

Abstract
The combination of cefepime with AAI101, a novel extended-spectrum β-lactamase inhibitor, possesses potent in vitro activity against many resistant Gram-negative pathogens. Against a panel of 20 mostly carbapenemase-producing cefepime-nonsusceptible strains of the family Enterobacteriaceae, we evaluated the MICs of cefepime in the presence of various fixed AAI101 concentrations (1, 2, 4, 8, and 16 mg/liter) and the in vivo efficacy of simulated human doses of cefepime and cefepime-AAI101 in a neutropenic murine thigh infection model. At 2 h after inoculation, mice were dosed with regimens that provided a profile mimicking the free drug concentration-time profile observed in humans given cefepime at 2 g every 8 h (q8h; as a 30-min infusion) or cefepime-AAI101 at 2 g/0.5 g q8h (as a 30-min infusion). Efficacy was determined by calculation of the change in thigh bacterial density (log10 number of CFU) after 24 h relative to the starting inoculum (0 h). After 24 h, bacterial growth of 2.7 ± 0.1 log10 CFU (mean ± standard error) was observed in control animals. Efficacy for cefepime monotherapy was observed against only 3 isolates, whereas increases in bacterial density similar to that in the control animals were noted for the remaining 17 strains (all with cefepime MICs of ≥64 mg/liter). The humanized cefepime-AAI101 dosing regimen resulted in bacterial reductions of ≥0.5 log10 CFU for 12 of the 20 strains. Evaluation of efficacy as a function of the fraction of the dosing interval during which free drug concentrations were above the MIC determined with different fixed concentrations of AAI101 suggested that a fixed concentration of 8 mg/liter AAI101 is most predictive of in vivo activity for the studied regimen.
INTRODUCTION
Production of β-lactamases by Gram-negative bacteria compromises the clinical utility of many diverse β-lactam antibiotics (1–3). The contemporary resistance profiles of clinical isolates are poorly met by the treatment options available, resulting in poor patient outcomes and placing a tremendous economic strain on health care systems. Historically, considerable success in circumventing the resistance caused by β-lactamases has been achieved by combining β-lactam antibiotics with β-lactamase inhibitors, such as amoxicillin-clavulanate, ampicillin-sulbactam, and piperacillin-tazobactam. However, neither clavulanate, sulbactam, nor tazobactam has activity against carbapenemases, enzymes that are gaining rapidly in prevalence all over the world.
AAI101 is a novel extended-spectrum β-lactamase (ESBL) inhibitor with activity against many β-lactamases, including some class A and class D carbapenemases (4, 5). This inhibitor is currently in clinical development as a combination with cefepime, a commonly utilized cephalosporin. We recently showed that addition of AAI101 to cefepime reduced the MIC50 against a collection of 223 cefepime-nonsusceptible isolates of the family Enterobacteriaceae from >64 mg/liter to 0.125 mg/liter (6). While these in vitro studies were conducted using a fixed AAI101 concentration of 8 mg/liter, it was unclear which concentration of AAI101 most appropriately predicts activity for this combination at the doses under investigation in the clinic. We sought to evaluate the efficacy of simulated human doses of cefepime-AAI101. Using the neutropenic murine thigh infection model and a selection of recent clinical multidrug-resistant isolates of the Enterobacteriaceae, we evaluated the pharmacodynamics of this β-lactam–β-lactamase inhibitor combination and obtained insights into the in vitro MIC methodology that best predicts the in vivo activity of the combination product.
MATERIALS AND METHODS
Antimicrobial test agent.
Commercially available cefepime (Bristol-Myers Squibb, Princeton, NJ) was obtained from the Hartford Hospital Pharmacy Department (Hartford, CT) and utilized for all in vivo studies. Analytical-grade cefepime (Sigma-Aldrich, St. Louis, MO) was utilized for MIC determinations. Analytical-grade AAI101 (weight purity, 95.7%) was supplied by Allecra Therapeutics SAS (St-Louis, France). Vials of cefepime for clinical use were reconstituted as described in the prescribing information and diluted as appropriate to achieve the desired concentrations; analytical cefepime and AAI101 powders were weighed in quantities sufficient to achieve the required concentrations and reconstituted immediately prior to use.
Bacterial isolates.
