reductions, however, was able to greatly increase the DAP fAUC0–24/MIC ratios among all the strains. The increase in the AUC0–24/MIC ratio provided by CPT is large, exceeding the resistance prevention cutoff AUC0–24/MIC ratio proposed by Werth and colleagues (38) 4-fold for E. faecalis and 8- to 16-fold for E. faecium. Owing to the concentration-dependent, AUC0–24-driven activity of DAP, this increase in exposure likely explains the pronounced, bactericidal effect the combination of DAP-CPT provides. The enhancement is not quite as pronounced with ERT or AMP, yet DAP-ERT was bactericidal against all strains, and DAP-AMP was bactericidal against one. Because the increases in the fAUC0–24/MIC ratios were similar between ERT and AMP, it is likely that the discrepancies in their activities are mediated by other means, perhaps the differences in the protein binding profiles between the agents.
The enhanced activity of DAP conferred by combination with CPT in these models may be explained by the unique binding affinity of CPT. β-Lactam resistance among enterococci is often mediated through mutations that result in altered penicillin binding protein (PBP) profiles. In particular, resistant enterococci frequently possess an abundance of PBP5, a PBP with low affinity for β-lactams that allows survival in the presence of β-lactam therapy (39). CPT binds to this resistant PBP, possibly allowing enhanced DAP efficacy (40). In addition to its binding affinity for PBP5, CPT binds to enterococcal PBPs 1 to 4 more strongly than several other cephalosporins, perhaps suggesting that saturation of several PBPs, not only resistant PBP5, is important for synergistic activity. Previous in vitro findings suggest that saturation of PBPs 1 to 5 with a combination β-lactam regimen increases activity against E. faecalis (41, 42). CRO and AMP together are active against PBPs 1 to 5, and recent clinical data describing the effective use of this combination further establish this possibility (43). Recent data have also demonstrated that ceftobiprole is synergistic with DAP against VRE that are both DAP susceptible and DAP nonsusceptible (44). Previous work has demonstrated that ceftobiprole possesses activity against enterococcal PBP5 and PBPs 1 to 4 similar to, although not quite as potent as, that of CPT (40). These data further suggest the importance of PBP binding for synergistic DAP activity against enterococci. To this end, the enhancement provided by ERT could possibly be attributed to similarly broad PBP binding. Against other species, ERT provides a relatively narrow spectrum of PBP binding compared to that of some other agents (45). However, its binding profile against enterococci is not well understood. Two studies have evaluated the PBP binding profile of imipenem, another carbapenem, against E. faecalis and E. faecium. Against E. faecalis, imipenem demonstrated low inhibitory concentration values against PBPs 1 to 5, demonstrating an ability similar to that of ceftaroline to broadly cover PBPs (46). Against E. faecium, imipenem demonstrated an inhibitory concentration >6,400-fold lower than that of ampicillin against PBP5 (47). Owing to the success of ERT against these VRE strains, it is possible that ERT behaves similarly to imipenem and possesses the correct, broad range of PBP coverage necessary to provide synergy with DAP. Complicating the PBP binding picture, recent, in vitro data demonstrate that CRO enhances DAP activity against E. faecalis and E. faecium (23). In that study, the DAP-CRO combination was evaluated against E. faecium 8019, and the activity of that combination was similar to the activity we observed against 8019 with the DAP-CPT, DAP-ERT, and DAP-AMP combinations. CRO, however, possesses a narrower PBP binding profile than CPT (40). Based on the available data, it appears that there is an important combination of PBPs that should be bound for optimal DAP activity, and further study is needed to evaluate the optimal PBP binding profile for DAP enhancement.
Our study demonstrates the ability of CPT, AMP, and ERT to enhance the antimicrobial activity of LL37 against a DAP-susceptible parent strain of E. faecium. CPT and ERT also enhanced LL37 killing against E. faecalis R6981 and E. faecium R6370, while AMP failed to provide as pronounced an effect. LL37 is a human, cationic, cathelicidin antimicrobial peptide that is active through bacterial membrane destabilization (48). LL37 is not only a marker of immune response but also an endogenous analogue to DAP (49). Our data echo previous findings demonstrating the synergistic effects of β-lactam agents on LL37 activity in E. faecalis, E. faecium, and Staphylococcus aureus (24, 25, 50, 51). From the available data, it appears that β-lactams have the ability to amplify both portions of the innate immune response (LL37) and DAP activity against enterococci and that this augmentation of DAP activity may explain the enhancement we have demonstrated. Further study is warranted to establish the effect of β-lactams on endogenous cationic peptides that mimic the effects of DAP.
