

Abstract
Daptomycin is increasingly used in combination with other antibiotics to enhance antimicrobial efficacy and/or to mitigate the emergence of daptomycin nonsusceptibility (DNS). This study used a clinical methicillin-resistant Staphylococcus aureus (MRSA) strain in which DNS emerged upon therapy to examine the influence of antibiotic combinations on the development of mutations in specific genes (mprF, rpoBC, dltA, cls2, and yycFG) previously associated with DNS. Whole genomes of bacteria obtained following 28 days of in vitro exposure to daptomycin with or without adjunctive clarithromycin, linezolid, oxacillin, or trimethoprim-sulfamethoxazole were sequenced, and the sequences were compared to that of the progenitor isolate. The addition of oxacillin to medium containing daptomycin prevented the emergence of mprF mutation but did not prevent rpoBC mutation (P < 0.01). These isolates maintained susceptibility to daptomycin during the combined exposure (median MIC, 1 mg/liter). Daptomycin plus clarithromycin or linezolid resulted in low-level (1.5 to 8 mg/liter) and high-level (12 to 96 mg/liter) DNS, respectively, and did not prevent mprF mutation. However, these same combinations prevented rpoBC mutation. Daptomycin alone or combined with linezolid or trimethoprim-sulfamethoxazole resulted in high-level DNS and mutations in mprF plus rpoBC, cls2, and yycFG. Combining daptomycin with different antimicrobials alters the mutational space available for DNS development, thereby favoring the development of predictable collateral susceptibilities.
INTRODUCTION
Daptomycin (DAP) is frequently used in the treatment of methicillin-resistant Staphylococcus aureus (MRSA) infection. However, DAP-nonsusceptible (DNS) strains are observed clinically and often associated with treatment failure (1). The development of DNS subpopulations is of particular concern in three clinical scenarios: (i) prolonged DAP therapy, particularly with underdosing, (ii) sequestered foci of infection where DAP penetration is reduced, such as osteomyelitis or infective endocarditis (2, 3), and (iii) refractory or recurrent infection where there has been previous exposure to host antimicrobial peptide defenses and/or glycopeptide antimicrobials (4, 5).
DNS MRSA emerges via the accumulation of single or multiple mutations, each resulting in a slight increase in the DAP MIC (6, 7). Several DNS MRSA strains have been characterized, with causal mutations identified in mprF, yycFG (walKR), dltA, rpoBC, and/or cls2 (8). These elements share a role in maintaining cell envelope homeostasis, either by directly modifying the cell membrane or as a regulatory response to cell surface perturbations. The identification of a direct mechanism for DNS development with any given mutation is frequently confounded by simultaneous changes in the expression and/or activity of the MprF, Cls2, and DltA enzymes (9). While there is significant strain-to-strain variability in the pathways affected, a frequent consequence of the development of DAP resistance is a reduced anionic cell membrane potential, resulting in reduced charge-charge attraction between Gram-positive membranes and either DAP or host cationic defense peptides (10). Although no single mutation is uniquely responsible for the DNS phenotype (11) and clinical DNS strains have emerged that have the wild-type mprF sequence (4), the first and most clinically relevant mutation to emerge typically occurs as a gain-of-function mutation in mprF (12). Thus, preventing or delaying the emergence of mprF mutation may result in a longer effective therapeutic window for DAP.
DAP has been increasingly used with additional antimicrobials either for empirical coverage of complex infection or targeted toward a specific infection type or organism to improve clinical efficacy (13, 14). It is unknown whether these adjunctive antibiotics exert a treatment-dependent influence on the pattern and/or frequency of mutation in genes associated with DNS development. Previous work in our laboratory demonstrated that combinations of DAP plus oxacillin or DAP plus clarithromycin suppressed the emergence of DNS (15). In this prior study, a DAP-susceptible clinical MRSA isolate from which DNS emerged upon therapy was exposed in vitro to escalating concentrations of DAP with or without the presence of additional antibiotics for a period of 28 consecutive days. This current study expands significantly on these initial results by comparing the frequencies of mutation in genes previously associated with DAP nonsusceptibility between strains and the efficiencies of different antibiotic combinations with DAP in preventing these mutations.
(Portions of this work were presented at the 54th Interscience Conference on Antimicrobial Agents and Chemotherapy, Washington, DC, 9 September 2014.)
MATERIALS AND METHODS
Bacterial strains.
MRSA strain J01 and the generation of DNS strains from it in vitro have been described in previous work (15). Briefly, DAP-susceptible clinical isolate J01 was collected from a patient presenting with left-sided endocarditis and multiple septic-embolic complications. A second clinical isolate, J03, was collected after 24 days of antimicrobial therapy and had become DNS. In order to explore mechanisms involved in the transition to a DNS phenotype, five independent replicates of J01 were exposed in liquid culture to escalating concentrations of DAP stepwise over a period of 28 days in the presence or absence of a static concentration of adjunctive antimicrobial at one-half the MIC. This current work assesses the five replicate strains obtained from each of the following exposures: (i) DAP monotherapy (D1 to D5), (ii) DAP plus clarithromycin (CLR; 100 mg/liter) (DC1 to DC5), (iii) DAP plus linezolid (LZD; 0.5 mg/liter) (DL1 to DL5), (iv) DAP plus oxacillin (OXA; 16 mg/liter) (DO1 to DO5), and (v) DAP plus trimethoprim-sulfamethoxazole (SXT; 6 mg/liter [sulfamethoxazole component]) (DS1 to DS5).
Antimicrobials and media.
