

Abstract
A multidrug-resistant Klebsiella pneumoniae isolate exhibiting heteroresistance to colistin was investigated. The colistin-resistant subpopulation harbored a single amino acid change (Asp191Tyr) in protein PhoP, which is part of the PhoPQ two-component system that activates pmrHFIJKLM expression responsible for l-aminoarabinose synthesis and polymyxin resistance. Complementation assays with a wild-type phoP gene restored full susceptibility to colistin. Then, analysis of the colistin-susceptible subpopulation showed a partial deletion (25 bp) in the phoP gene compared to that in the colistin-resistant subpopulation. That deletion disrupted the reading frame of phoP, leading to a longer and inactive protein (255 versus 223 amino acids long). This is the first report showing the involvement of mutation(s) in PhoP in colistin resistance. Furthermore, this is the first study to decipher the mechanisms leading to colistin heteroresistance in K. pneumoniae.
INTRODUCTION
Klebsiella pneumoniae is a Gram-negative bacterium often associated with nosocomial infections, including urinary tract infection, pneumonia, and bloodstream infection (1). While multidrug resistance is increasingly reported in that species due to acquisition of numerous resistance traits, including extended-spectrum β-lactamase and carbapenemase genes, colistin is increasingly used for treating infections due to multidrug-resistant isolates. Colistin and polymyxin B correspond to important therapeutic options for treating infections caused by multidrug-resistant K. pneumoniae, particularly in countries with a high prevalence of carbapenemase producers (2). Those drugs are bactericidal for Gram-negative bacteria, interacting with the lipid A moiety of lipopolysaccharide (LPS) and subsequently causing disorganization of the outer membrane (3).
In K. pneumoniae, colistin resistance is being increasingly reported, and some cases of heteroresistant K. pneumoniae isolates have also been observed (4, 5). Colistin heteroresistance is defined as “the emergence of resistance to colistin by a subpopulation from an otherwise susceptible (MIC of ≤2 mg/liter) population” (6) that may be related to exposure to a suboptimal polymyxin concentration (7). A comparison of the in vitro susceptibility testing methods for colistin showed that the Etest and agar dilution methods are reliable for detecting resistant subpopulations in contrast to some automatic methods; nonetheless, the broth microdilution method remains the reference test for determination of colistin MICs (8).
Several studies have shown that mutations in the PmrAB or PhoPQ regulatory systems may confer colistin resistance in K. pneumoniae (9–12). In addition, inactivation of the mgrB gene, which encodes the MgrB protein known to negatively regulate the PhoPQ signaling system, may also be the source of acquired resistance to colistin (13–16).
Acquired resistance to polymyxins is mediated by the addition of 4-deoxyaminoarabinose (LAra4N) and/or phosphoethanolamine (pEtN) to lipid A. LAra4N synthesis requires the products of the pmrE gene and pmrHFIJKLM operon, and pEtN synthesis is encoded by the pmrC gene. These modifications create a more positively charged lipopolysaccharide and thus reduce the affinity of LPS to positively charged polymyxins (7). Of note, mechanisms leading to heteroresistance in K. pneumoniae remain unknown.
PmrB and PhoQ are sensor cytoplasmic membrane-bound kinases activated by high concentrations of iron (Fe3+) and an acidic pH (pH 5.5) for the PmrAB system (17) and by low extracellular concentrations of magnesium (Mg2+) for the PhoPQ system (18). Upon activation, PmrB activates the response regulator PmrA, which in turn upregulates pmrC and pmrHFIJKLM. Likewise, PhoQ activates PhoP, which in turns activates the expression of the pmrHFIJKLM operon, directly by binding to the pmrHFIJKLM promoter and indirectly via PmrD-dependent activation of the PmrA protein also binding to the pmrHFIJKLM promoter (19).
The aim of this study was to determine the mechanism(s) responsible for heteroresistance to colistin in K. pneumoniae.