Three clinical isolates of Escherichia coli and 17 clinical isolates of Klebsiella pneumoniae were selected from the isolate inventory of the Center for Anti-Infective Research and Development, which contains organisms recently collected from 43 U.S. hospitals (7). All isolates were maintained in double-strength skim milk (BD Biosciences, Sparks, MD) at −80°C. Each isolate was subcultured twice on Trypticase soy agar with 5% sheep blood (BD Biosciences) prior to use.
Susceptibility testing.
MICs for cefepime and for cefepime-AAI101 were determined for each isolate by broth microdilution, as outlined by the Clinical and Laboratory Standards Institute (CLSI) (8). For cefepime-AAI101, doubling dilutions of cefepime were utilized in combination with a fixed AAI101 concentration of 1, 2, 4, 8, or 16 mg/liter. MIC values were obtained for a minimum of three replicates, and the modal MIC is reported.
All isolates were evaluated phenotypically for production of ESBLs using methods described by the CLSI (9). Briefly, ceftazidime and cefotaxime MICs were determined with and without clavulanate; those isolates that exhibited MIC shifts of ≥8-fold in the presence of clavulanate were classified as ESBL producers.
Isolates nonsusceptible to meropenem (≥4 mg/liter) were evaluated for carbapenemase production using the CarbaNP test (10).
Protein binding studies.
Free drug concentrations for cefepime were calculated on the basis of previously reported protein binding values of 0% for mice (11) and 20% for humans (12).
AAI101 protein binding studies were conducted as three independent tests using Amicon Centrifree micropartition devices (Millipore, Bedford, MA) with 30,000-molecular-weight-cutoff filters, according to the manufacturer's instructions. Nonspecific binding of AAI101 to the filter device was assessed using an aqueous solution of AAI101 at 50 mg/liter.
AAI101 concentrations of 200, 50, and 5 mg/liter were evaluated in freshly collected, pooled mouse plasma and human plasma. Solutions of AAI101 in plasma were heated at 37°C in a shaking water bath for 10 min, followed by centrifugation for 45 min at 10°C and 2,000 × g. Collection of blood for these purposes was reviewed and approved by the Hartford Hospital’s Institutional Review Board, and consent was obtained prior to collection.
Percent protein binding at each prepared concentration was calculated from the equation [(S − SUF)/S] × 100, where S is the AAI101 concentration in the initial plasma solutions and SUF is the AAI101 concentration in the filtrate.
Neutropenic thigh infection model.
Pathogen-free female ICR mice, each weighing approximately 25 g, were acquired from Harlan Sprague-Dawley, Inc. (Indianapolis, IN) and utilized throughout these experiments. Prior to experimentation, the protocol was reviewed and approved by the Hartford Hospital Institutional Animal Care and Use Committee. Animals were provided food and water ad libitum and maintained and used in accordance with National Research Council recommendations. Mice were rendered neutropenic with intraperitoneal injections of 150 and 100 mg/kg of body weight of cyclophosphamide (Cytoxan; Bristol-Myers Squibb, Princeton, NJ) at 4 days and 1 day, respectively, prior to infection. Three days before infection mice also received a single intraperitoneal injection of 5 mg/kg of uranyl nitrate, which produces a predictable degree of renal impairment to aid in humanizing the drug regimens (13). Two hours prior to initiation of antimicrobial therapy, each thigh was inoculated intramuscularly with 0.1 ml of a saline suspension containing approximately 106 CFU of the test isolate.
Development of humanized dosing regimens.
We determined a dosing regimen in mice simulating the fraction of the dosing interval during which free drug concentrations were above the MIC (fT > MIC) observed in humans given cefepime at 2 g every 8 h (q8h) as a 30-min infusion alone or combined with AAI101 at 0.5 g q8h as a 30-min infusion (cefepime-AAI101). Exposures to cefepime and to AAI101 represented the median concentration-time profiles observed in a study with healthy volunteers (14).
First, single-dose studies with cefepime and with cefepime-AAI101 were performed using thigh-infected neutropenic mice. Animals were dosed with a single weight-based subcutaneous injection (0.2 ml) of the study drug(s), and groups of six mice were euthanized at eight time points over the following 8 h. Blood samples were taken via cardiac puncture, and serum was stored at −80°C until analysis. Cefepime concentrations were analyzed at the Center for Anti-Infective Research and Development (Hartford, CT) using a validated high-performance liquid chromatography assay (15), whereas AAI101 concentrations were quantified using a validated liquid chromatography-tandem mass spectrometry protocol at the Aptuit Center for Drug Discovery & Development (Verona, Italy). Intraday and interday coefficients of variation for all high and low check samples for each assay were ≤5.9%.