CLR, erythromycin (ERY), OXA, and SXT were purchased from Sigma-Aldrich (Sigma, St. Louis, MO, USA). DAP was obtained from Cubist (Cubist Pharmaceuticals, Lexington, MA, USA). LZD was obtained from Pfizer (Pfizer, Inc., New York, NY, USA). Mueller-Hinton broth II (BD, Sparks, MD, USA) supplemented with 50 μg/ml calcium (as CaCl2) and 12.5 μg/ml magnesium (as MgCl2) was used to grow S. aureus in liquid culture. Isolates that exhibited poor growth in liquid culture due to aromatic compound auxotrophy were cultivated in medium supplemented with 1 mM shikimic acid.
Susceptibility testing.
The MICs of the study bacteria to DAP, OXA, and vancomycin (VAN) were determined by Etest as suggested by the manufacturer (bioMérieux, Marcy l'Etoile, France). The MICs to other antibiotics were determined by broth microdilution according to the Clinical and Laboratory Standards Institute guidelines (17). All samples were incubated at 35°C for 24 h. Reference strain ATCC 29213 was included in all MIC determination testing as an internal control. All values obtained were within the acceptable range for this organism.
Whole-genome sequencing.
Genomic DNA was extracted, and multiplex short-read DNA libraries were created with the Nextera XT DNA preparation kit (Illumina). Whole-genome sequencing was performed on a MiSeq instrument (Illumina) using 2× 300-bp chemistry; all steps were performed according to the manufacturer's recommendations.
A read-mapping approach was used to align the short-read sequences for all isolates to the assembled sequence of S. aureus FRP3757-USA300 (GenBank accession number NC_007793.1) using Bowtie2 (18). Single nucleotide polymorphisms, insertions/deletions up to 10 bp in length, and the predicted amino acid consequences were identified using Nesoni version 0.128 with a stringency threshold of 80% (http://bioinformatics.net.au). The effects of mutations in loci that have not previously been associated with the DNS phenotype will be explored in future work. This study focused exclusively on mutations identified in the following loci that have previously been associated with the DNS phenotype: SAUSA300_0020 (yycF), SAUSA300_0021 (yycG), SAUSA300_0527 (rpoB), SAUSA300_0528 (rpoC), SAUSA300_0835 (dltA), SAUSA300_1255 (mprF), and SAUSA300_2044 (cls2) (7, 12, 19). The full list of predicted changes to the products of genes of interest (mprF, rpoBC, dltA, yycFG, and cls2) is provided in Table 1.
TABLE 1.
Bacterial study strains, antibiotic susceptibilities, and genetic mutations in select loci
| Strain,a drug exposure | MIC ofb: |
Mutation(s)c in: |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| DAP | VAN | OXA | CLR | mprF | rpoB | rpoC | dltA | cls2 | yycF | yycG | |
| Clinical isolates | |||||||||||
| J01 | 0.5 | 2 | 32 | 256 | — | — | — | — | — | — | — |
| J03 | 3 | 2 | 16 | 256 | T345X | S464P | — | — | — | — | — |
| In vitro strains | |||||||||||
| DAP | |||||||||||
| D1 | 32 | 2 | 0.01 | 16 | L826F | — | N735K | — | R320L | — | — |
| D2 | 32 | 2 | 0.01 | 32 | L826I | — | N735K | — | R320S | — | 369delQ |
| D3 | 12 | 2 | 0.01 | 32 | L826I | — | N735K | — | R320S | — | 369delQ |
| D4 | 8 | 2 | 0.01 | 8 | L826F | — | N735K | — | — | — | — |
| D5 | 24 | 1.5 | 0.01 | 32 | P314L | — | — | — | F85X | — | — |
| DAP+CLR | |||||||||||
| DC1 | 3 | 2 | 0.01 | 64 | H376Y, W424C | — | — | — | — | — | — |
| DC2 | 1.5 | 1.5 | 0.01 | 64 | A302V | — | — | — | — | — | — |
| DC3 | 8 | 3 | 0.01 | 32 | P314L | — | — | — | — | — | — |
| DC4 | 3 | 1.5 | 0.02 | 32 | M347R | — | — | — | — | — | — |
| DC5 | 3 | 2 | 0.01 | 32 | S337L | — | — | — | — | — | — |
| DAP+LZD | |||||||||||
| DL1 | 32 | 2 | 0.01 | 32 | S337L | — | — | — | F85X | — | — |
| DL2 | 64 | 4 | 0.01 | 0.03 | 41insN | — | — | — | — | — | — |
| DL3 | 192 | 1.5 | 0.01 | 0.01 | L826F | — | — | — | L77F | — | G199E |
| DL4 | 64 | 3 | 0.03 | 1 | S295L | — | — | — | — | — | — |
| DL5 | 192 | 2 | 0.01 | 0.01 | L826F | — | — | — | L77F | — | G199E |
| DAP+OXA | |||||||||||
| DO1 | 0.02 | 1.5 | 256 | 2 | — | — | N735K | A426E | — | — | — |
| DO2 | 1 | 3 | 64 | 4 | — | — | N735K | — | — | — | — |
| DO3 | 3 | 3 | 128 | 4 | — | — | N735K | — | — | — | — |
| DO4 | 0.05 | 2 | 128 | 4 | — | — | N735K | — | — | — | — |
| DO5 | 3 | 3 | 128 | 0.06 | — | S366F | — | — | — | — | — |
| DAP+SXT | |||||||||||
| DS1 | 24 | 2 | 4 | 32 | L826I | — | N735K | — | — | — | — |
| DS2 | 96 | 2 | 4 | 32 | L826I | — | N735K | — | — | — | — |
| DS3 | 32 | 3 | 0.5 | 16 | L826I | — | N735K | — | R320L | — | — |
| DS4 | 12 | 3 | 32 | 8 | S295L | — | N735K | — | — | K151N | — |
| DS5 | 64 | 2 | 16 | 8 | L826I | V1130K, V1162I | — | — | A56G, T58N | — | — |
All in vitro strains were derived from J01 following 28 days of antibiotic exposure.