MATERIALS AND METHODS
Bacterial strains.
A multidrug-resistant K. pneumoniae clinical isolate named Kp75 was recovered in 2012 in South Africa. It produced the carbapenemase OXA-48, which has been reported elsewhere (20). The wild-type and colistin-susceptible K. pneumoniae ATCC 53153 strain was used as a control throughout the study.
Antimicrobial susceptibility assays.
MICs were determined using Etest strips (AB bioMérieux, La Balme-les-Grottes, France) on Mueller-Hinton agar plates (Bio-Rad, Marnes-la-Coquette, France) with a 0.5 McFarland inoculum. The MICs of colistin were also determined by a broth culture microdilution method as recommended by the CLSI (21), with Tween 80 being added at 0.002% as suggested in several studies (22, 23). According to the EUCAST guidelines, isolates with MICs of ≤2 μg/ml are categorized as susceptible, and those with MICs of >2 μg/ml are categorized as resistant (24).
PCR amplification and sequencing.
Whole-cell DNA was extracted using a commercially available kit (QIAquick; Qiagen, Valencia, CA) according to the manufacturer's instructions. The pmrA, pmrB, phoP, phoQ, and mgrB genes were amplified with specific oligonucleotide primers (the primers used in this study are listed in Table 1). Molecular detection of the β-lactam resistance genes was performed as reported previously (25). The amplified DNA fragments were purified with the QIAquick PCR purification kit (Qiagen, Courtaboeuf, France). Both strands of the amplification products obtained were sequenced with an ABI 3100 sequencer (Applied Biosystems, Foster City, CA). The nucleotide and deduced protein sequences were analyzed at the National Center for Biotechnology Information website (www.ncbi.nlm.nih.gov) by the Basic Local Alignment Search Tool (BLAST) program.
TABLE 1.
Oligonucleotides used as primers in this study
| Primer | Sequence (5′ to 3′) | Gene | Reference or study |
|---|---|---|---|
| pmrA ext F | CAT TTC CGC GCA CTG TCT GC | pmrA | 12 |
| pmrA ext R | CAG GTT TCA GTT GCA AAC AG | pmrA | 12 |
| pmrB ext F | ACC TAC GCG AAA AGA TTG GC | pmrB | 12 |
| pmrB ext R | GAT GAG GAT AGC GCC CAT GC | pmrB | 12 |
| phoP ext F | GAG CTT CAG ACT ACT ATC GA | phoP | 12 |
| phoP ext R | GGG AAG ATA TGC CGC AAC AG | phoP | 12 |
| phoQ ext F | ATA CCC ACA GGA CGT CAT CA | phoQ | 12 |
| phoQ ext R | CAG GTG TCT GAC AGG GAT TA | phoQ | 12 |
| mgrB ext F | TTA AGA AGG CCG TGC TAT CC | mgrB | 13 |
| mgrB ext R | AAG GCG TTC ATT CTA CCA CC | mgrB | 13 |
| mdh F | CCC AAC TCG CTT CAG GTT CAG | mdh | 26 |
| mdh R | CCG TTT TTC CCC AGC AGC AG | mdh | 26 |
| pmrC int F | GCG TGA TGA ATA TCC TCA CCA | pmrC | 12 |
| pmrC int R | CAC GCC AAA GTT CCA GAT GA | pmrC | 12 |
| pmrA int F | GAT GAA GAC GGG CTG CAT TT | pmrA | 12 |
| pmrA int R | ACC GCT AAT GCG ATC CTC AA | pmrA | 12 |
| pmrB int F | TGC CAG CTG ATA AGC GTC TT | pmrB | 12 |
| pmrB int R | TTC TGG TTG TTG TGC CCT TC | pmrB | 12 |
| pmrD int F | GAT CGC AGA GAT TGA AGC CT | pmrD | 12 |
| pmrD int R | GCG TTG CGG ATC TTC AAA GT | pmrD | 12 |
| pmrE int F | GCA TAC CGT AAT GCC GAC TA | pmrE | 12 |
| pmrE int R | GGG TTG ATC TCT GTG ACA TC | pmrE | 12 |
| pmrK int F | AGT ATC GGT CAG TGG CTG TT | pmrK | 12 |
| pmrK int R | CCG CTT ATC ACG AAA GAT CC | pmrK | 12 |
| phoP int F | GCG TCA CCA CCT CAA AGT TC | phoP | This study |
| phoP int R | GGC GAT ATC CGG GAG ATG TT | phoP | This study |
| phoQ int F | CTC AAG CGC AGC TAT ATG GT | phoQ | This study |
| phoQ int R | TCT TTG GCC AGC GAC TCA AT | phoQ | This study |
| rpsL int F | CCG TGG CGG TCG TGT TAA AGA | rpsL | 13 |
| rpsL int R | GCC GTA CTT GGA GCG AGC CTG | rpsL | 13 |
Analysis of the primary and secondary structures of the PhoP protein.
