

Abstract
The pressing need for better drugs against Chagas disease, African sleeping sickness, and schistosomiasis motivates the search for inhibitors of cruzain, rhodesain, and Schistosoma mansoni CB1 (SmCB1), the major cysteine proteases from Trypanosoma cruzi, Trypanosoma brucei, and S. mansoni, respectively. Thiosemicarbazones and heterocyclic analogues have been shown to be both antitrypanocidal and inhibitory against parasite cysteine proteases. A series of compounds was synthesized and evaluated against cruzain, rhodesain, and SmCB1 through biochemical assays to determine their potency and structure-activity relationships (SAR). This approach led to the discovery of 6 rhodesain, 4 cruzain, and 5 SmCB1 inhibitors with 50% inhibitory concentrations (IC50s) of ≤10 μM. Among the compounds tested, the thiosemicarbazone derivative of peracetylated galactoside (compound 4i) was discovered to be a potent rhodesain inhibitor (IC50 = 1.2 ± 1.0 μM). The impact of a range of modifications was determined; removal of thiosemicarbazone or its replacement by semicarbazone resulted in virtually inactive compounds, and modifications in the sugar also diminished potency. Compounds were also evaluated in vitro against the parasites T. cruzi, T. brucei, and S. mansoni, revealing active compounds among this series.
INTRODUCTION
New drugs for parasitic diseases are urgently needed, but these globally important infections are often “neglected” because they most commonly afflict poor and marginalized communities. Current therapies are limited by poor efficacy, toxicity, high costs, and parasite resistance. Chagas disease, African sleeping sickness, and schistosomiasis are examples of diseases for which new therapies are needed (1, 2). Among the most studied and exploited molecular targets for these diseases are cysteine proteases. These enzymes have essential roles in parasite nutrition, immune evasion, host cell invasion, and metacyclogenesis (3–6). Indeed, the cysteine proteases cruzain, rhodesain, and Schistosoma mansoni CB1 (SmCB1) from Trypanosoma cruzi, Trypanosoma brucei, and S. mansoni, respectively, are validated molecular targets and have been the subject of numerous medicinal chemistry projects (7–17) that have yielded trypanocidal inhibitors, both in parasite culture and in animal models of infection (13, 15, 18–21).
The diverse inhibitors of these enzymes comprise compound classes which bind noncovalently (11, 12) and scaffolds containing a “warhead” that binds covalently to the catalytic cysteine. Within the latter category, vinylsulfones (8, 22–25), oxy-methyl ketones (7, 26), nitriles (16), epoxides, and thiosemicarbazones (13–15, 27–29) have been described previously. Thiosemicarbazones present as advantages their low molecular weight, low cost of synthesis, and nonpeptidic nature (27). Greenbaum and coworkers synthesized and evaluated the cysteine protease inhibitory and antiparasitic activities of a library of thiosemicarbazones, with promising results (13). According to those authors, the thiosemicarbazones are regarded as validated drug leads capable of killing different species of protozoan parasites (T. cruzi, Plasmodium falciparum, and T. brucei) via inhibition of cysteine proteases.
Heterocyclic thiazole derivatives are also of great importance in medicinal chemistry due to their broad spectrum of biological activities (30–34). Also, many cysteine protease inhibitors bearing thiazole or isothiazolone ring systems have been described as promising compounds against parasitic diseases (35, 36). Because of the versatile approach to the synthesis of the thiazole scaffold from thiosemicarbazones, we synthesized and evaluated a series of thiazole analogues as potential inhibitors of cysteine proteases. The covalent attachment of the thiosemicarbazone or thiazole unit and a carbohydrate moiety was also designed to modulate solubility and properties of interaction (for example, by hydrogen bonding) with the molecular target (cysteine protease).
Here, we screened a series of thiosemicarbazones and cyclic analogues against rhodesain and discovered an acetylated derivative of galactose as a potent inhibitor (50% inhibitory concentration [IC50] = 1.2 ± 1.0 μM). This is the first case of a sugar moiety being present in an inhibitor from this chemical class, encouraging further structure-activity relationship (SAR) studies on these series. Here we report their synthesis and evaluation using the proteases cruzain, rhodesain, and SmCB1 and their in vitro bioactivities against T. cruzi, T. brucei, and S. mansoni.
MATERIALS AND METHODS
Chemistry.
All melting points (mp) were determined on a Microquímica MQAPF 301 apparatus. The infrared (IR) spectra were recorded using a PerkinElmer Spectrum One infrared spectrometer, and absorptions are reported as wave values (cm−1). The nuclear magnetic resonance (NMR) spectra were recorded on a Bruker Avance DRX 200 or Bruker Avance DRX 400 instrument, using tetramethylsilane (TMS) as the internal standard. Chemical shifts are given using the δ (ppm) scale, and J values are given in Hz. All reagents of analytical grade were obtained from commercial suppliers and used without further purification. Compounds 4c, 4 h, 4n to 4q, and 6a to 6f were synthesized according to a previously published procedure (36).
General procedure A, for the synthesis of aryl glycosides bearing a formyl group (1 to 3).
A solution of the corresponding peracetylglycosyl bromide (1 equivalent [equiv.]) dissolved in acetone was added to a solution of vanillin (3 equiv.) in water containing 2.8 equimolar amounts of lithium hydroxide. The reaction mixture was stirred at room temperature for 2 h. The progress of the reaction was followed by thin-layer chromatography (TLC) (1:1 hexane/ethyl acetate). The mixture was concentrated to remove acetone and then diluted with water (10 ml) and washed with dichloromethane. The organic layer was separated and washed with 10% (wt/vol) NaOH aqueous solution and water until pH 7 was reached. The resulting organic phase was dried over sodium sulfate, filtered, and concentrated to dryness under conditions of reduced pressure.
4-Formyl-2-methoxyphenyl 2,3,4,6-tetra-O-acetyl-β-d-galactopyranoside (compound 1). Obtained from general procedure A as a white solid, yield 60%; mp 123.1 to 123.8°C (123 to 124°C [37]); [α]D, −8.1 (c 0.49, CH2Cl2); IR (ν̄/cm−1), 2,988, 2,901 (C-H sp3), 1,752, 1,740 (C=O), 1,693 (C=O), 1,590, 1,514 (C=C), 1,370 (C-H sp3); 1H NMR (400 MHz; CDCl3), δ 9.89 (s, 1H, CHO); 7.43–7.40 (m, 2H); 7.25 (d, 1H, J = 8.4 Hz); 5.55 (t, 1H, J = 8.0 Hz); 5.46 (d, 1H, J = 2.8 Hz); 5.12 (dd, 1H, J = 8.0 Hz, J = 2.8 Hz); 5.05 (d, 1H, J = 8.0 Hz); 4.23 (dd, 1H, J = 11.8 Hz, J = 6.8 Hz); 4.16 (dd, 1H, J = 11.8 Hz, J = 6.4 Hz); 4.07–4.03 (m, 1H); 3.90 (s, 3H, OCH3); 2.17–2.02 (4s, 12H,COCH3); 13C NMR (100 MHz; CDCl3) δ 190.89 (CHO); 170.33–169.35 (4C, OCOCH3); 151.29; 150.97; 132.77; 125.39; 117.99; 110.78; 100.35; 71.28; 70.60; 68.48; 66.82; 61.31; 56.13 (OCH3); 20.69–20.58 (4C, COCH3).
4-Formyl-2-methoxyphenyl 2,3,4,6-tetra-O-acetyl-β-d-glucopyranoside (compound 2). Obtained from general procedure A as a white solid, yield 57%; mp 136.1 to 137.3°C (135 to 137°C [38]); [α]D −39.2 (c 0.51, CH2Cl2); IR (ν̄/cm−1): 2,988, 2,901 (C-H sp3), 1,753, 1,737 (C=O), 1,694 (C=O), 1,591, 1,510 (C=C), 1,378 (C-H sp3); 1H NMR (400 MHz; CDCl3) δ 9.89 (s, 1H, CHO); 7.43–7.40 (m, 2H); 7.21 (d, 1H, J = 8.0 Hz); 5.34–5.28 (m, 2H); 5.13 (t, 1H, J = 6.8 Hz); 5.09 (d, 1H, J = 6.4 Hz); 4.27 (dd, 1H, J = 12.4 Hz, J = 5.2 Hz); 4.18 (dd, 1H, J = 12.4 Hz, J = 2.4 Hz); 3.86 (s, 3H, OCH3); 3.85–3.70 (m, 1H); 2.07–2.04 (4s, 12H, COCH3); 13C NMR (100 MHz; CDCl3) δ 190.89 (CHO); 170.52–169.25 (4C, OCOCH3); 151.11; 151.03; 132.86; 125.34; 118.23; 110.85; 99.73; 72.41; 72.28; 71.06; 68.28; 61.90; 56.12 (OCH3); 20.67–20.59 (4C, COCH3).
4-Formyl-2-methoxyphenyl 2,3,4,6-tetra-O-acetyl-β-d-galactopyranosyl-(1→4)-2,3,6-tri-O-acetyl-β-d-glucopyranoside (compound 3). Obtained from general procedure A as a white solid, yield 52%; mp 120.0 to 122.1°C (121 to 124°C [39]); [α]D −12.5 (c 0.48, CH2Cl2); IR (ν̄/cm−1): 2,942 (C-H sp3), 1,741 (C=O), 1,687 (C=O), 1,592, 1,507 (C=C), 1,424, 1,370 (C-H sp3); 1H NMR (400 MHz; CDCl3) δ 9.88 (s, 1H, CHO); 7.42–7.39 (m, 2H); 7.17 (d, 1H, J = 8.0 Hz); 5.35 (d, 1H, J = 8.0 Hz); 5.32 (t, 1H, J = 8.8 Hz); 5.22 (t, 1H, J = 8.8 Hz); 5.14–5.08 (m, 2H); 4.97 (dd, 1H, J = 12.0 Hz, J = 3.2 Hz); 4.53–4.51 (m, 2H); 4.17–4.06 (m, 3H); 3.93–3.89 (m, 2H); 3.88 (s, 3H); 3,79–3.75 (m, 1H); 2.15–1.97 (s, 21H); 13C NMR (100 MHz; CDCl3) δ 190.89 (CHO); 170.38–169.10 (7C, COCH3); 151.16; 150.93; 132.72; 125.37; 117.87; 110.77; 101.13; 99.35; 76.05; 73.04; 72.47; 71.33; 70.94; 70.79; 69.14; 66.64; 61.84; 60.84; 56.11 (OCH3); 20.79–20.50 (7C, COCH3).