Pharmacokinetic parameters for single doses of cefepime and of cefepime-AAI101 were calculated by nonlinear least-squares techniques (WinNonlin, version 5.0.1; Pharsight, Mountain View, CA) using first-order input and elimination. Compartment model selection and weighting schemes were based on visual inspection of the fit and use of the correlation between observed and calculated concentrations.
Using the pharmacokinetic parameters derived in single-dose studies, regimens in mice were constructed to simulate the free drug exposure profile for humans given cefepime or cefepime-AAI101. Prior to use of these regimens in the pharmacodynamic analyses, confirmatory pharmacokinetic studies were undertaken in infected mice, and assessments of fT > MIC were made from the resulting concentration-time profiles. For these studies, infected neutropenic mice were dosed with the regimens calculated as described above, and groups of six mice were euthanized at four time points throughout the first dosing interval (i.e., 8 h) to confirm target exposures.
In vivo efficacy.
The 20 Enterobacteriaceae strains were used to infect groups of three mice each. At 2 h after inoculation, mice were treated with humanized regimens of cefepime or cefepime-AAI101. All doses were administered as 0.2-ml subcutaneous injections. To serve as control animals, an additional group of mice was administered normal saline at the same volume and frequency and by the same route. Thighs from all animals were harvested at 24 h after initiation of therapy. The harvesting procedure for all study mice began with euthanasia by CO2 exposure, followed by cervical dislocation. After sacrifice, thighs were removed and homogenized individually in normal saline. For determinations of the numbers of CFU, serial dilutions of thigh homogenates were spread onto Trypticase soy agar with 5% sheep blood using a spiral plater. In addition to the aforementioned treatment and control groups, another group of three infected but untreated mice was harvested at the initiation of dosing and served as a 0-h control. Efficacy, expressed as the change in bacterial density, was determined by calculation of the change in the log10 number of CFU obtained in mice after 24 h of treatment from the densities observed in the 0-h control animals.
Pharmacodynamic analyses.
In an effort to delineate which in vitro MIC methodology was most predictive of in vivo activity, plots were made of fT > MIC as a function of efficacy with MIC data for each fixed AAI101 concentration (1, 2, 4, 8, and 16 mg/liter). These plots were fitted to the sigmoidal maximum-effect inhibitory model (Phoenix 32 WinNonlin, version 6.3; Pharsight, Mountain View, CA), and coefficients of determination were evaluated. Efficacy targets identified from each profile were considered in the context of known values of cefepime monotherapy.
RESULTS
Bacterial isolates.
The phenotypic profiles of the 20 selected K. pneumoniae and E. coli strains toward cefepime and cefepime-AAI101 at various fixed concentrations of AAI101 and comparator agents are shown in Table 1. The studied strains were quite resistant to the comparator agents, and 16 of the 20 strains produced carbapenemases. Cefepime-AAI101 MICs decreased with increasing concentrations of AAI101 (over the range from 1 to 16 mg/liter) for most strains, demonstrating a concentration dependence of AAI101 on restoration of the antibacterial activity of the cephalosporin.