The primary structure of PhoP was analyzed by using the Pfam website (http://pfam.xfam.org/). The predicted secondary structures of the proteins were obtained by the GOR method using the EMBOSS 6.3.1 software available on the Mobyle@Pasteur portal (http://mobyle.pasteur.fr/cgi-bin/portal.py).
Complementation experiments.
The entire phoP and phoQ genes from the colistin-susceptible K. pneumoniae ATCC 53153 reference strain (MIC of colistin at 0.125 μg/ml) were amplified by PCR using 2× Phusion HF master mix (Thermo Scientific Finnzymes, Illkirch, France) and primers sets phoP ext F/phoP ext R and phoQ ext F/phoQ ext R, respectively. The noncoding sequence mdh was also amplified with primer set mdh F/mdh R as described previously (26) (the primers used are listed in Table 1). The amplified fragments were cloned into plasmid pCR-BluntII-TOPO (Applied Biosystems by Life Technologies, Carouge, Switzerland) and the resulting plasmids pTOPO-phoP, pTOPO-phoQ, and pTOPO-mdh were, respectively, introduced into the colistin-resistant isolate Kp75 by electroporation. Transformants were selected by overnight incubation at 37°C on Mueller-Hinton agar supplemented with 100 μg/ml of zeocin. The MICs of colistin were determined by Etest for all K. pneumoniae transformants.
Growth curves.
Growth kinetics were determined with subpopulations of isolate Kp75 in the absence of any selective pressure. Two hundred-milliliter volumes of Luria-Bertani broth were inoculated independently with 108 CFU of each strain, and cultures were grown for 24 h at 100 rpm and 37°C. Absorbance at 600 nm was measured every hour for the first 10 h and after 24 h. The colony counting was performed by serial dilution and final plating on solid medium.
Transcriptional analysis by qRT-PCR.
Quantitative real-time PCR (qRT-PCR) was used to measure the expression of the phoP, phoQ, pmrD, pmrC, pmrA, pmrB, and pmrK genes, using specific oligonucleotide primers (Table 1). A culture volume of 0.5 ml was taken during the mid-log phase of growth (optical density at 600 nm [OD600 nm] = 1.1) and combined with 1 ml of RNAprotect (Qiagen, Courtaboeuf, France). Total RNA was extracted from cell lysates using the RNeasy minikit (Qiagen), according to the manufacturer's instructions. qRT-PCR was carried out using a Rotor-Gene Q instrument (Qiagen), and QuantiFast SYBR green (Qiagen) was used as a signal reporter. In each run, a blank sample (distilled water) and a no reverse transcriptase control were included to exclude DNA contamination. Relative gene expression differences were calculated by first normalizing the CT values by subtracting the 12S rRNA control (rpsL gene) (13) and then comparing to the values obtained for the susceptible isolate.