General procedure B, for the synthesis of thiosemicarbazones.
Three drops of glacial acetic acid were added to a suspension of 1 equiv. of thiosemicarbazide and 1 equiv. of the corresponding aldehyde or ketone in ethanol. The reaction mixture was kept under conditions of reflux and magnetic stirring for 2 h. Then, the resulting suspension was vacuum filtered and washed with cold distilled water.
2-Phenylmethylenehydrazinecarbothioamide (compound 4a). Obtained from general procedure B as a white solid (91% yield); mp 157.3 to 158.6°C (literature values [lit.], 157 to 159°C [40]). IR (ν̄/cm−1): 3,418 (NH), 1,589 (C=N), 1,539, 1,447 (C=C aromatic). 1H NMR (200 MHz, dimethyl sulfoxide [DMSO] d6), δ/ppm: 11.43 (1 H, s, NH); 8.20 (1 H, s, NH2); 8.05 (1 H, s, CH=N); 7.99 (1 H, s, NH2); 7.78 (2 H, m, ArH); 7.39 (3 H, m, ArH).
2-[(4-Methylphenyl)methylene]hydrazinecarbothioamide (compound 4b). Obtained from general procedure B as a white solid (82% yield); mp 168.5 to 169.8°C (lit. 162 to 163°C [40]). IR (ν̄/cm−1): 3,398 (NH), 1,596 (C=N), 1,509, 1,462 (C=C aromatic).
2-(4-Pyridinylmethylene)hydrazinecarbothioamide (compound 4d). Obtained from general procedure B, as a pale yellow solid (80% yield); mp 235 to 236°C (lit. 240°C [41]). IR (ν̄/cm−1): 3,420 (NH), 1,591 (C=N), 1,536 (C=C aromatic). 1H NMR (200 MHz, DMSO-d6), δ/ppm: 11.69 (1 H, s, NH); 8.58 (2 H, d, H-2 pyridine); 8.04 (1 H, s, NH2); 8.21 (1 H, s, NH2); 8.00 (1 H, s, CH=N); 7.76 (2 H, d, H-3 pyridine).
2-(1H-Pyrrol-2-ylmethylene)hydrazinecarbothioamide (compound 4e). Obtained from general procedure B, as a solid (66% yield); mp 191.9 to 193.6°C (lit. 195 to 197°C [42]). IR (ν̄/cm−1): 3,445 (NH), 1,583 (C=N), 1,530, 1,550 (C=C aromatic).
1-Cyclopentylidenethiosemicarbazide (compound 4f). Obtained from general procedure B, as a solid (51% yield); mp 155.3 to 157°C (lit. 152 to 154°C [43]). IR (ν̄/cm−1): 3,375 (NH), 1,586 (C=N), 1,508, 1,448 (C=C aromatic). 1H NMR (200 MHz, CDCl3), δ/ppm: 8.61 (1 H, s, NH); 7.33 (1 H, s, NH2); 6.67 (1 H, s, NH2); 2.36 (4 H, m, CH2); 1.85 (4 H, m, CH2).
1-Cyclohexylidenethiosemicarbazide (compound 4g). Obtained from general procedure B, as a solid (65% yield); mp 160.2 to 161.1°C (lit. 154 to 155°C [43]). IR (ν̄/cm−1): 3,375 (NH), 1,583 (C=N), 1,505, 1,461 (C=C). 1H NMR (200 MHz, CDCl3), δ/ppm: 8.93 (1 H, s, NH); 7.30 (1 H, s, NH2); 6.60 (1 H, s, NH2); 2.32 (4 H, m, CH2); 1.67 (6 H, m, CH2).
2-[[3-Methoxy-4-[(2,3,4,6-tetra-O-acetyl-β-d-galactopyranosyl)oxy]phenyl]methylene] hydrazinecarbothioamide (compound 4i). Obtained from general procedure B as a solid, yield 76%; mp 127.2 to 129.9°C; [α]D −18.9 (c 0.53, EtOH); IR (ν̄/cm−1): 3,454 (NH), 1,743 (C=O), 1,597 (C=N), 1,504, 1,450 (C=C), 1,068 (C-O); 1H NMR (200 MHz; CDCl3) δ/ppm: 10.27 (1H, s, NH); 7.90 (1H, s, CH=N); 7.27–7.12 (4H, m, ArH); 6.64 (1H, s, NH2); 5.57–5.44 (m, 2H); 5.13 (1H, dd, J = 10.4 Hz, J = 3.2 Hz); 4.97 (1H, d, J = 8.0 Hz); 4.29–4.00 (3H, m); 3.86 (3H, s, OCH3); 2.17–2.02 (12H, 4s, COCH3); 13C NMR (50 MHz; CDCl3) δ/ppm: 177.55 (C=S); 170.02–169.09 (4C, OCOCH3); 150.38; 147.96; 143.33; 128.81; 121.22; 118.59; 109.59; 100.33; 70.65; 70.19; 68.10; 66.40; 60.86; 55.73 (OCH3); 20.34–20.25 (4C, COCH3); high-sensitivity mass spectrometry (HRMS) value (m/z), 556.1590 [M+H]+, calculated 556.1596 C23H30N3O11S+.
2-[[3-Methoxy-4-[(2,3,4,6-tetra-O-acetyl-β-d-glucopyranosyl)oxy]phenyl]methylene] hydrazinecarbothioamide (compound 4j). Obtained from general procedure B as a solid, yield 84%; mp 119.4 to 122.9°C; [α]D −28.6 (c 0.28, MeOH); IR (ν̄/cm−1): 3,278 (NH), 1,739 (C=O), 1,597 (C=N), 1,504, 1,450 (C=C), 1,030 (C-O); 1H NMR (200 MHz; CDCl3) δ/ppm: 10.37 (1H, s, NH); 7.91 (1H, s, CH=N); 7.21–7.10 (4H, m, ArH); 6.72 (1H, s, NH2); 5.31–5.00 (m, 4H); 4.32–4.15 (2H, m); 3.84 (4H, m, OCH3 + H-5); 2.07–2.04 (12H, 4s, COCH3); HRMS (m/z) 556.1591 [M+H]+, calculated 556.1596 C23H30N3O11S+.
2-[[3-Methoxy-4-[(2,3,4,6-tetra-O-acetyl-β-d-galactopyranosyl-(1→4)-2,3,6-tri-O-acetyl-β-d-glucopyranosyl)oxy]phenyl]methylene]hydrazinecarbothioamide (compound 4k). Obtained from general procedure B as a solid, yield 71%; mp 220.1 to 121.8°C; [α]D −12.0 (c 0.5, MeOH); IR (ν̄/cm−1): 3,469 (NH), 1,739 (C=O), 1,594 (C=N), 1,528, 1,505 (C=C), 1,046 (C-O); 1H NMR (200 MHz; CDCl3) δ/ppm: 10.0 (1H, s, NH); 7.86 (1H, s, CH=N); 7.27–7.08 (4H, m, ArH); 6.60 (1H, s, NH2); 5.37–4.98 (7H, m); 4.53 (2H, d, J = 7.8 Hz); 4.16–3.77 (8H, m); 2.16–1.97 (s, 21H); 13C NMR (50 MHz; CDCl3) δ 178 (C=S); 170.33–169.05 (7C, COCH3); 150.72; 148.20; 143.60; 129.08; 121.63; 118.88; 109.92; 100.99; 99.75; 75.97; 72.84; 72.36; 71.25; 70.85; 70.62; 69.01; 66.53; 61.72; 60.72; 56.07 (OCH3); 20.74–20.45 (7C, COCH3); HRMS (m/z) 844.2423 [M+H]+, calculated 844.2441 C35H46N3O19S+.
General procedure C, for the synthesis of thiazole derivatives.
One equiv. of 2-bromoacetophenone was added to a solution of 1 equiv. of thiosemicarbazone in isopropyl alcohol, and the resulting mixture was kept under conditions of reflux and magnetic stirring. The completion of the reaction was monitored by TLC (approximately 2 h). After cooling to room temperature, the formed precipitate was filtered and washed with a saturated solution of NaHCO3 followed by cold distilled water. The final product was recrystallized in ethanol.
Benzaldehyde-2-(4-phenyl-2-thiazolyl)hydrazone (compound 5a). Obtained from general procedure C as a pale solid, yield 63%; mp 187.6 to 188.8°C (lit. 186 to 187°C [44]). IR (ν̄/cm−1): 3,306 (NH), 1,557 (C=N), 1,482, 1,428 (C=C). 1H NMR (200 MHz, DMSO-d6), δ/ppm: 12.16 (1 H, s, NH); 7.88 (2 H, broad s, ArH); 7.61–7.08 (9 H, m, 8 × ArH and CH=N); 6.85 (1 H, s, H-thiazole).
4-Methylbenzaldehyde 2-(4-phenyl-2-thiazolyl)hydrazone (compound 5b).Obtained from general procedure C as a pale solid, yield 73%; mp 192.4 to 194.3°C (lit. 195 to 196°C [44]). IR (ν̄/cm−1): 3,279 (NH), 1,553 (C=N), 1,509, 1,480 (C=C).
4-N,N-Dimethylbenzaldehyde-2-(4-phenyl-2-thiazolyl)hydrazone (compound 5c). Obtained from general procedure C as a solid, yield 81%; mp 203.4 to 204.4°C (lit. 207 to 208°C [45]). IR (ν̄/cm−1): 3,288 (NH), 1,603 (C=N), 1,520, 1,480 (C=C). 1H NMR (200 MHz, DMSO-d6), δ/ppm: 11.85 (1 H, s, NH); 7.92–7.86 (3 H, m, ArH); 7.48–7.24 (6 H, m, ArH, and CH=N); 6.74 (2 H, broad s, ArH, and H-thiazole).