MICs to antibiotics are provided in mg/liter. Susceptibility breakpoints for S. aureus are as follows: DAP, ≤1 mg/liter; VAN, ≤2 mg/liter; OXA, ≤2 mg/liter; CLR, ≤2 mg/liter (17).
—, the strain has the wild-type sequence.
Cytochrome c binding.
The cytochrome c binding assay was adapted from previously published methods (20, 21). Briefly, overnight cultures of S. aureus grown in Trypticase soy broth (TSB) were diluted in fresh medium and allowed to regrow to logarithmic phase. Cells were harvested, washed twice with MOPS (morpholinepropanesulfonic acid) buffer (20 mM, pH 7.0), adjusted to an optical density at 600 nm (OD600) of 1.0, and collected from 1-ml aliquots via centrifugation. Cell pellets were resuspended in 200 μl MOPS buffer and combined with 50 μl of cytochrome c solution (equine heart, 2.5 mg/ml in MOPS buffer; Sigma). Samples were incubated for 10 min at room temperature and separated by centrifugation at top speed. Supernatants were recovered, and the OD530 was measured spectrophotometrically. The amount of cell-bound cytochrome c was determined using an extemporaneously prepared standard curve. The data presented are the average results of at least 3 independent replicates.
Statistical analysis.
Comparisons of the MIC values of different populations were conducted using the Mann-Whitney test. Comparisons of the development of specific mutations between populations exposed to different antibiotics were conducted using Fisher's exact test.
Nucleotide sequence accession number.
Sequence reads have been submitted to the European Nucleotide Archive under project accession number ERP009667.
RESULTS
In our data set of 175 loci previously associated with the DNS phenotype (seven target genes times 25 genomes analyzed), 49 (28%) had altered sequences compared to the genome sequence of S. aureus USA300-FPR3757, resulting from 26 unique mutations (Table 1). Of these, two involved insertion/deletion events (indels), while 24 involved single nucleotide polymorphisms (SNPs). Only one of the SNPs, in cls2, is predicted to result in the introduction of a premature stop codon; the remaining 25 SNPs and indels are predicted to result in single-amino-acid changes (substitutions, additions, or removals) in the protein product. None of the mutations are predicted to result in frameshift or synonymous changes. The end-of-study DAP MICs among the 25 strains analyzed were significantly increased in isolates with a genetic mutation in mprF and further increased when additional mutations existed within rpoBC, yycFG, or cls2 (Fig. 1).
FIG 1.
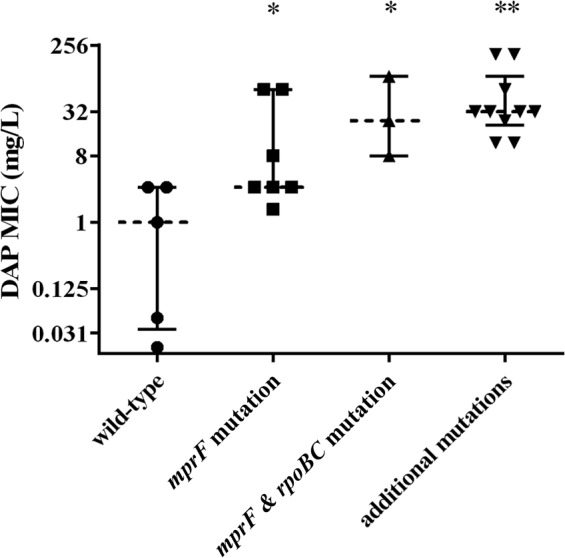
End-of-study DAP MICs grouped by patterns of mutation in genes that contribute to the DNS phenotype. Whiskers are the interquartile ranges, and the dashed line is the median for each group. Groups, left to right, comprise MICs for strains with the wild-type mprF sequence (n = 5), an mprF mutation only (n = 7), mprF and rpoBC mutations only (n = 3), or an mprF mutation and at least one additional mutation in either cls2 or yycFG (n = 10). ∗, P < 0.05 versus the results for the wild type; ∗∗, P < 0.001 versus the results for the wild type (Mann Whitney test).
Polymorphisms in mprF were the most prevalent mutation observed, occurring in 20/25 strains. Mutations were not localized to one specific region of MprF but did cluster within known DNS hot spots (22) in the bifunctional central transmembrane domain (S295L [an S-to-L change at position 295], A302V, P314L, S337L, M347R, H376Y, and W424C) or occurred to one specific residue near the C terminus of the cytosolic synthase domain (L826I and L826F) (7, 23). One strain contains a 3-nucleotide insertion and is predicted to incorporate an additional asparagine residue early in the MprF peptide sequence (D40DN). The combination of DAP and OXA prevented the development of an mprF mutation, whereas isolates from single-antibiotic exposure to DAP and from every other DAP combination displayed mprF mutations after 28 days (P < 0.01). A previous study had observed the establishment of a stable G-to-A transitional mutation in the noncoding region upstream from the AMP-dependent synthetase acsA in S. aureus MW2 (locus tag MW2528) prior to the development of an mprF mutation (12). S. aureus FPR3757 does not carry this transitional mutation, and none of the 25 strains analyzed in this study had any detectable sequence variation in the region upstream from acsA (locus tag SAUSA300_2542). Therefore, an intergenic transition upstream from acsA is not a necessary prerequisite for an mprF mutation.