Clinically, the successful augmentation of DAP by ERT is of interest. AMP is frequently employed in combination therapy for enterococcal infections, and CPT has been demonstrated to provide synergistic activity with DAP in clinical cases (6, 26). ERT provides a broad spectrum of bacterial coverage, which limits its utility when narrow, targeted therapy is desired (52). On its own, ERT lacks intrinsic activity against enterococci, so it is not used clinically in the setting of enterococcal infections. However, ERT may be advantageous in the setting of acute, polymicrobial infections, and its once-daily dosing regimen allows for simpler outpatient therapy than other β-lactams in the setting of prolonged antibiotic courses (53). Since enterococcal infections often require lengthy therapeutic regimens, ease of administration is an important factor to consider. Along with CRO, which has demonstrated similar in vitro efficacy in combination with DAP against VRE, ERT may present a viable, easy-to-administer option for long-term combination therapy in the setting of severe enterococcal infections.
There are some limitations to the current study. We studied only one strain of E. faecalis and two strains of E. faecium, possibly limiting the generalizability of the results. This is especially true of E. faecalis R6981 and E. faecium R6370, which showed little evidence of synergy in the presence of AMP, although AMP has demonstrated synergy in previous work. Also, our models were run over only 4 days, a much shorter regimen than what is frequently employed clinically. Further investigation is warranted to confirm the reproducibility of these results in other enterococcal strains.
Conclusion.
AMP and vancomycin resistance among enterococci necessitates novel therapeutic approaches. When deep-seated infections require prolonged, intensive therapy, the current available options are limited by bacteriostatic activity or adverse effects. DAP is frequently employed in this setting, but the emergence of DAP nonsusceptibility is concerning. The results of our study demonstrate the ability of several β-lactams, especially CPT and ERT, to provide synergistic activity with DAP and prevent the emergence of DAP nonsusceptibility. Our study provides promising evidence for the early use of high-dose DAP in combination with a β-lactam against VRE infections to prevent DAP nonsusceptibility, and we have also demonstrated that the DAP-CPT and DAP-ERT combinations provide synergistic activity and may present a viable therapeutic option. Further clinical research is warranted.
ACKNOWLEDGMENTS
This work was supported by internal funding and not funded from an external source.
We thank Forest Laboratories for the use of ceftaroline powder. Strain 8019 was graciously provided by George Sakoulas and his laboratory.
M.J.R. has received grant support from, consulted for, or provided lectures for Astellas, Cubist, Forest, Pfizer, and Novartis and is partially funded by NIH R21 AI109266-01. J.R.S., K.E.B., and A.R. have nothing to declare.
REFERENCES
- 1.Sievert DM, Ricks P, Edwards JR, Schneider A, Patel J, Srinivasan A, Kallen A, Limbago B, Fridkin S. 2013. Antimicrobial-resistant pathogens associated with healthcare-associated infections: summary of data reported to the National Healthcare Safety Network at the Centers for Disease Control and Prevention, 2009-2010. Infect Control Hosp Epidemiol 34:1–14. doi: 10.1086/668770. [DOI] [PubMed] [Google Scholar]
- 2.Rathnayake IU, Hargreaves M, Huygens F. 2012. Antibiotic resistance and virulence traits in clinical and environmental Enterococcus faecalis and Enterococcus faecium isolates. Syst Appl Microbiol 35:326–333. doi: 10.1016/j.syapm.2012.05.004. [DOI] [PubMed] [Google Scholar]
- 3.Hayakawa K, Marchaim D, Vidaillac C, Lephart P, Pogue JM, Sunkara B, Kotra H, Hasan A, Shango M, Yerramalla Y, Osunlana AM, Chopra T, Dhar S, Salimnia H, Rybak MJ, Kaye KS. 2011. Growing prevalence of vancomycin-resistant Enterococcus faecalis in the region with the highest prevalence of vancomycin-resistant Staphylococcus aureus. Infect Control Hosp Epidemiol 32:922–924. doi: 10.1086/661599. [DOI] [PubMed] [Google Scholar]