4-Pyridinylcarbaldehyde-2-(4-phenyl-2-thiazolyl)hydrazone (compound 5d). Obtained from general procedure C as an orange solid, yield 53%; mp 240.1 to 242.2°C (lit. 250 to 252°C [46]). IR (ν̄/cm−1): 3,450 (NH), 1,571 (C=N), 1,482, 1,441 (C=C). 1H NMR (200 MHz, DMSO-d6), δ/ppm: 12.57 (1 H, broad s, NH); 8.60 (1 H, broad s, ArH); 8.00 (1 H, s, CH=N); 7.85 (2 H, d, ArH); 7.61 (2 H, broad s, ArH); 7.38 (4 H, m, ArH, and H-thiazole).
Pyrrole-2-carboxaldehyde-(4-phenyl-1,3-thiazol-2-yl)hydrazone (compound 5e). Obtained from general procedure C as a dark solid, yield 99%; mp 126°C (lit. 125°C [47]). IR (ν̄/cm−1): 3,304 (NH), 1,623 (C=N), 1,498, 1,422 (C=C).
2-[(2-Cyclopentylmethylene)hydrazino]-4-phenyl-thiazole (compound 5f). Obtained from general procedure C as a solid, yield 98%, mp 170.2 to 172.4°C (lit. 156 to 157°C [48]). IR (ν̄/cm−1): 3,438 (NH), 1,626 (C=N), 1,560, 1,481 (C=C). 1H NMR (200 MHz, CDCl3), δ/ppm: 7.73 (2 H, dd, J = 7.8 Hz; J = 2.0 Hz, ArH); 7.51–7.36 (3H, m, ArH, and NH); 6.73 (1H, s, H-thiazole); 2.61–2.48 (4H, m, CH2); 1.99–1.79 (4H, m, CH2).
2-[(2-Cyclohexylmethylene)hydrazino]-4-phenyl-thiazole (compound 5g). Obtained from general procedure C as a solid, yield 51%, mp 149 to 151°C (lit. 148 to 149°C [48]). IR (ν̄/cm−1): 3,050 (NH), 1,610 (C=N), 1,476, 1,431 (C=C).
2-[(6,6-Dimethylbicyclo[3.1.1]hept-2-en-2-yl)methylene]hydrazino-4-phenyl-1,3-thiazole (compound 5h). Obtained from general procedure C as a solid, yield 29%, mp 152.4 to 154.2°C. IR (ν̄/cm−1): 3,363 (NH), 1,615 (C=N), 1,495, 1,470 (C=C). 1H NMR (200 MHz, DMSO-d6), δ/ppm: 7.87–7.69 (3H, m, ArH); 7.47–7.29 (4H, m, ArH, CH=N, and NH); 7.26 (1H, s, H-thiazole); 5.97 (1H, s, C=CH); 2.93–2.85 (1H, m, CH); 2.48–2.33 (3H, m, CH2, CH); 2.14 (1H, m, CH2); 1.33 (3H, s, CH3); 1.13–1.03 (1H, m, CH2); 0.78 (3H, s, CH3). HRMS (m/z) 324.1530 [M+H]+, calculated 324.1529 C19H22N3S+.
2-[[3-Methoxy-4-[(2,3,4,6-tetra-O-acetyl-β-d-galactopyranosyl)oxy]phenyl]methylene]hydrazino-4-phenyl-thiazole (compound 5i). Obtained from general procedure C as a solid, yield 41%; mp 111.4 to 113.7°C; [α]D −36 (c 0.5, MeOH); IR (ν̄/cm−1): 2,952 (NH), 1,748 (C=O), 1,602 (C=N), 1,567, 1,510 (C=C), 1,071 (C-O); 1H NMR (200 MHz; CDCl3) δ/ppm: 7.80 (2H, d, J = 7.0 Hz, ArH); 7.44–7.29 (4H, m, ArH, and N=CH); 7.05–6.97 (3H, m, ArH); 6.81–6.75 (2H, m, H-thiazole, and NH); 5.31–4.96 (4H, m); 4.33–4.13 (2H, m); 3.82 (4H, m, OCH3, and H-5); 2.08–2.04 (12H, 4s, COCH3); HRMS (m/z) 656.1905 [M+H]+, calculated 656.1909 C31H34N3O11S+.
Enzyme expression and purification.
Recombinant enzymes cruzain, rhodesain, and SmCB1 were expressed and purified as previously described (11, 49–51).
Assay measuring cruzain, rhodesain, and SmCB1 activity.
Cruzain, rhodesain, and SmCB1 activity were measured by monitoring the cleavage of the fluorogenic substrate Z-Phe-Arg-aminomethylcoumarin (Z-FR-AMC) in a Synergy 2 plate reader (Biotek) at the Center of Flow Cytometry and Fluorimetry at the Biochemistry and Immunology Department (Universidade Federal de Minas Gerais [UFMG]). All assays were performed in triplicate using 0.1 M sodium acetate (pH 5.5) in the presence of 1 mM beta-mercaptoethanol and 0.01% Triton X-100. The final concentrations of cruzain and rhodesain were 0.5 nM, and the substrate concentration was 2.5 μM (Km = 1 μM). For assays with SmCB1, the enzyme concentration was 8 nM and the substrate concentration was 5 μM. Enzyme kinetics was followed by continuous reading for 5 min at 12-s intervals, in the cases of cruzain and rhodesain, and for 30 min at 23-s intervals, in the case of SmCB1. Activity was calculated based on initial velocity rates compared to those seen with a DMSO control. For evaluation of time-dependent inhibition, percentages of enzyme inhibition by a compound with or without preincubation with enzyme for 10 min were compared. First, the inhibitory activity for all enzymes was screened at 100 μM compound. When the inhibition was higher than 80%, the IC50 was determined based on at least two IC50 curves. Each curve was determined on the basis of at least seven compound concentrations, in each case in triplicate, and the data were analyzed with GraphPad Prism 5.0, employing a nonlinear regression analysis of log (inhibitor) versus response with a variable slope and four parameters (data not shown). The values reported in Tables 1, 2, and 3 refer to averages and standard deviations of the results of comparisons of the values obtained for at least two curves.
TABLE 1.
Inhibition of rhodesain, cruzain, and SmCB1 by a series of thiosemicarbazones and cyclic analoguesa
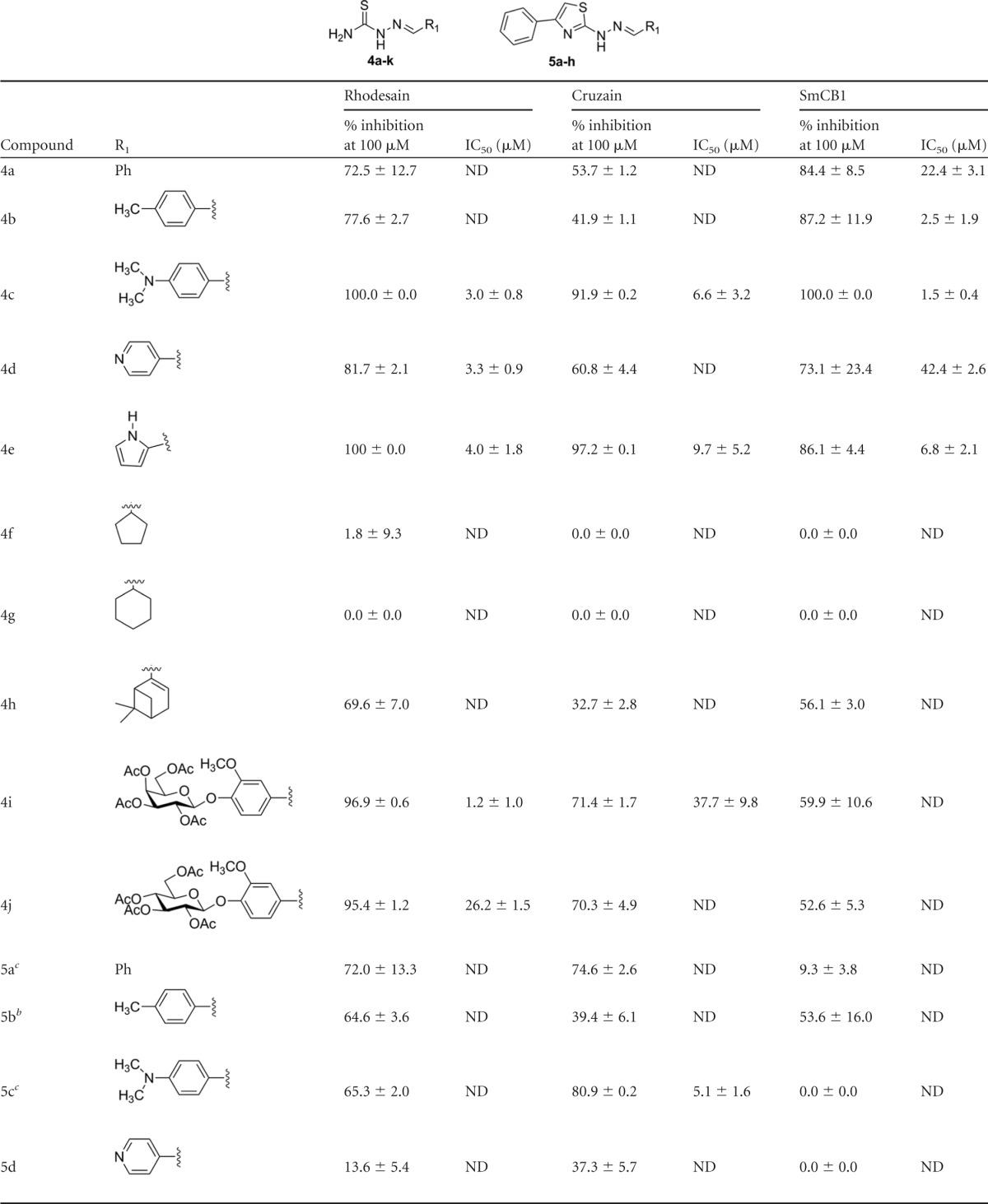
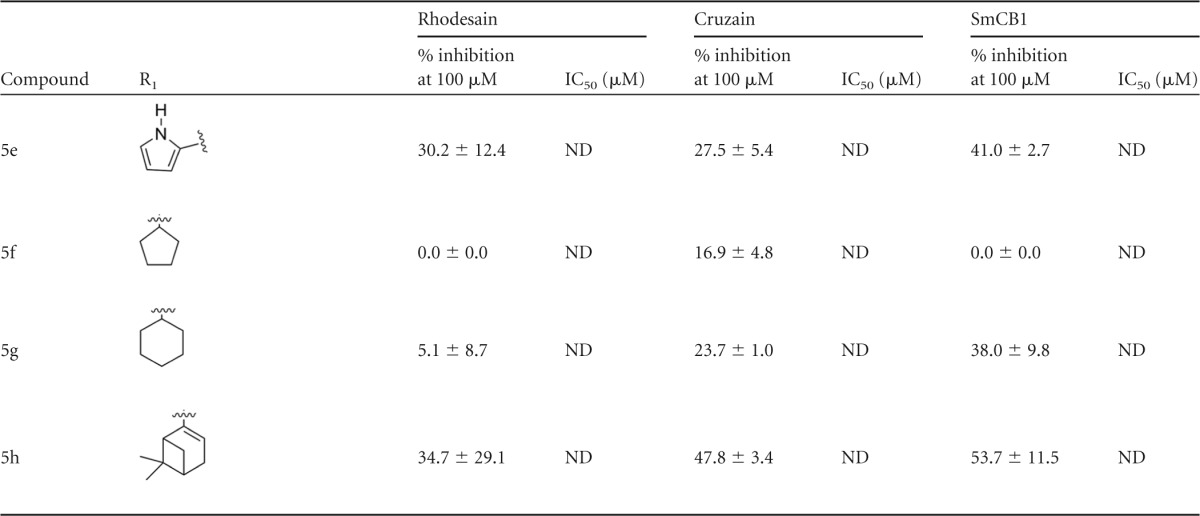
Percentages of inhibition are reported as averages and standard deviations of the results of at least two independent experiments, each performed in triplicate. IC50s represent averages and standard deviations of the results of at least two independent experiments. ND, not determined.