TABLE 1.
Phenotypic data for the E. coli and K. pneumoniae isolates studied during in vivo efficacy experimentsa
| Isolate | MIC (mg/liter) |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| FEP-AAIb |
FEP | TZP | MEM | CIP | TOB | |||||
| 1 mg/liter | 2 mg/liter | 4 mg/liter | 8 mg/liter | 16 mg/liter | ||||||
| K. pneumoniae C13-10c | 32 | 16 | 8 | 2 | 0.13 | 64 | >256 | 16 | >16 | 32 |
| K. pneumoniae C16-9d | >64 | 16 | 4 | 2 | 2 | >64 | >256 | 1 | >16 | 16 |
| K. pneumoniae C22-6 | 8 | 8 | 4 | 4 | 2 | 8 | >256 | 0.25 | 4 | 0.5 |
| E. coli C22-30c | 32 | 32 | 32 | 4 | 0.5 | 32 | >256 | 16 | >64 | 16 |
| E. coli C16-4c | >64 | >64 | 64 | 8 | 1 | >64 | >256 | 16 | 1 | 64 |
| K. pneumoniae C31-2c | >64 | 32 | 8 | 8 | 2 | >64 | >256 | 32 | >16 | >64 |
| K. pneumoniae C31-14c | 16 | 16 | 8 | 8 | 0.25 | >64 | >256 | 64 | >16 | 0.5 |
| K. pneumoniae C37-25c | 64 | 32 | 32 | 8 | 1 | >64 | >256 | 32 | >16 | 32 |
| K. pneumoniae C4-25c | 32 | 32 | 16 | 16 | 1 | 64 | >256 | 64 | >16 | 16 |
| K. pneumoniae C21-22c | >64 | >64 | 32 | 16 | 16 | >64 | >256 | 32 | >64 | 32 |
| K. pneumoniae C31-18c | >64 | >64 | 64 | 16 | 0.5 | >64 | >256 | 32 | >16 | 16 |
| K. pneumoniae C37-28c | >64 | >64 | 16 | 16 | 0.25 | >64 | >256 | 16 | >16 | 16 |
| K. pneumoniae C6-5c | >64 | >64 | 64 | 32 | 1 | >64 | >256 | 32 | >16 | 32 |
| K. pneumoniae C13-25 | 32 | 32 | 32 | 32 | 4 | >64 | 256 | >64 | 2 | 16 |
| K. pneumoniae C19-1c | 64 | 64 | 64 | 32 | 2 | >64 | >256 | 16 | >64 | 32 |
| K. pneumoniae C30-5c | 64 | 64 | 32 | 32 | 16 | >64 | >256 | 64 | >16 | 32 |
| E. coli C3-14 | >64 | >64 | >64 | 64 | 1 | >64 | >256 | 4 | 4 | 64 |
| K. pneumoniae C30-27c | >64 | >64 | >64 | 64 | 64 | >64 | >256 | >64 | >16 | 32 |
| K. pneumoniae C4-10c | >64 | >64 | >64 | >64 | >64 | >64 | >256 | >64 | >16 | 32 |
| K. pneumoniae C8-9c | >64 | >64 | >64 | >64 | 64 | >64 | >256 | >64 | >16 | 32 |
FEP-AAI, cefepime-AAI101; CIP, ciprofloxacin; FEP, cefepime; MEM, meropenem; TOB, tobramycin; TZP, piperacillin-tazobactam.
The fixed concentrations of AAI101 utilized during MIC determination are indicated.
Carbapenemase producer.
ESBL producer.
Protein binding studies.
Negligible protein binding was observed for AAI101 across the range of AAI101 concentrations used in both mouse and human plasma. Nonspecific binding studies revealed no binding of AAI101 to the filter device. On the basis of these findings, at all concentrations AAI101 was assumed to be 100% unbound.
Development of humanized dosing regimens.
The pharmacokinetics of cefepime and AAI101 were well described using a one-compartment model with first-order input and elimination. The final simulated human regimens consisted of two doses during each 8-h dosing interval (i.e., 6 doses total for the 24-h study duration). Figure 1 shows the free drug pharmacokinetic profile of cefepime alone in mice and humans, whereas the corresponding profile for cefepime-AAI101 is shown in Fig. 2. Of note, the addition of AAI101 did not alter the pharmacokinetics of cefepime. As anticipated on the basis of the similarities of the pharmacokinetic profiles, cefepime fT > MICs in mice and humans were comparable (Table 2).
FIG 1.
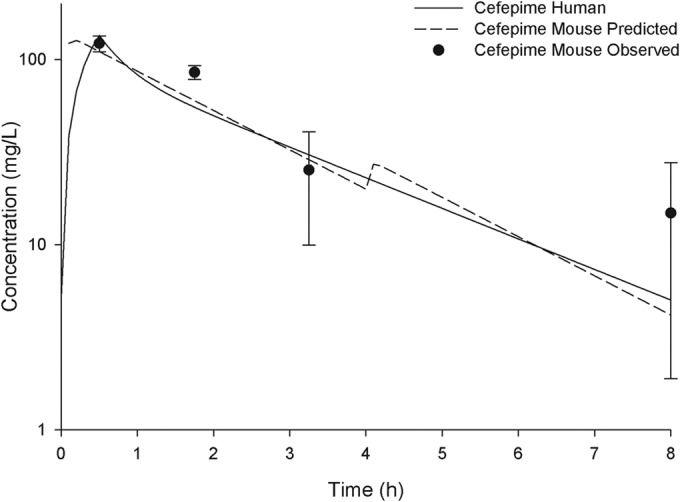
Free concentration-time profiles for the simulated human regimen of cefepime at 2 g every 8 h (30-min infusion) in mice compared with those in humans. Symbols represent means ± SDs.
FIG 2.