Nucleotide sequence accession numbers.
The nucleotide and protein sequences of the PhoP proteins of isolates Kp75a and Kp75b were registered in GenBank under accession numbers KP742843 and KP742844, respectively.
RESULTS AND DISCUSSION
Isolate Kp75 exhibits multidrug resistance, including resistance to colistin.
Isolate Kp75 was resistant to all β-lactams, including carbapenems (the MICs of imipenem, meropenem, and ertapenem were 3, 8, and >32 μg/ml, respectively). In addition, it was resistant to chloramphenicol, fluoroquinolones, gentamicin, tetracycline, and tobramycin. The MIC of rifampin was 16 μg/ml, and the MIC of nitrofurantoin was >128 μg/ml. It remained susceptible only to sulfonamides, tigecycline, and amikacin. Determination of the MIC of colistin by using the broth culture microdilution method showed that isolate Kp75 was resistant to colistin (MIC of 128 μg/ml). Molecular detection of the β-lactam resistance genes identified the narrow-spectrum β-lactamase blaTEM-1 gene, the expanded-spectrum β-lactamase blaCTX-M-15 gene, and the carbapenem-hydrolyzing class D β-lactamase blaOXA-48 gene.
Susceptibility testing by Etest revealed a heteroresistant phenotype.
When the MIC of colistin for isolate Kp75 was measured by using Etest strips, two subpopulations were identified close to the Etest strip (Fig. 1). Bacterial colonies located in the resistance and susceptible areas were picked up, and the two subpopulations were separated. The MICs for the two distinct subpopulations were again measured by Etest. The colistin-susceptible subpopulation (then termed Kp75a) and the colistin-resistant subpopulation (then termed Kp75b) had MICs of colistin at 0.12 and 12 μg/ml, respectively. With use of the microdilution method, the MICs were found to be 0.12 and 128 μg/ml, respectively. These data further highlight the discrepancies between values obtained by the two methods and reveal that the MICs obtained by the Etest are significantly lower than those obtained by the microdilution method for such a resistant isolate.
FIG 1.

MIC determination using a colistin Etest strip for K. pneumoniae Kp75. The arrow indicates the colistin-resistant subpopulation (Kp75b).
A single amino acid substitution in PhoP leads to colistin resistance in isolate Kp75b.
Total DNAs from isolate Kp75b and colistin-susceptible K. pneumoniae reference strain ATCC 53153 were used as the templates for PCRs to amplify the pmrA, pmrB, phoP, phoQ, and mgrB genes possibly involved in colistin resistance. A single base pair substitution was identified in the phoP gene of isolate Kp75b, leading to a single amino acid substitution (Asp191Tyr) compared to that of the PhoP proteins available in the GenBank databases and also compared to that of strain ATCC 53153 (Fig. 2). No substitution was identified in the pmrA, pmrB, phoQ, and mgrB genes.
FIG 2.

Alignment of a fragment of the phoP gene nucleotide sequences (A) and amino acid PhoP protein sequences (B) of the colistin-resistant isolate Kp75b and the colistin-susceptible isolate Kp75a, on the basis of that of the wild-type reference strain ATCC 53153. In panel A, dashes indicate absent nucleotides in Kp75a, and stars indicate conserved nucleotides; in panel B, dashes indicate amino acid residues identical to those of the reference sequence. In the lower portion of panel A, the shaded nucleotides indicate the deleted 25-bp fragment identified in Kp75a. The amino acid substitution (Asp191Tyr) involved in resistance to colistin in Kp75b is shaded in panel B.