Compound evaluated at 75 μM.
Compound evaluated at 50 μM.
TABLE 2.
Inhibition of rhodesain, cruzain, and SmCB1 by 4i analogues modified in the sugar moietya
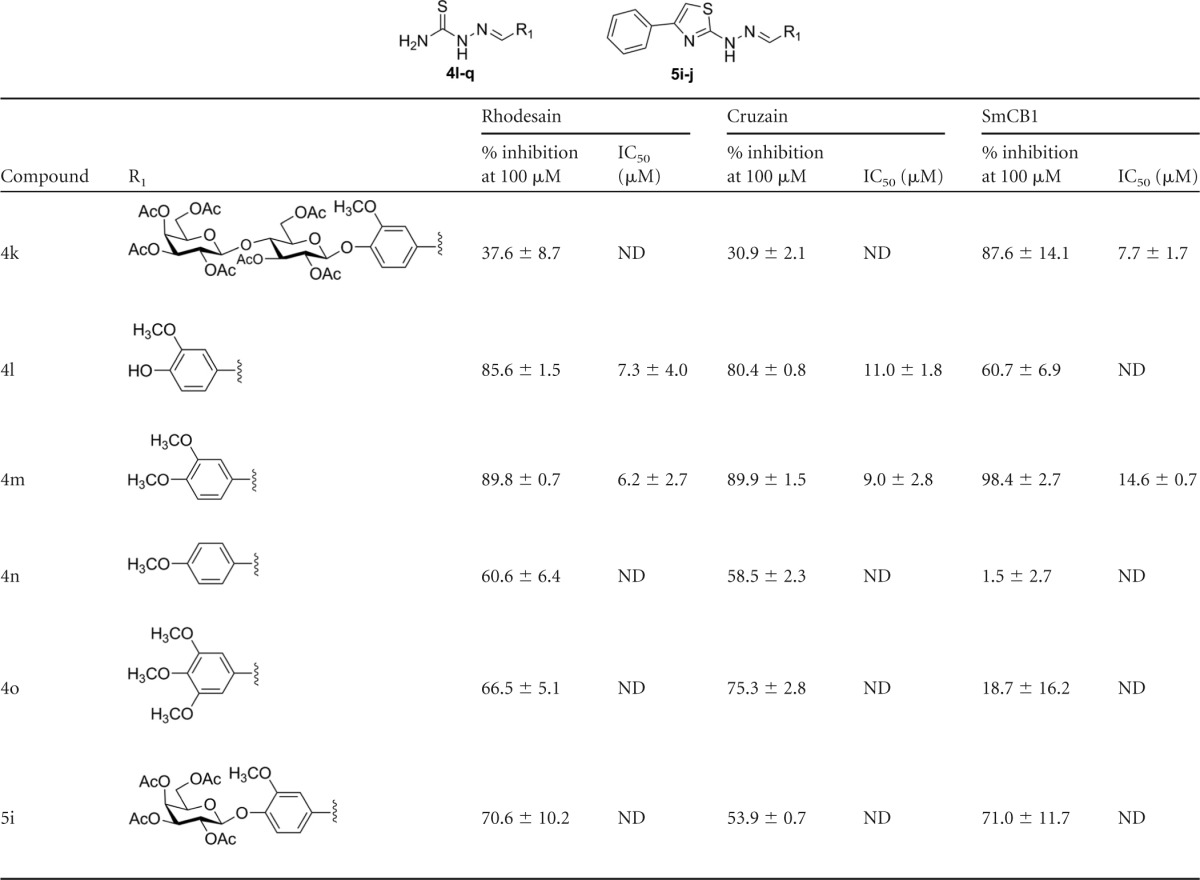
Percentages of inhibition are reported as averages and standard deviations of the results of at least two independent experiments, each performed in triplicate. IC50s represent averages and standard deviations of the results of at least two independent experiments. ND, not determined.
TABLE 3.
Inhibition of rhodesain, cruzain, and SmCB1 by 4i analogues without thiosemicarbazonesa

Percentages of inhibition are reported as averages and standard deviations of the results of at least two independent experiments, each performed in triplicate. IC50s represent averages and standard deviations of the results of at least two independent experiments. ND, not determined.
T. brucei brucei 221 maintenance.
The parasites were cultured in HMI-9 medium supplemented with 20% heat-inactivated fetal bovine serum (FBS) (Gibco) starting at a density of 2 × 104 parasites/ml and subculturing every other day.
Preparation of compound plates.
All compounds were stored in powder. Solutions at 10 mM in neat DMSO were prepared a few hours before assay experimentation and were seeded in row A of a 384-well plate (Greiner 784201). Serial dilutions with 2-fold factors (from 10 mM to 305 nM) were prepared until row P was reached. The compounds were pinned, and 50 nl of each well was transferred to the assay plate (Greiner 781091) containing 25 μl of HMI-9 media supplemented with 20% FBS.
T. brucei brucei screening assay.
Five thousand (5 × 103) parasites were seeded in a volume of 25 μl in the 384-well plate already containing the compounds in serial dilution, with the highest concentration tested being 10 μM. After 72 h of incubation at 37°C and 5% CO2, each well received 12.5 μl of Sybr green in lysis solution (30 mM Tris [pH 7.5], 7.5 mM EDTA, 0.012% saponin, 0.12% Triton X-100, 0.3 μl/ml of Sybr green). After addition of lysis solution, the plates were sealed with plastic film and the mixture was subjected to vortex mixing for 45 s at 1,700 rpm (MixMate). The mixture was incubated for 1 h at room temperature, and the plate was read in a Flexstation (Molecular Devices) to detect the fluorescence signal corresponding to parasite viability (excitation [Ex], 485 nm; emission [Em], 530 nm). Raw viability data consist of values of relative fluorescence units (RFU) obtained from the reading of Sybr green that binds to the viable parasite's DNA and include maximum (max) and minimum (min) controls and measured values. Thymerasol (2 μM) was used as the reference drug 100% effective concentration (EC100). The activity normalization was done based on the nontreated mixture (negative control, 0% activity) and the reference drug at the EC100 (positive control, 100% activity), with at least 16 wells for each control per plate.
T. cruzi screening assay.
The assay was performed using T. cruzi (Tulahuen strain) expressing Escherichia coli β-galactosidase as the reporter gene (52, 53). Infective trypomastigote forms were obtained through culture in monolayers of mouse L929 fibroblasts in RPMI 1640 medium (pH 7.2 to 7.4) without phenol red (Gibco BRL) plus 10% fetal bovine serum and 2 mM glutamine. For the bioassay, 4,000 L929 cells in 80 μl of supplemented medium were added to each well of a 96-well microtiter plate. After an overnight incubation, 40,000 trypomastigotes in 20 μl were added to the cells and incubated for 2 h. The medium containing extracellular parasites was replaced with 200 μl of fresh medium, and the plate was incubated for an additional 48 h to establish the infection. For IC50 determination, the cells were exposed to active samples at serial decreasing dilutions starting at 1,000 μM in DMSO (less than 1% in RPMI 1640 medium), and the plate was incubated for 96 h. After this period, 50 μl of 500 μM chlorophenol red β-d-galactopyranoside (CPRG)–0.5% Nonidet P40 was added to each well, and the plate was incubated for 16 to 20 h, after which the absorbance at 570 nm was measured. Controls with uninfected cells, untreated infected cells, and infected cells treated with benznidazole at 1 μg/ml (3.8 μM) (positive control) or 1% DMSO were used (54). The results were expressed as the percentage of T. cruzi growth inhibition in compound-tested cells compared to the infected cells and untreated cells. IC50s were calculated by linear interpolation. Four replicate experiments were run in the same plate, and the experiments were repeated at least once.