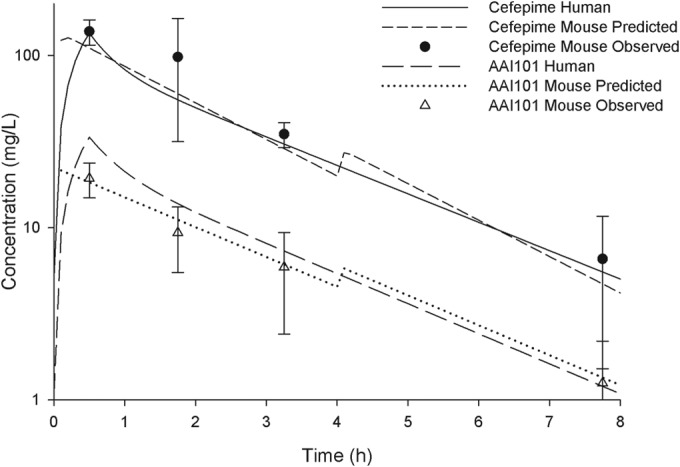
Free concentration-time profiles for the simulated human regimen of cefepime-AAI101 at 2 g/0.5 g every 8 h (30-min infusion) in mice compared with those in humans. Symbols represent means ± SDs.
TABLE 2.
Simulated human fT > MIC profile for cefepime at 2 g q8h in humans compared to that observed in mice
| MIC (mg/liter) | Cefepime fT > MIC (%) |
|
|---|---|---|
| Human | Mousea | |
| 4 | 100 | 100 |
| 8 | 84 | 83 |
| 16 | 61 | 65 |
| 32 | 39 | 38 |
| 64 | 16 | 20 |
| 128 | 1 | 1 |
This profile is similar in mice irrespective of AAI101 administration.
In vivo efficacy.
In these neutropenic animals, 0-h control mice displayed a mean ± standard error bacterial density of 5.9 ± 0.1 log10 CFU, which increased by an average of 2.7 ± 0.1 log10 CFU in untreated mice after 24 h. For cefepime monotherapy, efficacy (−0.6 ± 0.1 log10 CFU) was observed for three isolates with cefepime MICs of 8, 32, and >64 mg/liter, whereas increases in bacterial density similar to those in control animals (2.3 ± 0.2 log10 CFU) were noted for the remaining 17 strains, all with cefepime MICs of ≥64 mg/liter (Fig. 3). Cefepime-AAI101 treatment resulted in reductions in bacterial density of ≥0.5 log10 CFU for 12 of the 20 strains tested and reductions of ≥1 log10 CFU for 6 of these. Increases in bacterial density were observed for only four strains, three of which had cefepime-AAI101 MICs of ≥64 mg/liter, irrespective of the AAI101 concentration. Figure 3 gives a representation of these data, in which MICs were determined using a fixed AAI101 concentration of 8 mg/liter.
FIG 3.

Comparative efficacy of simulated human doses of cefepime at 2 g q8h as monotherapy or in combination with AAI101 against Enterobacteriaceae in the neutropenic thigh infection model. Bars represent means ± SDs. KP, K. pneumoniae; EC, E. coli; FEP, cefepime; AAI, AAI101.
Pharmacodynamic analyses.
The results of the pharmacodynamic analyses are shown in Fig. 4A to E. Coefficients of determination (R2 values) increased with increasing fixed concentrations of AAI101 (R2 range, 0.319 to 0.722), suggesting that MICs determined with lower fixed concentrations of AAI101 were not predictive of the in vivo activity of a cefepime-AAI101 regimen of 2 g/0.5 g q8h. There was no appreciable difference in the coefficient of determination between fixed AAI101 concentrations of 8 mg/liter and 16 mg/liter.
FIG 4.

Pharmacodynamic profile of humanized cefepime-AAI101 (FEP/AAI) at 2 g/0.5 g against a distribution of Enterobacteriaceae sorted by cefepime-AAI101 MICs determined with fixed AAI101 concentrations of 1 mg/liter (A), 2 mg/liter (B), 4 mg/liter (C), 8 mg/liter (D), or 16 mg/liter (E). Symbols represent means ± SDs.