- 4.Billington EO, Phang SH, Gregson DB, Pitout JD, Ross T, Church DL, Laupland KB, Parkins MD. 2014. Incidence, risk factors, and outcomes of Enterococcus spp blood stream infections: a population-based study. Int J Infect Dis 26:76–82. doi: 10.1016/j.ijid.2014.02.012. [DOI] [PubMed] [Google Scholar]
- 5.Carmeli Y, Eliopoulos G, Mozaffari E, Samore M. 2002. Health and economic outcomes of vancomycin-resistant enterococci. Arch Intern Med 162:2223–2228. doi: 10.1001/archinte.162.19.2223. [DOI] [PubMed] [Google Scholar]
- 6.Baddour LM, Wilson WR, Bayer AS, Fowler VG Jr, Bolger AF, Levison ME, Ferrieri P, Gerber MA, Tani LY, Gewitz MH, Tong DC, Steckelberg JM, Baltimore RS, Shulman ST, Burns JC, Falace DA, Newburger JW, Pallasch TJ, Takahashi M, Taubert KA. 2005. Infective endocarditis: diagnosis, antimicrobial therapy, and management of complications: a statement for healthcare professionals from the Committee on Rheumatic Fever, Endocarditis, and Kawasaki Disease, Council on Cardiovascular Disease in the Young, and the Councils on Clinical Cardiology, Stroke, and Cardiovascular Surgery and Anesthesia, American Heart Association: endorsed by the Infectious Diseases Society of America. Circulation 111:e394–e434. doi: 10.1161/CIRCULATIONAHA.105.165564. [DOI] [PubMed] [Google Scholar]
- 7.Casapao AM, Kullar R, Davis SL, Levine DP, Zhao JJ, Potoski BA, Goff DA, Crank CW, Segreti J, Sakoulas G, Cosgrove SE, Rybak MJ. 2013. Multicenter study of high-dose daptomycin for treatment of enterococcal infections. Antimicrob Agents Chemother 57:4190–4196. doi: 10.1128/AAC.00526-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Falagas ME, Manta KG, Ntziora F, Vardakas KZ. 2006. Linezolid for the treatment of patients with endocarditis: a systematic review of the published evidence. J Antimicrob Chemother 58:273–280. doi: 10.1093/jac/dkl219. [DOI] [PubMed] [Google Scholar]
- 9.Humphries RM, Pollett S, Sakoulas G. 2013. A current perspective on daptomycin for the clinical microbiologist. Clin Microbiol Rev 26:759–780. doi: 10.1128/CMR.00030-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pogliano J, Pogliano N, Silverman JA. 2012. Daptomycin-mediated reorganization of membrane architecture causes mislocalization of essential cell division proteins. J Bacteriol 194:4494–4504. doi: 10.1128/JB.00011-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Straus SK, Hancock RE. 2006. Mode of action of the new antibiotic for Gram-positive pathogens daptomycin: comparison with cationic antimicrobial peptides and lipopeptides. Biochim Biophys Acta 1758:1215–1223. doi: 10.1016/j.bbamem.2006.02.009. [DOI] [PubMed] [Google Scholar]
- 12.Akins RL, Rybak MJ. 2001. Bactericidal activities of two daptomycin regimens against clinical strains of glycopeptide intermediate-resistant Staphylococcus aureus, vancomycin-resistant Enterococcus faecium, and methicillin-resistant Staphylococcus aureus isolates in an in vitro pharmacodynamic model with simulated endocardial vegetations. Antimicrob Agents Chemother 45:454–459. doi: 10.1128/AAC.45.2.454-459.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hall AD, Steed ME, Arias CA, Murray BE, Rybak MJ. 2012. Evaluation of standard- and high-dose daptomycin versus linezolid against vancomycin-resistant Enterococcus isolates in an in vitro pharmacokinetic/pharmacodynamic model with simulated endocardial vegetations. Antimicrob Agents Chemother 56:3174–3180. doi: 10.1128/AAC.06439-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kullar R, Casapao AM, Davis SL, Levine DP, Zhao JJ, Crank CW, Segreti J, Sakoulas G, Cosgrove SE, Rybak MJ. 2013. A multicentre evaluation of the effectiveness and safety of high-dose daptomycin for the treatment of infective endocarditis. J Antimicrob Chemother 68:2921–2926. doi: 10.1093/jac/dkt294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sader HS, Farrell DJ, Flamm RK, Jones RN. 2014. Daptomycin activity tested against 164457 bacterial isolates from hospitalised patients: summary of 8 years of a Worldwide Surveillance Programme (2005-2012). Int J Antimicrob Agents 43:465–469. doi: 10.1016/j.ijantimicag.2014.01.018. [DOI] [PubMed] [Google Scholar]