In vitro assay for analysis of cell viability.
The compounds active against T. cruzi were tested in vitro for cytotoxicity using L-929 cells and alamarBlue dye. The same cell number, time of the cell development, and time of compound exposure were used as had been used for the beta-galactosidase assay. The cells were exposed to compounds at increasing concentrations starting at the IC50 of T. cruzi. The compounds were tested in four replicate experiments. After 96 h of compound exposure, alamarBlue was added and the absorbance at 570 and 600 nm measured after 4 to 6 h. The cell viability was expressed as the percentage of the difference in the levels of reduction seen with the treated and untreated cells (53). IC50s were calculated by linear interpolation, and the selectivity index (SI) was determined by the ratio between the 50% cytotoxic concentration (CC50) and the IC50 against the parasite for each compound.
S. mansoni screening assay.
The acquisition, preparation, and in vitro maintenance of newly transformed S. mansoni schistosomula (derived from infection-stage cercariae) and adult parasites have been described previously by us (55, 56). We employed a Puerto Rican isolate of S. mansoni that had been cycled between Biomphalaria glabrata snails and female Golden Syrian hamsters (infected at 4 to 6 weeks of age) as the intermediate and definitive hosts, respectively. Maintenance and handling of small mammals were carried out in accordance with a protocol approved by the Institutional Animal Care and Use Committee (IACUC) of the University of California, San Francisco. For schistosomula, 200 to 300 parasites were incubated in 96-well flat-bottomed plates (Corning Costar 3599) containing 200 μl of Basch medium 169 (57) supplemented with 2.5% FBS and 1× penicillin-streptomycin solution in a 5% CO2 atmosphere at 37°C. For adult parasites, 5 male worms were incubated in 2 ml of the medium described above in 24-well flat-bottomed plates (Corning Costar 3526) under the same conditions. The compound, 100% DMSO, was added at the final concentrations indicated in Table 4. Controls employed the equivalent volume of DMSO at a final concentration never exceeding 0.5%. Parasite responses to chemical insult were adjudicated visually each day using a constrained descriptive nomenclature (55, 58). The types and number of phenotypic responses recorded were then converted into a “severity score” ranging from 0 (no effect) to 4 (severely compromised). Thus, for schistosomula and adults, alterations in shape (e.g., “rounding”), motility (“slow” or “overactive”), and density (“darkening”) were each awarded a score of from 1 to the maximum of 4. In addition, for adults, the inability to adhere to the bottom of the well was awarded a score of 1; damage to the integrity of the outer surface (tegument) is considered lethal to the parasite and was awarded the maximum score of 4.
TABLE 4.
Activity against Schistosoma mansoni schistosomula and adult worms
| Compound | Severity score against schistosomula on indicated day at: |
Severity score against adult worms on indicated day at 5 μM |
|||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 0.1 μM |
1 μM |
10 μM |
|||||||||||||
| 1 | 2 | 3 | 4 | 1 | 2 | 3 | 4 | 1 | 2 | 3 | 4 | 1 | 2 | 5 | |
| 4a | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 1 | NTb | NT | NT |
| 4b | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | NT | NT | NT |
| 4c | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | NT | NT | NT |
| 4d | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | NT | NT | NT |
| 4e | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | NT | NT | NT |
| 4f | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | NT | NT | NT |
| 4g | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | NT | NT | NT |
| 4h | 0 | 0 | 0 | 0 | 0 | 0 | 3 | 2 | 0 | 0 | 2 | 2 | 0 | 0 | 0 |
| 4i | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | NT | NT | NT |
| 4j | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | NT | NT | NT |
| 4k | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | NT | NT | NT |
| 4l | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | NT | NT | NT |
| 4n | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | NT | NT | NT |
| 4o | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | NT | NT | NT |
| 4q | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | NT | NT | NT |
| 5aa | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 1 | 2 | 0 | 4 | 4 | 0 | 0 | 0 |
| 5b | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 1 | 4 | 0 | 0 | 0 |
| 5c | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 4 | 0 | 0 | 4 | 4 | 0 | 0 | 0 |
| 5d | 1 | 0 | 1 | 4 | 2 | 2 | 3 | 4 | 1 | 4 | 4 | 4 | 0 | 1 | 2 |
| 5e | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 1 | 1 | NT | NT | NT |
| 5f | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | NT | NT | NT |
| 5g | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | NT | NT | NT |
| 5h | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | NT | NT | NT |
| 6a | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | NT | NT | NT |
| 6b | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | NT | NT | NT |
| 6c | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | NT | NT | NT |
| 6d | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | NT | NT | NT |
| 6f | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | NT | NT | NT |
| Galactosyl 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 2 | NT | NT | NT |
| Lactosyl 3 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | NT | NT | NT |
Compound evaluated at 50 μM due to solubility limitations.
NT, not tested.
RESULTS AND DISCUSSION
Chemistry.
The sugar derivatives (compounds 1, 2, and 3) were synthesized by reaction of vanillin with the corresponding peracetylglycosyl bromide using lithium hydroxide as a base, according to a method previously described (Fig. 1) (59). A series of thiosemicarbazones was synthesized by classical methods from an aldehyde or ketone and thiosemicarbazide, with yields in the range of 51% to 95%. Then, the thiosemicarbazones obtained were subjected to reaction with α-bromo-acetophenone, giving the corresponding thiazole heterocycles (yield, 29% to 98%) (Fig. 2). The stereochemistry at the C=N bond of the thiosemicarbazone derivatives was established by 1H NMR spectroscopy. The value of the chemical shift of the NH (9 to 12 ppm) is indicative of (E) configuration (60). Some semicarbazones were also synthesized, according to a previously described procedure (61), for comparison of the activity of semicarbazones with that of the thiosemicarbazones.
FIG 1.
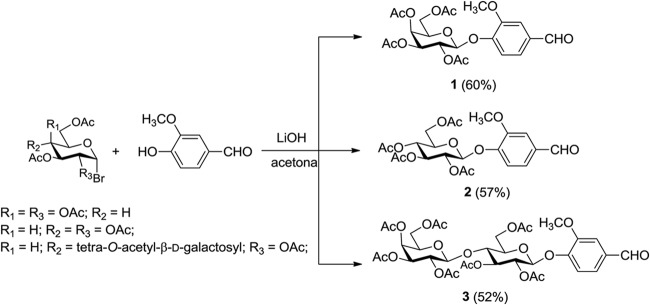
Synthesis of glycosides 1, 2, and 3.
FIG 2.
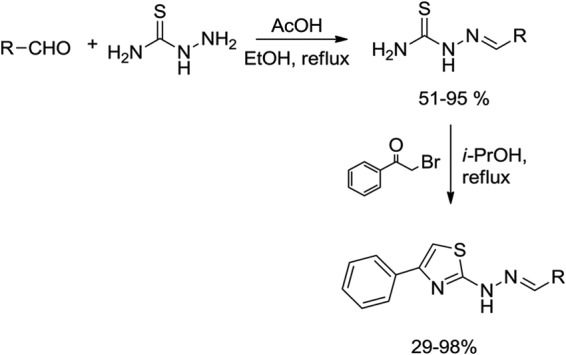
General synthetic route for preparation of thiosemicarbazones and their corresponding thiazole heterocycles.
Fifteen thiosemicarbazones and 9 thiazole analogues containing an aryl or heteroaryl, and/or cycloalkyl or glycosyl moieties, were prepared (Fig. 3). The introduction of the carbohydrate moiety (compounds 4i to 4k and 5i) was designed to modulate solubility and the properties of interaction (for example, by hydrogen bonding) with the molecular target (cysteine protease).
FIG 3.

Chemical structure of synthesized thiosemicarbazones 4a to 4o and cyclic analogues 5a to 5i.
Discovery of potent rhodesain, cruzain, and SmCB1 inhibitors.
An initial screen of 18 compounds, mostly thiosemicarbazones, was performed against rhodesain. Compounds were screened under two conditions: with and without a 10-min preincubation with the enzyme (Table 1). A clear time dependence was observed for the active compounds, as expected on the basis of the formation of a covalent bond between rhodesain and the inhibitor. To verify whether the differences in the percentages of inhibition observed were statistically significant, we applied an unpaired t test comparing the results of assays with and without preincubation. This analysis revealed that, for all compounds which inhibited rhodesain by more than 80% in the screen, the percentage of inhibition was higher when the enzyme was preincubated with the compound, and this difference is statistically significant, with P values of <0.0002 (data available from the authors upon request). Therefore, all subsequent assays were performed with a 10-min preincubation against the enzymes; the SAR discussion refers to the inhibition observed under this condition.
Eleven compounds inhibited rhodesain by at least 50% at 100 μM; IC50s were determined for five of them. Several trends were observed based on this initial screen. Comparison of compounds 4a and 4g indicates the importance of the aromatic ring. Addition of a methyl substituent at the para position did not affect inhibition (compound 4b), whereas a dimethylamine at the same position resulted in a more potent compound (compound 4c; IC50 = 3.0 ± 0.8 μM), as did the replacement of the phenyl by a pyridine (compound 4d; IC50 = 3.3 ± 0.9 μM) or by an imidazol (compound 4e; IC50 = 4.0 ± 1.8 μM). Three cyclic analogues were less soluble and in a few cases could not be evaluated at 100 μM. They were therefore assayed at 50 μM (compound 5c) or 75 μM (compounds 5a and 5b). Assay results for several pairs of compounds suggested that replacement of the thiosemicarbazones by a cyclized analogue decreases potency against the enzyme (compound 4d versus 5d, compound 4h versus 5h, compound 4e versus 5e, and compound 4j versus 5i), except for compound 4a versus 5a, which showed similar potencies. This initial screen resulted in the discovery of a potent rhodesain inhibitor, compound 4i, with an IC50 of 1.2 ± 1.0 μM. Comparison to compound 4j shows the importance of the sugar, since replacement of the acetylated galactose by an acetylated glucose, representing a difference in only one chiral center, resulted in a 20-fold decrease in potency (compound 4j; IC50 = 26.2 ± 1.5 μM).