DISCUSSION
In light of increasing resistance to currently available antimicrobials, new therapeutic options for Gram-negative organisms are urgently needed. For Enterobacteriaceae, the high prevalence of β-lactamase production makes addition of a novel β-lactamase inhibitor to a new or marketed β-lactam an attractive option for combating these organisms. AAI101 is one such β-lactamase inhibitor with an extended spectrum of activity and currently is in clinical development as a combination with cefepime. Although coverage of cefepime-AAI101 against Gram-negative organisms is not restricted to isolates in the Enterobacteriaceae (4), this particular study focused on an ensemble of predominantly carbapenemase-producing, multidrug-resistant K. pneumoniae and E. coli strains. Using the murine thigh infection model, we found that treatment with cefepime-AAI101 was effective against most of the strains examined, many of which were identified phenotypically to be carbapenemase producers, whereas cefepime monotherapy was not effective.
The MICs of cefepime-AAI101 were determined with various fixed concentrations of AAI101. Although cefepime MICs for 18 of the 20 strains were ≥64 mg/liter, addition of AAI101, even at a concentration as low as 1 mg/liter, enhanced cefepime potency. As expected on the basis of the high prevalence of β-lactamase production by these strains, cefepime-AAI101 MICs decreased with increasing fixed concentrations of AAI101. Severalfold decreases in the MICs were observed for most strains when AAI101 concentrations were ≥4 mg/liter, concordant with the findings of previous studies using genotyped isogenic strains of E. coli K-12 (5) and clinical isolates of Gram-negative bacilli (4). When evaluating cefepime-AAI101 MICs with fixed AAI101 concentrations of 4 and 8 mg/liter and assuming a susceptibility breakpoint of 2 mg/liter, Mushtaq and colleagues (4) reported 75% and 80% susceptibility, respectively, for a challenge panel of 61 clinical isolates (producers of ESBLs and KPC, AmpC, and OXA enzymes; porin mutants; upregulated effluxers), whereas the rate of susceptibility to cefepime alone was 28%. In our study, 5 organisms retained cefepime-AAI101 MICs of ≥16 mg/liter, despite increasing AAI101 concentrations. Although each of these organisms was characterized phenotypically as a carbapenemase producer, their specific genotypic profiles have not been confirmed.
The focus of the current study was to cover a broad range of MICs encompassing the pharmacodynamic spectrum of drug activity and drug failure. Therefore, the cefepime or cefepime-AAI101 MIC distributions for the strain panel used in this study are not representative of the cefepime or cefepime-AAI101 MIC distributions that would be encountered in the clinic. To identify organisms with cefepime-AAI101 MICs in the range of 2 to >64 mg/liter, we concentrated on strains that were cefepime and/or meropenem nonsusceptible. As such, of the 20 clinical isolates examined in vivo, 19 (95%) were resistant to cefepime and 18 (90%) were resistant to meropenem. Compared with recent data on the surveillance of respiratory E. coli and K. pneumoniae isolates collected from the United States, Europe, and the Mediterranean region showing cefepime and meropenem resistance ranges of 9.1 to 9.6% and 0.5 to 6.9%, respectively, the strains selected here fell at the extreme upper end of the MIC distribution.
These high rates of cefepime resistance in vitro translated into minimal in vivo activity of this cephalosporin alone, as cefepime treatment resulted in reductions in bacterial density after 24 h for only three strains. Addition of 0.5 g of AAI101 to cefepime resulted in decreased bacterial densities for 16 of the 20 strains. An effort was made to correlate in vivo efficacy with in vitro cefepime-AAI101 MIC determinations under different testing conditions. When evaluating efficacy as a function of fT > MIC, the correlation was enhanced with increasing fixed AAI101 concentrations in vitro (Fig. 4). Given that the target peak and trough concentrations of humanized 0.5 g AAI101 were 33.5 mg/liter and 1.1 mg/liter, respectively, a fixed concentration of 1 mg/liter and, perhaps, one of 2 mg/liter, for which coefficients of determination (R2) were 0.319 and 0.422, respectively, would not be expected to be predictive of activity, as these data suggest (Fig. 4A and B). On the opposite end of the AAI101 concentration range studied, AAI101 concentrations of >16 mg/liter were present for only 1.2 h of the 8-h dosing interval. When this is coupled with the observation that, because of the considerable decrease in in vitro MICs observed, 100% fT > MIC yielded changes in bacterial density ranging from of an increase of 1.1 log10 CFU to a decrease of 1.9 log10 CFU, this suggests that a fixed concentration of 16 mg/liter likewise is not highly predictive of in vivo activity for this cefepime-AAI101 dose, even though it has a marginally higher R2 value (Fig. 4E). Fixed AAI101 concentrations of 4 mg/liter and 8 mg/liter (Fig. 4C and D) had relatively high coefficients of determination (0.642 and 0.722, respectively) and pharmacodynamic profiles similar to what might be expected for cefepime alone. Previous studies with other β-lactam–β-lactamase inhibitor combinations have indicated that, once threshold β-lactamase inhibitor concentrations have been achieved, pharmacodynamics are predicated by the parent β-lactam (16–19). All things considered, these data suggest that the MICs determined using a fixed concentration of 8 mg/liter of AAI101 correlate well with the in vivo activity of the studied regimen.