- 16.CLSI. 2012. Performance standards for antimicrobial susceptibility testing; 22nd informational supplement. CLSI, Wayne, PA. [Google Scholar]
- 17.Edelsberg J, Weycker D, Barron R, Li X, Wu H, Oster G, Badre S, Langeberg WJ, Weber DJ. 2014. Prevalence of antibiotic resistance in US hospitals. Diagn Microbiol Infect Dis 78:255–262. doi: 10.1016/j.diagmicrobio.2013.11.011. [DOI] [PubMed] [Google Scholar]
- 18.Munita JM, Panesso D, Diaz L, Tran TT, Reyes J, Wanger A, Murray BE, Arias CA. 2012. Correlation between mutations in liaFSR of Enterococcus faecium and MIC of daptomycin: revisiting daptomycin breakpoints. Antimicrob Agents Chemother 56:4354–4359. doi: 10.1128/AAC.00509-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Humphries RM, Kelesidis T, Tewhey R, Rose WE, Schork N, Nizet V, Sakoulas G. 2012. Genotypic and phenotypic evaluation of the evolution of high-level daptomycin nonsusceptibility in vancomycin-resistant Enterococcus faecium. Antimicrob Agents Chemother 56:6051–6053. doi: 10.1128/AAC.01318-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bayer AS, Mishra NN, Sakoulas G, Nonejuie P, Nast CC, Pogliano J, Chen KT, Ellison SN, Yeaman MR, Yang SJ. 2014. Heterogeneity of mprF sequences in methicillin-resistant Staphylococcus aureus clinical isolates: role in cross-resistance between daptomycin and host defense antimicrobial peptides. Antimicrob Agents Chemother 58:7462–7467. doi: 10.1128/AAC.03422-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mishra NN, Yang SJ, Chen L, Muller C, Saleh-Mghir A, Kuhn S, Peschel A, Yeaman MR, Nast CC, Kreiswirth BN, Cremieux AC, Bayer AS. 2013. Emergence of daptomycin resistance in daptomycin-naive rabbits with methicillin-resistant Staphylococcus aureus prosthetic joint infection is associated with resistance to host defense cationic peptides and mprF polymorphisms. PLoS One 8:e71151. doi: 10.1371/journal.pone.0071151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mishra NN, Bayer AS, Moise PA, Yeaman MR, Sakoulas G. 2012. Reduced susceptibility to host-defense cationic peptides and daptomycin coemerge in methicillin-resistant Staphylococcus aureus from daptomycin-naive bacteremic patients. J Infect Dis 206:1160–1167. doi: 10.1093/infdis/jis482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hall Snyder A, Werth BJ, Barber KE, Sakoulas G, Rybak MJ. 2014. Evaluation of the novel combination of daptomycin plus ceftriaxone against vancomycin-resistant enterococci in an in vitro pharmacokinetic/pharmacodynamic simulated endocardial vegetation model. J Antimicrob Chemother 56:2148–2154. doi: 10.1093/jac/dku113. [DOI] [PubMed] [Google Scholar]
- 24.Sakoulas G, Bayer AS, Pogliano J, Tsuji BT, Yang SJ, Mishra NN, Nizet V, Yeaman MR, Moise PA. 2012. Ampicillin enhances daptomycin- and cationic host defense peptide-mediated killing of ampicillin- and vancomycin-resistant Enterococcus faecium. Antimicrob Agents Chemother 56:838–844. doi: 10.1128/AAC.05551-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sakoulas G, Rose W, Nonejuie P, Olson J, Pogliano J, Humphries R, Nizet V. 2014. Ceftaroline restores daptomycin activity against daptomycin-nonsusceptible vancomycin-resistant Enterococcus faecium. Antimicrob Agents Chemother 58:1494–1500. doi: 10.1128/AAC.02274-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sakoulas G, Nonejuie P, Nizet V, Pogliano J, Crum-Cianflone N, Haddad F. 2013. Treatment of high-level gentamicin-resistant Enterococcus faecalis endocarditis with daptomycin plus ceftaroline. Antimicrob Agents Chemother 57:4042–4045. doi: 10.1128/AAC.02481-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sierra-Hoffman