The compounds were also evaluated against the T. cruzi and S. mansoni cysteine proteases cruzain and SmCB1. Like rhodesain, cruzain is a cathepsin L-like protease, and only two active residues differ between the two proteins. The similarity between the cathepsin B-like SmCB1 and rhodesain active sites is lower; however, common inhibitors have been reported for these enzymes (19). The SAR for the three enzymes showed several similarities and also interesting differences. The importance of the aromatic ring was confirmed (for compound 4a versus 4g), and, as observed for rhodesain, both the addition of a dimethylamine in this ring and its replacement by a pyrol increased potency against cruzain (for compound 4c, IC50 = 6.6 ± 3.2 μM; for compound 4e, IC50 = 9.7 ± 5.2 μM) and SmCB1 (for compound 4c, IC50 = 1.5 ± 0.4 μM; for compound 4e, IC50 = 6.8 ± 2.1 μM). However, in contrast to what was measured for rhodesain, against cruzain and SmCB1, the pyridine analogue (compound 4d) was not as potent as the compound containing a phenyl ring (compound 4a). Interestingly, although addition of the methyl substituent influenced the inhibition of neither cruzain nor rhodesain, potency against SmCB1 was increased 10-fold (IC50 = 22.4 ± 3.1 μM for compound 4a versus 2.5 ± 1.9 μM for compound 4b). Overall, thiosemicarbazones 4c and 4e were the most potent inhibitors of the three enzymes.
The most significant difference in potency was observed for compound 4i. Although this compound had low micromolar potency against rhodesain (IC50 = 1.2 ± 1.0 μM), it was approximately 35-fold less potent against cruzain (IC50 = 37.7 ± 9.8 μM) and essentially inactive against SmCB1 (the IC50 could not be determined). Despite the similarity of the active sites of cruzain and rhodesain, the bottom of the S2 pocket in cruzain and SmCB1 contains glutamates (Glu208 and Glu316, respectively), whereas rhodesain has an alanine in the equivalent position. The S2 pocket is therefore considerably more open in rhodesain, possibly providing an explanation for the ability of this enzyme to bind larger scaffolds. The 4j epimer showed lower potency against cruzain and SmCB1 than against rhodesain. Nevertheless, we observed that the two compounds (4i and 4j) were only modest inhibitors of these two enzymes.
SAR.
The SAR for compound 4i was exploited on the basis of the impact of removing or modifying the sugar (Table 2) and of removing the thiosemicarbazone or modifying it to a semicarbazone (Table 3). Significant differences were observed in the SAR for the three enzymes regarding modifications in the sugar. Removal of the sugar moiety decreased potency against rhodesain by at least 5-fold (for compound 4l, IC50 = 7.3 ± 4.0 μM; for compound 4m, IC50 = 6.2 ± 2.7 μM) or more, depending on the pattern of phenyl substitution (compounds 4n and 4o). On the other hand, this modification increased potency against cruzain by 6-fold, and compound 4l and compound 4m had IC50s of approximately 10 μM. It is worth noting that, in the analogues which do not contain a sugar (4l to 4o), the potencies against cruzain and rhodesain were similar.
Despite the sugar not being essential for binding, it could drastically affect inhibition. For example, addition of another sugar monomer (compound 4k) resulted in a compound inactive against both cruzain and rhodesain but active against SmCB1 (compound 4k; IC50 = 7.7 ± 1.7 μM).
Removal of the thiosemicarbazone (galactosyl 1 and lactosyl 3) or its replacement by semicarbazone (compound 6a versus 4n, 6b versus 4m, 6c versus 4l, 6d versus 4o, and 6f versus 4 h) resulted in compounds inactive against the three enzymes, the only exception being compound 6f against SmCB1 (IC50 = 5.2 ± 2.8 μM). This effect has also been reported for a related compound series (27) and can be explained by the more electrophilic nature of the thiosemicarbazones and by the mechanism of cysteine protease inhibition by these compounds.
Assays against parasites in vitro.
Compounds were evaluated against T. brucei, T. cruzi, and S. mansoni. To assess the antiparasitic activity of the 24 compounds against the bloodstream form of T. brucei brucei, we used a viability assay based on the fluorescence of the parasite's nucleic acid (54). Parasites were coincubated for 72 h with compound 2-fold serially diluted from 10 μM to 305 pM. At 10 μM, none of the 24 tested compounds showed more than 60% bioactivity, defined as the reduction in parasite numbers compared to those seen with the untreated control (data not shown).
For T. cruzi, compounds were tested against both the amastigote and trypomastigote forms of the Tulahuen strain (52). Weak trypanocidal activity was observed for this class, and five compounds generated IC50s under 100 μM. On the basis of the difference between the IC50s for parasites and the L929 mouse fibroblast cell line, a selectivity index (SI) could be determined for each compound. The SI ranged from 1.3 to 10.7 (data available from the authors upon request). Importantly, even though the trypanocidal IC50s were high, the trypanocidal concentration was not toxic to fibroblasts, and in the case of compounds 4b and 4 h, the IC50 values for fibroblasts were an order of magnitude greater than those for T. cruzi (data available from the authors upon request). Although those compounds with SI values of ≥10 might be considered prototypes for new trypanocidal drugs, they are not recommended for in vivo tests as the SI values do not cross the decision gate threshold of 50 (53).
For S. mansoni, screens were performed against postinvasion larvae (schistosomula) and adult parasites, as previously described (55, 56). Parasite responses to chemical insult were adjudicated visually each day using a constrained nomenclature (55, 58) that accounts for changes in shape, motility, and density. The types and numbers of phenotypic responses recorded are then converted into a severity score ranging from 0 (no effect) to 4 (severely compromised). Five compounds (5a, 5b, 5c, 5d, and 4h) showed activity against schistosomula. Among these compounds, only 5d was active against adult worms.
Conclusion.
Here we report the discovery of compound 4i, a sugar-containing thiosemicarbazone which showed low micromolar potency against rhodesain (IC50 = 1.2 ± 1.0 μM) and modest potency against cruzain (IC50 = 37.7 ± 9.8 μM). Synthesis of a series of analogues allowed determination of the SAR in this series and resulted in the identification of six rhodesain, four cruzain, and five SmCB1 inhibitors with IC50 values of ≤10 μM. Only three thiosemicarbazones (compounds 4c, 4e, and 4m) showed similar potencies against rhodesain, cruzain, and SmCB1, a result that demonstrates that considerable differences in the SAR for the three enzymes exist. In a few cases, using larger scaffolds, higher potency was observed against rhodesain. Direct assays of the most potent inhibitors of the parasites T. cruzi, T. brucei, and S. mansoni showed some antiparasitic activity but also suggested that further SAR modifications will be needed to produce lead compounds.
ACKNOWLEDGMENTS
R.S.F. acknowledges CNPq (grant 477435/2012-2), Capes (grants A118/2013 and Edital Biocomputacional AUXPE 3379/2013), and FAPEMIG (grant PPM-00357-14), and R.B.D.O. acknowledges CNPq (grant 4041130/2012-0) for financial support. G.A.N.P. acknowledges a postdoctoral fellowship from CAPES (grant A118/2013). J.H.M. and J.L.D.S.N. received funding from the European Community's 7th Framework Programme (602777) Project Kindred. Research by C.R.C. is supported in part by NIH-NIAD R21AI107390 and R01AI089896 awards.
We also thank the Center of Flow Cytometry and Fluorimetry at the Biochemistry and Immunology Department (UFMG) and the Program for Technological Development of Tools for Health-PDTIS-FIOCRUZ for use of its facilities and Plataforma de Bioprospecção RPT10A-PDTIS-CPqRR-Fiocruz for HRMS measurements.