When considering the pharmacodynamic profile for cefepime-AAI101 when MICs were determined with a fixed AAI101 concentration of 8 mg/liter, humanized regimens achieved bacterial reductions against all isolates with MICs of ≤16 mg/liter (fT > MIC, ≥65%) and for four of eight isolates with MICs of ≥32 mg/liter (fT > MIC, ≤38%). This break in activity is consistent with the pharmacodynamics of cephalosporin monotherapy, where fT > MIC targets of approximately 60% are required for optimal outcomes (20, 21).
In conclusion, AAI101 is an extended-spectrum β-lactamase inhibitor that restores the in vitro and in vivo activity of cefepime. While the appropriate fixed concentration of AAI101 to be used for MIC studies had not been established previously, in vivo pharmacodynamics suggest that a fixed concentration of 8 mg/liter of AAI101 is most predictive of efficacy for a human dose of 2 g/0.5 g cefepime-AAI101 administered q8h. Given the high prevalence of β-lactamase production among Gram-negative bacteria, these results support the continued development of cefepime-AAI101 for the treatment of infections caused by these challenging pathogens.
ACKNOWLEDGMENTS
We acknowledge Henry Christenson, Kimelyn Greenwood, Jennifer Hull, Cindy Lamb, Shawn MacVane, Sara Robinson, Debora Santini, Wonhee So, and Pam Tessier for their assistance in the in vivo studies. We also thank Christina Sutherland (Center for Anti-Infective Research and Development, Hartford, CT) and Andrea Bertasini (Aptuit Center for Drug Discovery & Development, Verona, Italy) for cefepime and AAI101 concentration determinations, respectively.
This study was sponsored by a grant from Allecra Therapeutics SAS (St-Louis, France).
REFERENCES
- 1.Rotstein C, Evans G, Born A, Grossman R, Light RB, Magder S, McTaggart B, Weiss K, Zhanel GG. 2008. Clinical practice guidelines for hospital-acquired pneumonia and ventilator-associated pneumonia in adults. Can J Infect Dis Med Microbiol 19:19–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hooton TM, Bradley SF, Cardenas DD, Colgan R, Geerlings SE, Rice JC, Saint S, Schaeffer AJ, Tambayh PA, Tenke P, Nicolle LE. 2010. Diagnosis, prevention, and treatment of catheter-associated urinary tract infection in adults: 2009 international clinical practice guidelines from the Infectious Diseases Society of America. Clin Infect Dis 50:625–663. doi: 10.1086/650482. [DOI] [PubMed] [Google Scholar]
- 3.Stevens DL, Bisno AL, Chambers HF, Everett ED, Dellinger P, Goldstein EJ, Gorbach SL, Hirschmann JV, Kaplan EL, Montoya JG, Wade JC. 2005. Practice guidelines for the diagnosis and management of skin and soft-tissue infections. Clin Infect Dis 41:1373–1406. doi: 10.1086/497143. [DOI] [PubMed] [Google Scholar]
- 4.Mushtaq S, Chaudhry A, Adkin R, Woodford N, Benedict N, Pypstra R, Shapiro S. 2014. In-vitro activity of diverse β-lactam/AAI101 combinations vs. multidrug-resistant Gram-negative clinical strains, abstr P452. Abstr 24th Eur Congr Clin Microbiol Infect Dis, Barcelona, Spain. [Google Scholar]
- 5.Nordmann P, Girlich D, Benedict N, Pypstra R, Shapiro S. 2014. Characterization of β-lactamase inhibition by AAI101, abstr P451. Abstr 24th Eur Congr Clin Microbiol Infect Dis, Barcelona, Spain. [Google Scholar]
- 6.Crandon JL, Nicolau D. 2014. Comparative potency of cefepime and cefepime/AAI101 against highly resistant Enterobacteriaceae, abstr 490. Abstr 54th Intersci Conf Antimicrob Agents Chemother. American Society for Microbiology, Washington, DC. [Google Scholar]
- 7.Sutherland C, Nicolau D. 2014. Susceptibility profile of commonly utilized parenteral antimicrobials against E. coli, K. pneumoniae and P. aeruginosa from US hospitals, abstr C119. Abstr 54th Intersci Conf Antimicrob Agents Chemother. American Society for Microbiology, Washington, DC. [Google Scholar]