M, Iznaola O, Goodwin M, Mohr J. 2012. Combination therapy with ampicillin and daptomycin for treatment of Enterococcus faecalis endocarditis. Antimicrob Agents Chemother 56:6064. doi: 10.1128/AAC.01760-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Smith JR, Barber KE, Raut A, Aboutaleb M, Sakoulas G, Rybak MJ. 1 February 2015. β-Lactam combinations with daptomycin provide synergy against vancomycin-resistant Enterococcus faecalis and Enterococcus faecium. J Antimicrob Chemother doi: 10.1093/jac/dkv007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Benvenuto M, Benziger DP, Yankelev S, Vigliani G. 2006. Pharmacokinetics and tolerability of daptomycin at doses up to 12 milligrams per kilogram of body weight once daily in healthy volunteers. Antimicrob Agents Chemother 50:3245–3249. doi: 10.1128/AAC.00247-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Drusano GL. 2010. Pharmacodynamics of ceftaroline fosamil for complicated skin and skin structure infection: rationale for improved anti-methicillin-resistant Staphylococcus aureus activity. J Antimicrob Chemother 65(Suppl 4):iv33–iv39. doi: 10.1093/jac/dkq253. [DOI] [PubMed] [Google Scholar]
- 31.Majumdar AK, Musson DG, Birk KL, Kitchen CJ, Holland S, McCrea J, Mistry G, Hesney M, Xi L, Li SX, Haesen R, Blum RA, Lins RL, Greenberg H, Waldman S, Deutsch P, Rogers JD. 2002. Pharmacokinetics of ertapenem in healthy young volunteers. Antimicrob Agents Chemother 46:3506–3511. doi: 10.1128/AAC.46.11.3506-3511.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Meyers BR, Wilkinson P, Mendelson MH, Walsh S, Bournazos C, Hirschman SZ. 1991. Pharmacokinetics of ampicillin-sulbactam in healthy elderly and young volunteers. Antimicrob Agents Chemother 35:2098–2101. doi: 10.1128/AAC.35.10.2098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Soto E, Shoji S, Muto C, Tomono Y, Marshall S. 2014. Population pharmacokinetics of ampicillin and sulbactam in patients with community-acquired pneumonia: evaluation of the impact of renal impairment. Br J Clin Pharmacol 77:509–521. doi: 10.1111/bcp.12232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wiskirchen DE, Housman ST, Quintiliani R, Nicolau DP, Kuti JL. 2013. Comparative pharmacokinetics, pharmacodynamics, and tolerability of ertapenem 1 gram/day administered as a rapid 5-minute infusion versus the standard 30-minute infusion in healthy adult volunteers. Pharmacotherapy 33:266–274. doi: 10.1002/phar.1197. [DOI] [PubMed] [Google Scholar]
- 35.Blaser J. 1985. In-vitro model for simultaneous simulation of the serum kinetics of two drugs with different half-lives. J Antimicrob Chemother 15(Suppl A):125–130. [DOI] [PubMed] [Google Scholar]
- 36.Dvorchik BH, Brazier D, DeBruin MF, Arbeit RD. 2003. Daptomycin pharmacokinetics and safety following administration of escalating doses once daily to healthy subjects. Antimicrob Agents Chemother 47:1318–1323. doi: 10.1128/AAC.47.4.1318-1323.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Diaz L, Tran TT, Munita JM, Miller WR, Rincon S, Carvajal LP, Wollam A, Reyes J, Panesso D, Rojas NL, Shamoo Y, Murray BE, Weinstock GM, Arias CA. 2014. Whole genome analyses of Enterococcus faecium with diverse daptomycin minimal inhibitory concentrations. Antimicrob Agents Chemother 58:4527–4534. doi: 10.1128/AAC.02686-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Werth BJ, Steed ME, Ireland CE, Tran TT, Nonejuie P, Murray BE, Rose WE, Sakoulas G, Pogliano J, Arias CA, Rybak MJ. 2014. Defining daptomycin resistance prevention exposures in vancomycin-resistant Enterococcus faecium and E. faecalis. Antimicrob Agents Chemother 58:5253–5261. doi: 10.1128/AAC.00098-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rice LB, Carias LL, Hutton-Thomas R, Sifaoui F, Gutmann L, Rudin SD. 2001. Penicillin-binding protein 5 and expression of ampicillin resistance in Enterococcus faecium. Antimicrob Agents Chemother 45:1480–1486. doi: 10.1128/AAC.45.5.1480-1486.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Henry X, Verlaine O, Amoroso A, Coyette J, Frere JM, Joris B. 2013. Activity of ceftaroline against Enterococcus faecium PBP5. Antimicrob Agents Chemother 57:6358–6360. doi: 10.1128/AAC.00923-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Liao CH, Huang YT, Tsai HY, Hsueh PR. 2014. In vitro synergy of ampicillin with gentamicin, ceftriaxone and ciprofloxacin against Enterococcus faecalis. Int J Antimicrob Agents 44:85–86. doi: 10.1016/j.ijantimicag.2014.03.011. [DOI] [PubMed] [Google Scholar]