The amino acid substitution was identified at position 191 of the PhoP protein, which is located in its C-terminal domain. We therefore speculate that the replacement of a polar negative amino acid by an aromatic amino acid partially hydrophobic in the C-terminal domain of PhoP might induce a constitutive activation of PhoP, disregulating the two-component regulatory system PhoPQ. The predicted secondary structures of two PhoP proteins, namely that of a wild-type strain and that of isolate Kp75b, were compared, and it was revealed that the Asp191Tyr amino acid substitution significantly modified the secondary structure of the protein, with an interruption of the α-helix (see Fig. S1 in the supplemental material). It was therefore likely that resistance to colistin in isolate Kp75b was related to that single substitution in PhoP. To further assess this hypothesis, an experiment was conducted in order to complement isolate Kp75b with the entire phoP gene obtained from the wild-type strain ATCC 53153. The entire phoP and phoQ genes, together with a fragment of the mdh gene (a noncoding sequence as control), were amplified from the reference strain ATCC 53153 and cloned in plasmid pTOPO-BluntII, giving rise to, respectively, pTOPO-phoP, pTOPO-phoQ, and pTOPO-mdh and then transformed into the resistant-colistin clinical isolate Kp75b. The MIC determinations showed a complete reversion to susceptibility only for the transformant complemented with pTOPO-phoP (MIC at 0.125 μg/ml), whereas the MICs of transformants complemented with recombinant plasmids pTOPO-phoQ or pTOPO-mdh remained unchanged. This result strongly suggested that the amino acid substitution identified in PhoP was indeed responsible for the colistin resistance in Kp75b (Fig. 3).
FIG 3.
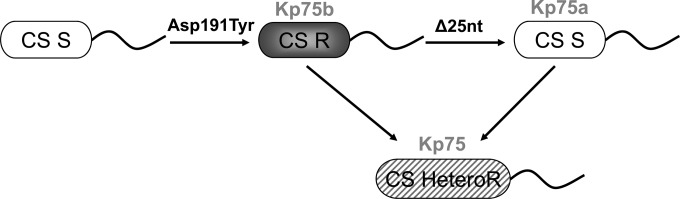
Schematic representation of the mechanisms leading to heteroresistance in isolate Kp75. The colistin-susceptible (CS S) subpopulation (Kp75a) is represented in white, the colistin-resistant (CS R) subpopulation (Kp75b) in gray, and the mixed (CS HeteroR) population (Kp75) in gray stripes. The PhoP amino acid substitution (Asp191Tyr) and the deletion of the 25-bp fragment (Δ25 nucleotides [nt]) are indicated.
A partial deletion of the phoP gene is responsible for a reversion in colistin susceptibility.
Similarly to what was performed with isolate Kp75b, PCRs followed by sequencing were performed to amplify the pmrA, pmrB, phoP, phoQ, and mgrB genes of the colistin-susceptible isolate Kp75a. All sequences were found to be identical with those obtained with Kp75b, except for the phoP gene. Indeed, the phoP gene harbored not only a substitution leading to the Asp191Tyr amino acid substitution in the corresponding protein sequence but also a partial deletion (25 bp long) in the phoP gene of Kp75a, disrupting its reading frame and consequently giving rise to a longer protein of 255 amino acids instead of 223 amino acids as found in Kp75b and all other PhoP sequences in the databases (Fig. 2). This deletion was therefore considered to be responsible for the reversion into a colistin-susceptible phenotype. Figure 3 provides a summary of the evolution process observed in Kp75. This mechanism of partial gene deletion involved in reversion to colistin susceptibility was previously described in the PmrAB two-component system of Acinetobacter baumannii (27).
Comparative growth rates of colistin-susceptible and colistin-resistant isogenic strains.
The growth kinetics of colistin-resistant (Kp75b) and colistin-susceptible (Kp75a) isogenic strains were determined in the absence of selective pressure. During a 24-h period, no significant difference was observed in growth rates between those two strains (data not shown).
Upregulation of phoP, phoQ, pmrD, and pmrK gene expression in colistin-resistant strain Kp75b.