Polymorphisms in rpoC were frequent in the study population, occurring in 14/25 strains. Mutations in rpoB were less common (2/25) and did not appear to localize to a single region of the gene or to a single domain of the encoded protein (S746F in replicate DO5, and S464P, V1130K [nucleotide change, GT3388AA], and V1162I in replicate DS5). However, each mutation in rpoB did map to one of the three regions shown to alter the response of RNA polymerase to transcriptional pausing and termination signals in Escherichia coli (nucleotides 500 to 575, 740 to 840, and 1225 to 1342 in the E. coli RpoB sequence) (24). Changes in S. aureus RpoB have already been described elsewhere as contributing to the DNS phenotype (12), potentially via increased DltA activity (9); however, in contrast to the mutations identified in this study, only one (A1086V) of the three RpoB changes associated with the DNS phenotype in previous work (A621E, I953S, and A1086V) localizes to any of the regions associated with modified termination signal recognition. Unlike the mutations in MprF and RpoB, all mutation events in rpoC resulted in the same single-amino-acid change in domain 4 (N735K). This region constitutes the funnel domain and is a site involved in binding of transcription factors (25). While changes in S. aureus RpoC have already been described elsewhere as contributing to the DNS phenotype (12), the F632S and Q961K modifications identified in that work occur outside domain 4. No strains exposed to either CLR or LZD during DAP selection developed one of the otherwise nearly ubiquitous rpoBC mutations (P < 0.001).
Our study population occasionally contained polymorphisms in cardiolipin synthase (cls2), which occurred in 9/25 strains. Mutation events localized to either the linker region between the transmembrane domain and the first phospholipase D domain (A56G, T58N, L77F, or F85X) or to one residue in the N-terminal region of the second phospholipase D domain (R320L or R320S) (26). No strains exposed to either CLR or OXA during DAP selection developed any cls2 mutation (P = 0.028). Furthermore, strains that had developed a mutation resulting in a change to amino acid residue 826 of MprF (i.e., L826I or L826F) were strongly associated with the development of an additional mutation in cls2 (Fisher's exact test, P = 0.009). Mutations in yycFG or dltA were infrequent, occurring in only five and one strain, respectively, and did not correlate with exposure to any particular antibiotic. We do note that none of the strains exposed to either CLR or OXA during DAP selection developed any yycFG mutation, but the trend was not statistically significant (P = 0.061).
Treatment-dependent changes in the MICs of DAP, VAN, OXA, and CLR are reported in Table 1. The addition of either OXA or CLR during the 28 days of DAP selection resulted in a significantly lower end-of-study DAP MIC than selection with DAP monotherapy (P < 0.05). Furthermore, these combinations delayed the development of the DNS phenotype compared to its development during selection with DAP monotherapy or, in some replicates, prevented the emergence of the DNS phenotype altogether. In contrast, the addition of either LZD or SXT did not result in any difference in the end-of-study DAP MIC or any delay in DNS development compared to the results for selection with DAP monotherapy (P > 0.05), as reported previously (15). VAN MICs remained stable during selection, with no treatment-dependent variability and no MIC changes greater than one doubling dilution. While changes to VAN MICs were modest overall, the progenitor strain is at the borderline of VAN susceptibility (MIC, 2 mg/liter) and one end-of-study strain developed intermediate VAN susceptibility (MIC, 4 mg/liter). In contrast, end-of-study strains developed markedly (>3 log10) increased susceptibilities to OXA, from an initial MIC value of 32 mg/liter to an end-of-study average of approximately 0.01 mg/liter. Exceptions to this trend occurred when OXA or SXT was added during DAP selection, resulting in end-of-study median OXA MICs of 128 mg/liter and 4 mg/liter, respectively (P < 0.05). An increase in end-of-study OXA MICs in strains exposed to OXA during DAP selection can be attributed to the selective pressure exerted by continuous OXA exposure. The mechanism by which exposure to SXT could result in a partial reversal of the OXA MIC decreases observed during DNS selection remains unclear. Of note, a wild-type mecA sequence was confirmed in 23/25 strains, indicating that OXA susceptibility is not dependent on loss of the staphylococcal cassette chromosome mec (SCCmec) element, as has been discussed elsewhere (27). In contrast, the mecA sequence was not detectable in DC4 and DL3, suggesting that loss of the cassette chromosome may contribute to OXA susceptibility in these isolates.
Analogous to the overall decreases observed for OXA MICs, all end-of-study strains developed increased sensitivity to CLR (0.01 to 64 mg/liter), although the absolute change in CLR MICs was less pronounced. The addition of LZD, OXA, or SXT during DAP selection did not affect the magnitude or the direction of changes in CLR MICs, although there were higher end-of-study CLR MICs in strains exposed to DAP and CLR than in other combination treatment arms, again attributable to selective pressure exerted by continuous CLR exposure (P < 0.01).
The initial characterization of the yycFG two-component regulator system in S. aureus identified a marked reduction in the MICs of macrolides upon yycF disruption (28). Since perturbations in the yycFG system have also been associated with the development of the DNS phenotype (4) and DNS study strains frequently show reduced CLR MICs (Table 1), one consequence of DNS development may be altered yycFG signaling, resulting in increased macrolide sensitivity. However, in the population of MRSA strains containing yycFG disruption, there was no significant difference in CLR or ERY MICs compared to those of DNS strains that maintain a wild-type yycFG system (P > 0.05 for both comparisons). Therefore, while the MICs to macrolide antibiotics were frequently reduced in DNS study strains, that reduction was not dependent on yycFG mutation.