REFERENCES
- 1.Caffrey CR, Secor WE. 2011. Schistosomiasis: from drug deployment to drug development. Curr Opin Infect Dis 24:410–417. doi: 10.1097/QCO.0b013e328349156f. [DOI] [PubMed] [Google Scholar]
- 2.Coura JR, Vinas PA. 2010. Chagas disease: a new worldwide challenge. 465:S6–S7. doi: 10.1038/nature09221. [DOI] [PubMed] [Google Scholar]
- 3.McKerrow JH. 1999. Development of cysteine protease inhibitors as chemotherapy for parasitic diseases: insights on safety, target validation, and mechanism of action. Int J Parasitol 29:833–837. doi: 10.1016/S0020-7519(99)00044-2. [DOI] [PubMed] [Google Scholar]
- 4.Sajid M, McKerrow JH. 2002. Cysteine proteases of parasitic organisms. Mol Biochem Parasitol 120:1–21. doi: 10.1016/S0166-6851(01)00438-8. [DOI] [PubMed] [Google Scholar]
- 5.Renslo AR, McKerrow JH. 2006. Drug discovery and development for neglected parasitic diseases. Nat Chem Biol 2:701–710. doi: 10.1038/nchembio837. [DOI] [PubMed] [Google Scholar]
- 6.McKerrow JH, Doyle PS, Engel JC, Podust LM, Robertson SA, Ferreira R, Saxton T, Arkin M, Kerr ID, Brinen LS, Craik CS. 2009. Two approaches to discovering and developing new drugs for Chagas disease. Mem Inst Oswaldo Cruz 104(Suppl 1):263–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Brak K, Kerr ID, Barrett KT, Fuchi N, Debnath M, Ang K, Engel JC, McKerrow JH, Doyle PS, Brinen LS, Ellman JA. 2010. Nonpeptidic tetrafluorophenoxymethyl ketone cruzain inhibitors as promising new leads for Chagas disease chemotherapy. J Med Chem 53:1763–1773. doi: 10.1021/jm901633v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bryant C, Kerr ID, Debnath M, Ang KKH, Ratnam J, Ferreira RS, Jaishankar P, Zhao D, Arkin MR, McKerrow JH, Brinen LS, Renslo AR. 2009. Novel non-peptidic vinylsulfones targeting the S2 and S3 subsites of parasite cysteine proteases. Bioorg Med Chem Lett 19:6218–6221. doi: 10.1016/j.bmcl.2009.08.098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Choe Y, Brinen LS, Price MS, Engel JC, Lange M, Grisostomi C, Weston SG, Pallai PV, Cheng H, Hardy LW, Hartsough DS, McMakin M, Tilton RF, Baldino CM, Craik CS. 2005. Development of alpha-keto-based inhibitors of cruzain, a cysteine protease implicated in Chagas disease. Bioorg Med Chem Lett 13:2141–2156. doi: 10.1016/j.bmc.2004.12.053. [DOI] [PubMed] [Google Scholar]
- 10.Ferreira RS, Bryant C, Ang KKH, McKerrow JH, Shoichet BK, Renslo AR. 2009. Divergent modes of enzyme inhibition in a homologous structure-activity series. J Med Chem 52:5005–5008. doi: 10.1021/jm9009229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ferreira RS, Simeonov A, Jadhav A, Eidam O, Mott BT, Keiser MJ, McKerrow JH, Maloney DJ, Irwin JJ, Shoichet BK. 2010. Complementarity between a docking and a high-throughput screen in discovering new cruzain inhibitors. J Med Chem 53:4891–4905. doi: 10.1021/jm100488w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ferreira RS, Dessoy MA, Pauli I, Souza ML, Krogh R, Sales AIL, Oliva G, Dias LC, Andricopulo AD. 2014. Synthesis, biological evaluation, and structure-activity relationships of potent noncovalent and nonpeptidic cruzain inhibitors as anti-Trypanosoma cruzi agents. J Med Chem 57:2380–2392. doi: 10.1021/jm401709b. [DOI] [PubMed] [Google Scholar]
- 13.Greenbaum DC, Mackey Z, Hansell E, Doyle P, Gut J, Caffrey CR, Lehrman J, Rosenthal PJ, McKerrow JH, Chibale K. 2004. Synthesis and structure-activity relationships of parasiticidal thiosemicarbazone cysteine protease inhibitors against Plasmodium falciparum, Trypanosoma brucei, and Trypanosoma cruzi. J Med Chem 47:3212–3219. doi: 10.1021/jm030549j. [DOI] [PubMed] [Google Scholar]
- 14.Magalhaes Moreira DR, de Oliveira ADT, Teixeira de Moraes Gomes PA, de Simone CA, Villela FS, Ferreira RS, da Silva AC, dos Santos TAR, Brelaz de Castro MCA, Pereira VRA, Leite ACL. 2014. Conformational restriction of aryl thiosemicarbazones produces potent and selective anti-Trypanosoma cruzi compounds which induce apoptotic parasite death. Eur J Med Chem 75:467–478. doi: 10.1016/j.ejmech.2014.02.001. [DOI] [PubMed] [Google Scholar]
- 15.Mallari JP, Shelat A, Kosinski A, Caffrey CR, Connelly M, Zhu F, McKerrow JH, Guy RK. 2008. Discovery of trypanocidal thiosemicarbazone inhibitors of rhodesain and TbcatB. Bioorg Med Chem Lett 18:2883–2885. doi: 10.1016/j.bmcl.2008.03.083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mott BT, Ferreira RS, Simeonov A, Jadhav A, Ang KK-H, Leister W, Shen M, Silveira JT, Doyle PS, Arkin MR, McKerrow JH, Inglese J, Austin CP, Thomas CJ, Shoichet BK, Maloney DJ. 2010. Identification and optimization of inhibitors of trypanosomal cysteine proteases: cruzain, rhodesain, and TbCatB. J Med Chem 53:52–60. doi: 10.1021/jm901069a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.dos Santos Filho JM, Moreira DRM, de Simone CA, Ferreira RS, McKerrow JH, Meira CS, Guimarães ET, Soares MBP. 2012. Optimization of anti-Trypanosoma cruzi oxadiazoles leads to identification of compounds with efficacy in infected mice. Bioorg Med Chem 20:6423–6433. doi: 10.1016/j.bmc.2012.08.047. [DOI] [PubMed] [Google Scholar]
- 18.Abdulla MH, O'Brien T, Mackey ZB, Sajid M, Grab DJ, McKerrow JH. 2008. RNA interference of Trypanosoma brucei cathepsin B and L affects disease progression in a mouse model. PLoS Negl Trop Dis 2:e298. doi: 10.1371/journal.pntd.0000298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Abdulla M-H, Lim K-C, Sajid M, McKerrow JH, Caffrey CR. 2007. Schistosomiasis mansoni: novel chemotherapy using a cysteine protease inhibitor. PLoS Med 4:e14. doi: 10.1371/journal.pmed.0040014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Doyle PS, Zhou YM, Engel JC, McKerrow JH. 2007. A cysteine protease inhibitor cures Chagas disease in an immunodeficient-mouse model of infection. Antimicrob Agents Chemother 51:3932–3939. doi: 10.1128/AAC.00436-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Engel JC, Doyle PS, Hsieh I, McKerrow JH. 1998. Cysteine protease inhibitors cure an experimental Trypanosoma cruzi infection. J Exp Med 188:725–734. doi: 10.1084/jem.188.4.725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Palmer JT, Rasnick D, Klaus JL, Bromme D. 1995. Vinyl sulfones as mechanism-based cysteine protease inhibitors. J Med Chem 38:3193–3196. doi: 10.1021/jm00017a002. [DOI] [PubMed] [Google Scholar]
- 23.Roush WR, Gwaltney SL II, Cheng J, Scheidt KA, McKerrow JH, Hansell E. 1998. Vinyl sulfonate esters and vinyl sulfonamides: potent, irreversible inhibitors of cysteine proteases. J Am Chem Soc 120:10994–10995. doi: 10.1021/ja981792o. [DOI] [Google Scholar]
- 24.Roush WR, Cheng J, Knapp-Reed B, Alvarez-Hernandez A, McKerrow JH, Hansell E, Engel JC. 2001. Potent second generation vinyl sulfonamide inhibitors of the trypanosomal cysteine protease cruzain. Bioorg Med Chem Lett 11:2759–2762. doi: 10.1016/S0960-894X(01)00566-2. [DOI] [PubMed] [Google Scholar]
- 25.Kerr ID, Lee JH, Farady CJ, Marion R, Rickert M, Sajid M, Pandey KC, Caffrey CR, Legac J, Hansell E, McKerrow JH, Craik CS, Rosenthal PJ, Brinen LS. 2009. Vinyl sulfones as antiparasitic agents and a structural basis for drug design. J Biol Chem 284:25697–25703. doi: 10.1074/jbc.M109.014340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Brak K, Doyle PS, McKerrow JH, Ellman JA. 2008. Identification of a new class of nonpeptidic inhibitors of cruzain. J Am Chem Soc 130:6404–6410. doi: 10.1021/ja710254m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Du X, Guo C, Hansell E, Doyle PS, Caffrey CR, Holler TP, McKerrow JH, Cohen FE. 2002. Synthesis and structure-activity relationship study of potent trypanocidal thio semicarbazone inhibitors of the trypanosomal cysteine protease cruzain. J Med Chem 45:2695–2707. doi: 10.1021/jm010459j. [DOI] [PubMed] [Google Scholar]
- 28.Caputto ME, Fabian LE, Benítez D, Merlino A, Ríos N, Cerecetto H, Moltrasio GY, Moglioni AG, González M, Finkielsztein LM. 2011. Thiosemicarbazones derived from 1-indanones as new anti-Trypanosoma cruzi agents. Bioorg Med Chem 19:6818–6826. doi: 10.1016/j.bmc.2011.09.037. [DOI] [PubMed] [Google Scholar]
- 29.Fujii N, Mallari JP, Hansell EJ, Mackey Z, Doyle P, Zhou YM, Gut J, Rosenthal PJ, McKerrow JH, Guy RK. 2005. Discovery of potent thiosemicarbazone inhibitors of rhodesain and cruzain. Bioorg Med Chem Lett 15:121–123. doi: 10.1016/j.bmcl.2004.10.023. [DOI] [PubMed] [Google Scholar]
- 30.Siddiqui N, Ahsan W. 2011. Synthesis, anticonvulsant and toxicity screening of thiazolyl–thiadiazole derivatives. Med Chem Res 20:261–268. doi: 10.1007/s00044-010-9313-6. [DOI] [Google Scholar]
- 31.Bharti SK, Nath G, Tilak R, Singh SK. 2010. Synthesis, anti-bacterial and anti-fungal activities of some novel Schiff bases containing 2,4-disubstituted thiazole ring. Eur J Med Chem 45:651–660. doi: 10.1016/j.ejmech.2009.11.008. [DOI] [PubMed] [Google Scholar]