- 8.Clinical and Laboratory Standards Institute. 2011. Methods for dilution antimicrobial susceptibility tests for bacteria that grow aerobically; approved standard, 8th ed CLSI publication M07-A8. Clinical and Laboratory Standards Institute, Wayne, PA. [Google Scholar]
- 9.Clinical and Laboratory Standards Institute. 2014. Performance standards for antimicrobial susceptibility testing; 24th informational supplement. CLSI publication M100-S24. Clinical and Laboratory Standards Institute, Wayne, PA. [Google Scholar]
- 10.Dortet L, Poirel L, Nordmann P. 2012. Rapid identification of carbapenemase types in Enterobacteriaceae and Pseudomonas spp. by using a biochemical test. Antimicrob Agents Chemother 56:6437–6440. doi: 10.1128/AAC.01395-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mattie H, Sekh BA, van Ogtrop ML, van Strijen E. 1992. Comparison of the antibacterial effects of cefepime and ceftazidime against Escherichia coli in vitro and in vivo. Antimicrob Agents Chemother 36:2439–2443. doi: 10.1128/AAC.36.11.2439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hospira, Inc. 2014. Maxipime package insert. Hospira, Inc, Lake Forest, IL. [Google Scholar]
- 13.Andes D, Craig WA. 1998. In vivo activities of amoxicillin and amoxicillin-clavulanate against Streptococcus pneumoniae: application to breakpoint determinations. Antimicrob Agents Chemother 42:2375–2379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Homery MC, Denot C, Pypstra R, Benedict N, Patat A. 2014. Safety, tolerability and pharmacokinetics of AAI101, an extended-spectrum β-lactamase inhibitor, in healthy adult males, abstr A-1339. Abstr 54th Intersci Conf Antimicrob Agents Chemother. American Society for Microbiology, Washington, DC. [Google Scholar]
- 15.Burgess DS, Hastings RW, Hardin TC. 2000. Pharmacokinetics and pharmacodynamics of cefepime administered by intermittent and continuous infusion. Clin Ther 22:66–75. doi: 10.1016/S0149-2918(00)87978-3. [DOI] [PubMed] [Google Scholar]
- 16.Crandon JL, Nicolau DP. 2013. Human simulated studies of aztreonam and aztreonam-avibactam to evaluate activity against challenging Gram-negative organisms, including metallo-beta-lactamase producers. Antimicrob Agents Chemother 57:3299–3306. doi: 10.1128/AAC.01989-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Crandon JL, Schuck VJ, Banevicius MA, Beaudoin ME, Nichols WW, Tanudra MA, Nicolau DP. 2012. Comparative in vitro and in vivo efficacies of human simulated doses of ceftazidime and ceftazidime-avibactam against Pseudomonas aeruginosa. Antimicrob Agents Chemother 56:6137–6146. doi: 10.1128/AAC.00851-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dudley MN. 1995. Combination beta-lactam and beta-lactamase-inhibitor therapy: pharmacokinetic and pharmacodynamic considerations. Am J Health Syst Pharm 52:S23–S28. [DOI] [PubMed] [Google Scholar]
- 19.VanScoy B, Mendes RE, Nicasio AM, Castanheira M, Bulik CC, Okusanya OO, Bhavnani SM, Forrest A, Jones RN, Friedrich LV, Steenbergen JN, Ambrose PG. 2013. Pharmacokinetics-pharmacodynamics of tazobactam in combination with ceftolozane in an in vitro infection model. Antimicrob Agents Chemother 57:2809–2814. doi: 10.1128/AAC.02513-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Crandon JL, Bulik CC, Kuti JL, Nicolau DP. 2010. Clinical pharmacodynamics of cefepime in patients infected with Pseudomonas aeruginosa. Antimicrob Agents Chemother 54:1111–1116. doi: 10.1128/AAC.01183-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Turnidge JD. 1998. The pharmacodynamics of beta-lactams. Clin Infect Dis 27:10–22. doi: 10.1086/514622. [DOI] [PubMed] [Google Scholar]