One well-characterized consequence of DAP exposure is the evolution of a less anionic bacterial envelope (10). Mutations associated with DNS MRSA may contribute to this phenotype via direct membrane modification (MprF) (29), modified proton gradient homeostasis (Cls2) (30), altered membrane biosynthesis regulation (YycFG) (31), or increased lipoteichoic acid modification (RpoBC) (9). Consistent with this model, the study strains overall demonstrated a reduced anionic membrane potential, as shown by the results in Fig. 2. The binding of cationic cytochrome c to bacterial membranes was reduced for all study strains relative to its binding to the progenitor strain (P < 0.05), with the exception of strains exposed to DAP and OXA, where the binding was overall similar to that of the parent strain (P = 0.54) and consistent with the low daptomycin susceptibility compared to the results for other combinations.
FIG 2.
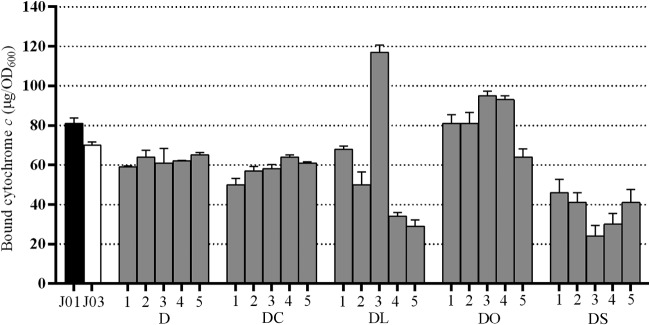
Cytochrome c binding affinities of study strains. Isolates J01 and J03 were derived in vivo during patient treatment. The in vitro-exposure strains (gray bars) were derived over 28 days of exposure to daptomycin plus an adjunctive antibiotic. Higher values correspond to a more anionic membrane. Data represent the mean values ± standard deviations from three independent replicates for each of the five replicate strains obtained for each drug exposure. DC, DAP+CLR; DL, DAP+LZD; DO, DAP+OXA; DS, DAP+SXT.
DISCUSSION
Due to the complexities and treatment failures associated with serious S. aureus infection, clinicians are increasingly interested in alternatives to standardized therapy. Many practitioners prescribe DAP with an adjunctive antibiotic for complex or prolonged infection, to enhance antimicrobial efficacy that results in improved therapeutic outcomes (32). It is not known whether these additional antimicrobials alter the metabolic cost associated with establishing and maintaining various mutations that result in DNS development. Our group and others have previously demonstrated that the addition of antistaphylococcal β-lactams during DAP exposure prevents the phenotypic emergence of DNS subpopulations (15, 21). This combination has gained acceptance among practitioners and has been used in patients, resulting in clinical successes (33–35). This has been attributed partially to β-lactam-mediated enhancement of DAP binding to the bacterial surface, but increased binding is not essential for improved activity, and other mechanisms are being explored (36).
In this study, we analyzed the DNA sequences of known determinants of DAP susceptibility in a collection of in vitro-derived MRSA strains to determine whether adjunctive treatment can alter patterns of DNS mutation. We have summarized these findings in Fig. 3, which provides a conceptual map delineating the complex contributions of genetic mutations and phenotypic adaptations associated with DNS development. Our primary findings were that (i) strains exposed to OXA during 28 days of DAP selective pressure uniquely maintained wild-type mprF sequences and remained DAP susceptible and (ii) strains exposed to CLR or LZD during 28 days of DAP selective pressure uniquely maintained wild-type rpoBC sequences but did not develop DNS. Because strains exposed to OXA maintained DAP susceptibility even after rpoBC mutation developed, we conclude that the primary mediator of the DNS phenotype in the larger collection of strains is MprF derangement.
FIG 3.

Biological model of the contributions of genetic mutations and phenotypic adaptations to the development of daptomycin-nonsusceptible Staphylococcus aureus. Genes in which mutations emerged following treatment-dependent exposure conditions in the current study are represented by dark circles, and light-gray circles indicate wild-type sequences. Implications of mutations for the downstream elements (regulated gene or phenotype) are indicated by a plus or minus sign in the pathway. SXT, trimethoprim-sulfamethoxazole.
MprF was first identified in 2001 as FmtC, a membrane protein that confers OXA susceptibility when inactivated (37), and later was characterized as a factor that confers resistance to host cationic peptides via membrane lysinylation (38). Soon after FDA approval and introduction of DAP into clinical practice, mutations in mprF were found to be associated with reduced susceptibility to DAP (12) and later shown to be causal of the phenotype (39). Recent work shows that depletion of MprF using antisense RNA technology not only restores the DAP-susceptible phenotype in DNS MRSA but also decreases the susceptibility to OXA (40). Thus, MprF activity has consistently been associated with both OXA and DAP susceptibility, usually with an inverse relationship.
Study strains exposed over 28 days to both DAP and OXA showed heterogeneous cytochrome c binding profiles, but overall, the binding was similar to that of the parent strain. Two replicates (DO1 and DO2) displayed progenitor strain-level cytochrome binding (P > 0.85). Two replicates (DO3 and DO4) displayed enhanced cytochrome c binding (P < 0.01), consistent with previous findings where the addition of β-lactam antibiotics during DAP exposure resulted in more anionic membranes and enhanced the binding of the cationic peptide (and presumably DAP) (21). These two strains (DO3 and DO4) share a unique frameshift mutation that results in truncation of a hypothetical membrane protein (nt334delA; SAUSA300_0694, accession number CP000255.1). The full-length hypothetical membrane protein is predicted to be highly basic, with an isoelectric point of 9.63 and a net charge at pH 7 of +6. The loss of such a protein would potentially result in a more anionic membrane and enhanced cytochrome binding. One strain (DO5) displayed reduced cytochrome binding, analogous to the cytochrome affinity of the other DAP exposure strains (P < 0.01, J01 versus DO5). This heterogeneity in binding profiles in strains exposed to both DAP and OXA, rather than the enhanced binding reported previously, is perhaps a consequence of the more prolonged exposure of study strains to both agents.