- 42.Mainardi JL, Gutmann L, Acar JF, Goldstein FW. 1995. Synergistic effect of amoxicillin and cefotaxime against Enterococcus faecalis. Antimicrob Agents Chemother 39:1984–1987. doi: 10.1128/AAC.39.9.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fernandez-Hidalgo N, Almirante B, Gavalda J, Gurgui M, Pena C, de Alarcon A, Ruiz J, Vilacosta I, Montejo M, Vallejo N, Lopez-Medrano F, Plata A, Lopez J, Hidalgo-Tenorio C, Galvez J, Saez C, Lomas JM, Falcone M, de la Torre J, Martinez-Lacasa X, Pahissa A. 2013. Ampicillin plus ceftriaxone is as effective as ampicillin plus gentamicin for treating enterococcus faecalis infective endocarditis. Clin Infect Dis 56:1261–1268. doi: 10.1093/cid/cit052. [DOI] [PubMed] [Google Scholar]
- 44.Werth BJ, Barber KE, Tran KN, Nonejuie P, Sakoulas G, Pogliano J, Rybak MJ. 2015. Ceftobiprole and ampicillin increase daptomycin susceptibility of daptomycin-susceptible and -resistant VRE. J Antimicrob Chemother 70:489–493. doi: 10.1093/jac/dku386. [DOI] [PubMed] [Google Scholar]
- 45.Zhanel GG, Wiebe R, Dilay L, Thomson K, Rubinstein E, Hoban DJ, Noreddin AM, Karlowsky JA. 2007. Comparative review of the carbapenems. Drugs 67:1027–1052. doi: 10.2165/00003495-200767070-00006. [DOI] [PubMed] [Google Scholar]
- 46.Tsuchimori N, Okonogi K. 1996. Penicillin-binding protein 5 as an inhibitory target of cefozopran in Enterococcus faecalis. J Antimicrob Chemother 37:605–609. doi: 10.1093/jac/37.3.605. [DOI] [PubMed] [Google Scholar]
- 47.Mainardi JL, Hugonnet JE, Rusconi F, Fourgeaud M, Dubost L, Moumi AN, Delfosse V, Mayer C, Gutmann L, Rice LB, Arthur M. 2007. Unexpected inhibition of peptidoglycan LD-transpeptidase from Enterococcus faecium by the beta-lactam imipenem. J Biol Chem 282:30414–30422. doi: 10.1074/jbc.M704286200. [DOI] [PubMed] [Google Scholar]
- 48.Turner J, Cho Y, Dinh NN, Waring AJ, Lehrer RI. 1998. Activities of LL-37, a cathelin-associated antimicrobial peptide of human neutrophils. Antimicrob Agents Chemother 42:2206–2214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mensa B, Howell GL, Scott R, DeGrado WF. 2014. Comparative mechanistic studies of brilacidin, daptomycin and the antimicrobial peptide LL16. Antimicrob Agents Chemother 58:5136–5145. doi: 10.1128/AAC.02955-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sakoulas G, Okumura CY, Thienphrapa W, Olson J, Nonejuie P, Dam Q, Dhand A, Pogliano J, Yeaman MR, Hensler ME, Bayer AS, Nizet V. 2014. Nafcillin enhances innate immune-mediated killing of methicillin-resistant Staphylococcus aureus. J Mol Med (Berl) 92:139–149. doi: 10.1007/s00109-013-1100-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Werth BJ, Sakoulas G, Rose WE, Pogliano J, Tewhey R, Rybak MJ. 2013. Ceftaroline increases membrane binding and enhances the activity of daptomycin against daptomycin-nonsusceptible vancomycin-intermediate Staphylococcus aureus in a pharmacokinetic/pharmacodynamic model. Antimicrob Agents Chemother 57:66–73. doi: 10.1128/AAC.01586-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Odenholt I. 2001. Ertapenem: a new carbapenem. Expert Opin Invest Drugs 10:1157–1166. doi: 10.1517/13543784.10.6.1157. [DOI] [PubMed] [Google Scholar]
- 53.Senneville E, Nguyen S. 2013. Current pharmacotherapy options for osteomyelitis: convergences, divergences and lessons to be drawn. Expert Opin Pharmacother 14:723–734. doi: 10.1517/14656566.2013.780596. [DOI] [PubMed] [Google Scholar]