- 32.Singh N, Bhati SK, Kumar A. 2008. Thiazolyl/oxazolyl formazanyl indoles as potent anti-inflammatory agents. Eur J Med Chem 43:2597–2609. doi: 10.1016/j.ejmech.2007.12.024. [DOI] [PubMed] [Google Scholar]
- 33.Havrylyuk D, Mosula L, Zimenkovsky B, Vasylenko O, Gzella A, Lesyk R. 2010. Synthesis and anticancer activity evaluation of 4-thiazolidinones containing benzothiazole moiety. Eur J Med Chem 45:5012–5021. doi: 10.1016/j.ejmech.2010.08.008. [DOI] [PubMed] [Google Scholar]
- 34.Rawal RK, Tripathi R, Katti SB, Pannecouque C, De Clercq E. 2008. Design and synthesis of 2-(2,6-dibromophenyl)-3-heteroaryl-1,3-thiazolidin-4-ones as anti-HIV agents. Eur J Med Chem 43:2800–2806. doi: 10.1016/j.ejmech.2007.12.015. [DOI] [PubMed] [Google Scholar]
- 35.Wisastra R, Ghizzoni M, Maarsingh H, Minnaard AJ, Haisma HJ, Dekker FJ. 2011. Isothiazolones; thiol-reactive inhibitors of cysteine protease cathepsin B and histone acetyltransferase PCAF. Org Biomol Chem 9:1817. doi: 10.1039/c0ob00464b. [DOI] [PubMed] [Google Scholar]
- 36.Goud PM, Sheri A, Desai PV, Watkins EB, Tekwani B, Sabnis Y, Gut J, Rosenthal PJ, Avery MA. 2005. Design, synthesis and evaluation of trisubstituted thiazoles targeting Plasmodium falciparum cysteine proteases. Med Chem Res 14:74–105. doi: 10.1007/s00044-005-0126-y. [DOI] [Google Scholar]
- 37.Yuen C-T, Price RG, Chattagoon L, Richardson AC, Praill PFG. 1982. Colorimetric assays for N-acetyl-β-d-glucosaminidase and β-d-galactosidase in human urine using newly-developed ω-nitrostyryl substrates. Clin Chim Acta 124:195–204. doi: 10.1016/0009-8981(82)90387-4. [DOI] [PubMed] [Google Scholar]
- 38.Mohri K, Watanabe Y, Yoshida Y, Satoh M, Isobe K, Sugimoto N, Tsuda Y. 2003. Synthesis of glycosylcurcuminoids. Chem Pharm Bull (Tokyo) 51:1268–1272. doi: 10.1248/cpb.51.1268. [DOI] [PubMed] [Google Scholar]
- 39.Chen C-Y, Sun J-G, Liu F-Y, Fung K-P, Wu P, Huang Z-Z. 2012. Synthesis and biological evaluation of glycosylated psoralen derivatives. Tetrahedron 68:2598–2606. doi: 10.1016/j.tet.2012.01.090. [DOI] [Google Scholar]
- 40.Rajak H, Agarawal A, Parmar P, Thakur BS, Veerasamy R, Sharma PC, Kharya MD. 2011. 2,5-Disubstituted-1,3,4-oxadiazoles/thiadiazole as surface recognition moiety: design and synthesis of novel hydroxamic acid based histone deacetylase inhibitors. Bioorg Med Chem Lett 21:5735–5738. doi: 10.1016/j.bmcl.2011.08.022. [DOI] [PubMed] [Google Scholar]
- 41.Konig HB, Seifken W, Hans O. 1954. Sulfur-containing derivatives of pyridinecarboxylic acids and compounds derived therefrom. Chem Ber 87:825–834. doi: 10.1002/cber.19540870607. [DOI] [Google Scholar]
- 42.Anderson FE, Duca CJ, Scudi JV. 1951. Some heterocyclic thiosemicarbazones. J Am Chem Soc 73:4967–4968. doi: 10.1021/ja01154a501. [DOI] [Google Scholar]
- 43.Liu J, Cao R, Yi W, Ma C, Wan Y, Zhou B, Ma L, Song H. 2009. A class of potent tyrosinase inhibitors: alkylidenethiosemicarbazide compounds. Eur J Med Chem 44:1773–1778. doi: 10.1016/j.ejmech.2008.04.002. [DOI] [PubMed] [Google Scholar]
- 44.Shih M-H, Su Y-S, Wu C-L. 2007. Syntheses of aromatic substituted hydrazino-thiazole derivatives to clarify structural characterization and antioxidant activity between 3-arylsydnonyl and aryl substituted hydrazino-thiazoles. Chem Pharm Bull (Tokyo) 55:1126–1135. doi: 10.1248/cpb.55.1126. [DOI] [PubMed] [Google Scholar]
- 45.Alam MS, Liu L, Lee Y-E, Lee D-U. 2011. Synthesis, antibacterial activity and quantum-chemical studies of novel 2-arylidenehydrazinyl-4-arylthiazole analogues. Chem Pharm Bull (Tokyo) 59:568–573. doi: 10.1248/cpb.59.568. [DOI] [PubMed] [Google Scholar]
- 46.Bilinski S, Tyburczyk W, Urban T. 1961. Condensation of thiosemicarbazones of nicotinic and isonicotinic aldehydes with halo ketones. Ann Univ Mariae Curie-Sklodowska 15:123–128. [Google Scholar]
- 47.Yurttaş L, Özkay Y, Kaplancıklı ZA, Tunalı Y, Karaca H. 2013. Synthesis and antimicrobial activity of some new hydrazone-bridged thiazole-pyrrole derivatives. J Enzyme Inhib Med Chem 28:830–835. doi: 10.3109/14756366.2012.688043. [DOI] [PubMed] [Google Scholar]
- 48.Zhang D-N, Li J-T, Song Y-L, Liu H-M, Li H-Y. 2012. Efficient one-pot three-component synthesis of N-(4-arylthiazol-2-yl) hydrazones in water under ultrasound irradiation. Ultrason Sonochem 19:475–478. doi: 10.1016/j.ultsonch.2011.10.017. [DOI] [PubMed] [Google Scholar]
- 49.Caffrey CR, Hansell E, Lucas KD, Brinen LS, Alvarez Hernandez A, Cheng J, Gwaltney SL, Roush WR, Stierhof Y-D, Bogyo M, Steverding D, McKerrow JH. 2001. Active site mapping, biochemical properties and subcellular localization of rhodesain, the major cysteine protease of Trypanosoma brucei rhodesiense. Mol Biochem Parasitol 118:61–73. doi: 10.1016/S0166-6851(01)00368-1. [DOI] [PubMed] [Google Scholar]
- 50.Jílková A, Rezácová P, Lepsík M, Horn M, Váchová J, Fanfrlík J, Brynda J, McKerrow JH, Caffrey CR, Mares M. 2011. Structural basis for inhibition of cathepsin B drug target from the human blood fluke, Schistosoma mansoni. J Biol Chem 286:35770–35781. doi: 10.1074/jbc.M111.271304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sajid M, McKerrow JH, Hansell E, Mathieu MA, Lucas KD, Hsieh I, Greenbaum D, Bogyo M, Salter JP, Lim KC, Franklin C, Kim J-H, Caffrey CR. 2003. Functional expression and characterization of Schistosoma mansoni cathepsin B and its trans-activation by an endogenous asparaginyl endopeptidase. Mol Biochem Parasitol 131:65–75. doi: 10.1016/S0166-6851(03)00194-4. [DOI] [PubMed] [Google Scholar]
- 52.Buckner FS, Verlinde CL, La Flamme AC, Van Voorhis WC. 1996. Efficient technique for screening drugs for activity against Trypanosoma cruzi using parasites expressing beta-galactosidase. Antimicrob Agents Chemother 40:2592–2597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Romanha AJ, Castro SL, Soeiro Mde N, Lannes-Vieira J, Ribeiro I, Talvani A, Bourdin B, Blum B, Olivieri B, Zani C, Spadafora C, Chiari E, Chatelain E, Chaves G, Calzada JE, Bustamante JM, Freitas-Junior LH, Romero LI, Bahia MT, Lotrowska M, Soares M, Andrade SG, Armstrong T, Degrave W, Andrade Zde A. 2010. In vitro and in vivo experimental models for drug screening and development for Chagas disease. Mem Inst Oswaldo Cruz 105:233–238. doi: 10.1590/S0074-02762010000200022. [DOI] [PubMed] [Google Scholar]
- 54.Faria J, Moraes CB, Song R, Pascoalino BS, Lee N, Siqueira-Neto JL, Cruz DJ, Parkinson T, Ioset JR, Cordeiro-da-Silva A, Freitas-Junior LH. 2015. Drug discovery for human African trypanosomiasis: identification of novel scaffolds by the newly developed HTS SYBR Green assay for Trypanosoma brucei. J Biomol Screen 20:70–81. doi: 10.1177/1087057114556236. [DOI] [PubMed] [Google Scholar]
- 55.Abdulla M-H, Ruelas DS, Wolff B, Snedecor J, Lim K-C, Xu F, Renslo AR, Williams J, McKerrow JH, Caffrey CR. 2009. Drug discovery for schistosomiasis: hit and lead compounds identified in a library of known drugs by medium-throughput phenotypic screening. PLoS Negl Trop Dis 3:e478. doi: 10.1371/journal.pntd.0000478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Stefanić S, Dvořák J, Horn M, Braschi S, Sojka D, Ruelas DS, Suzuki B, Lim KC, Hopkins SD, McKerrow JH, Caffrey CR. 2010. RNA interference in Schistosoma mansoni schistosomula: selectivity, sensitivity and operation for larger-scale screening. PLoS Negl Trop Dis 4:e850. doi: 10.1371/journal.pntd.0000850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Basch PF. 1981. Cultivation of Schistosoma mansoni in vitro. I. Establishment of cultures from cercariae and development until pairing. J Parasitol 67:179–185. [PubMed] [Google Scholar]
- 58.Rojo-Arreola L, Long T, Asarnow D, Suzuki BM, Singh R, Caffrey CR. 2014. Chemical and genetic validation of the statin drug target to treat the helminth disease, schistosomiasis. PLoS One 9:e87594. doi: 10.1371/journal.pone.0087594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Conchie J, Levvy GA, Marsh CA. 1957. Methyl and phenyl glycosides of the common sugars. Adv Carbohydr Chem 12:157–187. [DOI] [PubMed] [Google Scholar]
- 60.Tenchiu Deleanu AC, Kostas ID, Kovala-Demertzi D, Terzis A. 2009. Synthesis and characterization of new aromatic aldehyde/ketone 4-(β-d-glucopyranosyl)thiosemicarbazones. Carbohydr Res 344:1352–1364. doi: 10.1016/j.carres.2009.05.010. [DOI] [PubMed] [Google Scholar]
- 61.de Oliveira RB, de Souza-Fagundes EM, Soares RPP, Andrade AA, Krettli AU, Zani CL. 2008. Synthesis and antimalarial activity of semicarbazone and thiosemicarbazone derivatives. Eur J Med Chem 43:1983–1988. doi: 10.1016/j.ejmech.2007.11.012. [DOI] [PubMed] [Google Scholar]