Based upon these results, it is tempting to speculate on why exposure to OXA prevents the development of DNS. The establishment of multiple mutations resulting in the DNS phenotype may be facilitated by an initial derangement in MprF function. Therefore, prevention of mprF mutation with OXA may prevent the first committed step during the development of DNS. Alternatively, the DNS phenotype itself may reduce the ability of the cell to withstand β-lactam-mediated stresses regardless of the causative genetic mechanism. In this scenario, MprF does not play a gatekeeper role but, rather, is one of many mutations that experiences negative selection in the presence of OXA. With therapeutic concentrations in the patient setting, β-lactams increase DAP killing and effectiveness (33, 41); therefore, this may leave fewer bacterial cells available to develop mprF mutation and DAP resistance. Further work is needed to understand the clinical effects of prolonged exposure to this antibiotic combination.
While this work has focused on the use of adjunctive therapy to reduce the emergence of DNS subpopulations, clinicians often consider combining antimicrobials to take advantage of synergistic activity. Alternatively referred to as the seesaw effect or collateral susceptibility, it has been reported that improved activity of OXA improves as the MRSA strain becomes DNS. This appears to be a β-lactam class effect, although the phenomenon appears to be more pronounced for β-lactams that preferentially target PBP1 (41). This study suggests that adjunctive therapy with OXA is reasonable for both synergy and prevention of the development of resistance, possibly by maintaining wild-type mprF. This effect may extend to additional antistaphylococcal β-lactams and perhaps to ceftaroline, but so far it has only been demonstrated experimentally with OXA.
To our knowledge, this is the first study to identify the potential utility of therapy using DAP plus an antistaphylococcal β-lactam both for increased antibiotic activity and for the prevention of specific genetic changes associated with DNS development during treatment. While the adjunctive antibiotic exposures were at one-half the MIC and not therapeutic exposures, this approach provides a useful direct comparison among antibiotics to identify different phenotypic and genotypic pathways for the suppression or emergence of daptomycin resistance with combination exposures. Further studies are warranted to identify the clinical significance and risks of long-term combination therapy using DAP plus antistaphylococcal β-lactams to treat prolonged infection. This study is limited by the analysis of only previously known mediators of the DNS phenotype. Future work will examine the influence of novel mutations in other loci on the development of DNS. The results of this study indicate that clinicians can affect the mutational space permitted for DNS development by using adjunctive antibiotic therapy.
ACKNOWLEDGMENTS
This work was supported by internal funding. A.D.B. is supported by a Pharmacy Practice research fellowship at the University of Wisconsin—Madison School of Pharmacy. This work was supported by the National Health and Medical Research Council, Australia (fellowship APP1023526).
REFERENCES
- 1.Gasch O, Camoez M, Domínguez MA, Padilla B, Pintado V, Almirante B, Martín C, López-Medrano F, de Gopegui ER, Blanco JR, García-Pardo G, Calbo E, Montero M, Granados A, Jover A, Dueñas C, Pujol M, REIPI/GEIH study groups. 2014. Emergence of resistance to daptomycin in a cohort of patients with methicillin-resistant Staphylococcus aureus persistent bacteraemia treated with daptomycin. J Antimicrob Chemother 69:568–571. doi: 10.1093/jac/dkt396. [DOI] [PubMed] [Google Scholar]
- 2.Parra-Ruiz J, Vidaillac C, Rose WE, Rybak MJ. 2010. Activities of high-dose daptomycin, vancomycin, and moxifloxacin alone or in combination with clarithromycin or rifampin in a novel in vitro model of Staphylococcus aureus biofilm. Antimicrob Agents Chemother 54:4329–4334. doi: 10.1128/AAC.00455-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Traunmüller F, Schintler MV, Metzler J, Spendel S, Mauric O, Popovic M, Konz KH, Scharnagl E, Joukhadar C. 2010. Soft tissue and bone penetration abilities of daptomycin in diabetic patients with bacterial foot infections. J Antimicrob Chemother 65:1252–1257. doi: 10.1093/jac/dkq109. [DOI] [PubMed] [Google Scholar]
- 4.Howden BP, McEvoy CR, Allen DL, Chua K, Gao W, Harrison PF, Bell J, Coombs G, Bennett-Wood V, Porter JL, Robins-Browne R, Davies JK, Seemann T, Stinear TP. 2011. Evolution of multidrug resistance during Staphylococcus aureus infection involves mutation of the essential two component regulator WalKR. PLoS Pathog 7:e1002359. doi: 10.1371/journal.ppat.1002359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mishra NN, Bayer AS, Moise PA, Yeaman MR, Sakoulas G. 2012. Reduced susceptibility to host-defense cationic peptides and daptomycin coemerge in methicillin-resistant Staphylococcus aureus from daptomycin-naive bacteremic patients. J Infect Dis 206:1160–1167. doi: 10.1093/infdis/jis482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Patel D, Husain M, Vidaillac C, Steed ME, Rybak MJ, Seo SM, Kaatz GW. 2011. Mechanisms of in vitro-selected daptomycin nonsusceptibility in Staphylococcus aureus. Int J Antimicrob Agents 38:442–446. doi: 10.1016/j.ijantimicag.2011.06.010. [DOI] [PubMed] [Google Scholar]
- 7.Bayer AS, Schneider T, Sahl HG. 2013. Mechanisms of daptomycin resistance in Staphylococcus aureus: role of the cell membrane and cell wall. Ann N Y Acad Sci 1277:139–158. doi: 10.1111/j.1749-6632.2012.06819.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Peleg AY, Miyakis S, Ward DV, Earl AM, Rubio A, Cameron DR, Pillai S, Moellering RC, Eliopoulos GM. 2012. Whole genome characterization of the mechanisms of daptomycin resistance in clinical and laboratory derived isolates of Staphylococcus aureus. PLoS One 7:e28316. doi: 10.1371/journal.pone.0028316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cui L, Isii T, Fukuda M, Ochiai T, Neoh HM, Camargo IL, Watanabe Y, Shoji M, Hishinuma T, Hiramatsu K. 2010. An RpoB mutation confers dual heteroresistance to daptomycin and vancomycin in Staphylococcus aureus. Antimicrob Agents Chemother 54:5222–5233. doi: 10.1128/AAC.00437-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jones T, Yeaman MR, Sakoulas G, Yang SJ, Proctor RA, Sahl HG, Schrenzel J, Xiong YQ, Bayer AS. 2008. Failures in clinical treatment of Staphylococcus aureus infection with daptomycin are associated with alterations in surface charge, membrane phospholipid asymmetry, and drug binding. Antimicrob Agents Chemother 52:269–278. doi: 10.1128/AAC.00719-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mishra NN, Rubio A, Nast CC, Bayer AS. 2012. Differential adaptations of methicillin-resistant Staphylococcus aureus to serial in vitro passage in daptomycin: evolution of daptomycin resistance and role of membrane carotenoid content and fluidity. Int J Microbiol 2012:683450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Friedman L, Alder JD, Silverman JA. 2006. Genetic changes that correlate with reduced susceptibility to daptomycin in Staphylococcus aureus. Antimicrob Agents Chemother 50:2137–2145. doi: 10.1128/AAC.00039-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gould IM, Miró JM, Rybak MJ. 2013. Daptomycin: the role of high-dose and combination therapy for Gram-positive infections. Int J Antimicrob Agents 42:202–210. doi: 10.1016/j.ijantimicag.2013.05.005. [DOI] [PubMed] [Google Scholar]
- 14.Kelesidis T, Humphries R, Ward K, Lewinski MA, Yang OO. 2011. Combination therapy with daptomycin, linezolid, and rifampin as treatment option for MRSA meningitis and bacteremia. Diagn Microbiol Infect Dis 71:286–290. doi: 10.1016/j.diagmicrobio.2011.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Berti AD, Wergin JE, Girdaukas GG, Hetzel SJ, Sakoulas G, Rose WE. 2012. Altering the proclivity towards daptomycin resistance in methicillin-resistant Staphylococcus aureus using combinations with other antibiotics. Antimicrob Agents Chemother 56:5046–5053. doi: 10.1128/AAC.00502-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Reference deleted.
- 17.Clinical and Laboratory Standards Institute CLSI. 2010. Performance standards for antimicrobial susceptibility testing; approved standard, 20th informational supplement M100-S20 CLSI, Wayne, PA. [Google Scholar]
- 18.Langmead B, Salzberg SL. 2012. Fast gapped-read alignment with Bowtie 2. Nat Methods 9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cafiso V, Bertuccio T, Purrello S, Campanile F, Mammina C, Sartor A, Raglio A, Stefani S. 2014. dltA overexpression: a strain-independent keystone of daptomycin resistance in methicillin-resistant Staphylococcus aureus. Int J Antimicrob Agents 43:26–31. doi: 10.1016/j.ijantimicag.2013.10.001. [DOI] [PubMed] [Google Scholar]
- 20.Gasch O, Pillai S, Dakos J, Miyakis S, Moellering R, Eliopoulos G. 2013. Daptomycin in vitro activity against methicillin-resistant Staphylococcus aureus is enhanced by d-cycloserine in a mechanism associated with a decrease in cell surface charge. Antimicrob Agents Chemother 57:4537–4539. doi: 10.1128/AAC.00799-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mehta S, Singh C, Plata KB, Chanda PK, Paul A, Riosa S, Rosato RR, Rosato AE. 2012. β-Lactams increase the antibacterial activity of daptomycin against clinical methicillin-resistant Staphylococcus aureus strains and prevent selection of daptomycin-resistant derivatives. Antimicrob Agents Chemother 56:6192–6200. doi: 10.1128/AAC.01525-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bayer AS, Mishra NN, Sakoulas G, Nonejuie P, Nast CC, Pogliano J, Chen KT, Ellison SN, Yeaman MR, Yang SJ. 2014. Heterogeneity of mprF sequences in methicillin-resistant Staphylococcus aureus clinical isolates: role in cross-resistance between daptomycin and host defense antimicrobial peptides. Antimicrob Agents Chemother 58:7462–7467. doi: 10.1128/AAC.03422-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ernst CM, Peschel A. 2011. Broad-spectrum antimicrobial peptide resistance by MprF-mediated aminoacylation and flipping of phospholipids. Mol Microbiol 80:290–299. doi: 10.1111/j.1365-2958.2011.07576.x. [DOI] [PubMed] [Google Scholar]
- 24.Landick R, Stewart J, Lee DN. 1990. Amino acid changes in conserved regions of the beta-subunit of Escherichia coli RNA polymerase alter transcription pausing and termination. Genes Dev 4:1623–1636. doi: 10.1101/gad.4.9.1623. [DOI] [PubMed] [Google Scholar]
- 25.García-López MC, Navarro F. 2011. RNA polymerase II conserved protein domains as platforms for protein-protein interactions. Transcription 2:193–197. doi: 10.4161/trns.2.4.16786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Davlieva M, Zhang W, Arias CA, Shamoo Y. 2013. Biochemical characterization of cardiolipin synthase mutations associated with daptomycin resistance in enterococci. Antimicrob Agents Chemother 57:289–296. doi: 10.1128/AAC.01743-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yang SJ, Xiong YQ, Boyle-Vavra S, Daum R, Jones T, Bayer AS. 2010. Daptomycin-oxacillin combinations in treatment of experimental endocarditis caused by daptomycin-nonsusceptible strains of methicillin-resistant Staphylococcus aureus with evolving oxacillin susceptibility (the “seesaw effect”). Antimicrob Agents Chemother 54:3161–3169. doi: 10.1128/AAC.00487-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Martin PK, Li T, Sun D, Biek DP, Schmid MB. 1999. Role in cell permeability of an essential two-component system in Staphylococcus aureus. J Bacteriol 181:3666–3673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ernst CM, Staubitz P, Mishra NN, Yang SJ, Hornig G, Kalbacher H, Bayer AS, Kraus D, Peschel A. 2009. The bacterial defensin resistance protein MprF consists of separable domains for lipid lysinylation and antimicrobial peptide repulsion. PLoS Pathog 5:e1000660. doi: 10.1371/journal.ppat.1000660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Haines TH, Dencher NA. 2002. Cardiolipin: a proton trap for oxidative phosphorylation. FEBS Lett 528:35–39. doi: 10.1016/S0014-5793(02)03292-1. [DOI] [PubMed] [Google Scholar]
- 31.Mohedano ML, Overweg K, de la Fuente A, Reuter M, Altabe S, Mulholland F, de Mendoza D, López P, Wells JM. 2005. Evidence that the essential response regulator YycF in Streptococcus pneumoniae modulates expression of fatty acid biosynthesis genes and alters membrane composition. J Bacteriol 187:2357–2367. doi: 10.1128/JB.187.7.2357-2367.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nadrah K, Strle F. 2011. Antibiotic combinations with daptomycin for treatment of Staphylococcus aureus infections. Chemother Res Pract 2011:619321. doi: 10.1155/2011/619321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dhand A, Bayer AS, Pogliano J, Yang SJ, Bolaris M, Nizet V, Wang G, Sakoulas G. 2011. Use of antistaphylococcal beta-lactams to increase daptomycin activity in eradicating persistent bacteremia due to methicillin-resistant Staphylococcus aureus: role of enhanced daptomycin binding. Clin Infect Dis 53:158–163. doi: 10.1093/cid/cir340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sakoulas G, Moise PA, Casapao AM, Nonejuie P, Olson J, Okumura CY, Rybak MJ, Kullar R, Dhand A, Rose WE, Goff DA, Bressler AM, Lee Y, Pogliano J, Johns S, Kaatz GW, Ebright JR, Nizet V. 2014. Antimicrobial salvage therapy for persistent staphylococcal bacteremia using daptomycin plus ceftaroline. Clin Ther 36:1317–1333. doi: 10.1016/j.clinthera.2014.05.061. [DOI] [PubMed] [Google Scholar]
- 35.Moise PA, Amodio-Groton M, Rashid M, Lamp KC, Hoffman-Roberts HL, Sakoulas G, Yoon MJ, Schweitzer S, Rastogi A. 2013. Multicenter evaluation of the clinical outcomes of daptomycin with and without concomitant β-lactams in patients with Staphylococcus aureus bacteremia and mild to moderate renal impairment. Antimicrob Agents Chemother 57:1192–1200. doi: 10.1128/AAC.02192-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rose W, Berti A, Sauer J, Nizet V, Pogliano J, Sakoulas G. 2013. β-Lactams targeting PBP1 induce cell morphology changes in methicilin-resistant Staphylococcus aureus to selectively enhance daptomycin activity, abstr A021. Abstr 53rd Intersci Conf Antimicrob Agents Chemother. American Society for Microbiology, Washington, DC. [Google Scholar]
- 37.Komatsuzawa H, Ohta K, Fujiwara T, Choi GH, Labischinski H, Sugai M. 2001. Cloning and sequencing of the gene, fmtC, which affects oxacillin resistance in methicillin-resistant Staphylococcus aureus. FEMS Microbiol Lett 203:49–54. doi: 10.1111/j.1574-6968.2001.tb10819.x. [DOI] [PubMed] [Google Scholar]
- 38.Peschel A, Jack RW, Otto M, Collins LV, Staubitz P, Nicholson G, Kalbacher H, Nieuwenhuizen WF, Jung G, Tarkowski A, van Kessel KP, van Strijp JA. 2001. Staphylococcus aureus resistance to human defensins and evasion of neutrophil killing via the novel virulence factor MprF is based on modification of membrane lipids with L-lysine. J Exp Med 193:1067–1076. doi: 10.1084/jem.193.9.1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yang SJ, Mishra NN, Rubio A, Bayer AS. 2013. Causal role of single nucleotide polymorphisms within the mprF gene of Staphylococcus aureus in daptomycin resistance. Antimicrob Agents Chemother 57:5658–5664. doi: 10.1128/AAC.01184-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rubio A, Conrad M, Haselbeck RJ, Kedar GC, Brown-Driver V, Finn J, Silverman JA. 2011. Regulation of mprF by antisense RNA restores daptomycin susceptibility to daptomycin-resistant isolates of Staphylococcus aureus. Antimicrob Agents Chemother 55:364–367. doi: 10.1128/AAC.00429-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Berti AD, Sakoulas G, Nizet V, Tewhey R, Rose WE. 2013. β-Lactam antibiotics targeting PBP1 selectively enhance daptomycin activity against methicillin-resistant Staphylococcus aureus. Antimicrob Agents Chemother 57:5005–5012. doi: 10.1128/AAC.00594-13. [DOI] [PMC free article] [PubMed] [Google Scholar]