Expression of the pmr and pho genes was investigated to determine whether the expression of those genes involved in the PmrAB or PhoPQ regulatory systems might differ between the two Kp75 subpopulations. RT-PCR identified upregulation of the phoP, phoQ, pmrD, and pmrK genes in the resistant isolate Kp75b compared with that of the isogenic susceptible isolate Kp75a. In contrast, expression of the pmrC, pmrA, and pmrB genes did not differ significantly (Fig. 4).
FIG 4.

Relative expression of the phoP, phoQ, pmrD, pmrK, pmrC, pmrA, and pmrB genes considering the colistin-resistant isolate Kp75b compared to the colistin-susceptible isolate Kp75a. Values and standard deviations are the means from three independent experiments.
We may therefore consider that the mutated protein PhoP activates the transcription of the pmrHFIJKLM operon, the product of which leads to synthesis of l-amino-arabinose and ultimately to colistin resistance in K. pneumoniae. We also observed that PhoP activates the expression of the PmrD-encoding gene and that PmrD did not activate the expression of the pmrA gene, contrary to what was expected (19).
In conclusion, our study suggests that colistin heteroresistance in K. pneumoniae may be caused by alterations in the PhoPQ regulatory system. To the best of our knowledge, this is the first study investigating the genetic basis of colistin heteroresistance in that bacterial species. We first demonstrate here the involvement of the PhoP protein in resistance to colistin in K. pneumoniae. This additional mechanism adds to the diversity of genetic events that may lead to acquisition of resistance to colistin in that species. Interestingly, our data showed that the heteroresistance observed here was related to a reversion of resistance into susceptibility and not the opposite. Indeed, the amino acid substitution identified in PhoP to which the resistance was attributed was still identified in the susceptible isolate.
In addition, we want to emphasize that the phenomenon of colistin heteroresistance remains very likely underestimated, considering that it may not be seen when the method (broth microdilution) recommended by the CLSI is used for the evaluation of susceptibility to polymyxins. We showed here that heteroresistance may be well demonstrated when only Etest strips are used. Notably, the clinical significance of colistin heteroresistance in K. pneumoniae is still unknown and remains to be evaluated.
Supplementary Material
ACKNOWLEDGMENTS
This work was funded by the University of Fribourg, Switzerland, and by a grant from the European Community (R-GNOSIS, FP7/HEALTH-F3-2011-282512).
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AAC.05055-14.
REFERENCES
- 1.Podschun R, Ullmann U. 1998. Klebsiella spp. as nosocomial pathogens: epidemiology, taxonomy, typing methods, and pathogenicity factors. Clin Microbiol Rev 11:589–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Falagas ME, Lourida P, Poulikakos P, Rafailidis PI, Tansarli GS. 2014. Antibiotic treatment of infections due to carbapenem-resistant Enterobacteriaceae: systematic evaluation of the available evidence. Antimicrob Agents Chemother 58:654–663. doi: 10.1128/AAC.01222-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hancock RE. 1997. Peptide antibiotics. Lancet 349:418–422. doi: 10.1016/S0140-6736(97)80051-7. [DOI] [PubMed] [Google Scholar]
- 4.Meletis G, Tzampaz E, Sianou E, Tzavaras I, Sofianou D. 2011. Colistin heteroresistance in carbapenemase-producing Klebsiella pneumoniae. J Antimicrob Chemother 66:946–947. doi: 10.1093/jac/dkr007. [DOI] [PubMed] [Google Scholar]
- 5.Poudyal A, Howden BP, Bell JM, Gao W, Owen RJ, Turnidge JD, Nation RL, Li J. 2008. In vitro pharmacodynamics of colistin against multidrug-resistant Klebsiella pneumoniae. J Antimicrob Chemother 62:1311–1318. doi: 10.1093/jac/dkn425. [DOI] [PubMed] [Google Scholar]
- 6.Li J, Nation RL, Turnidge JD, Milne RW, Coulthard K, Rayner CR, Paterson DL. 2006. Colistin: the re-emerging antibiotic for multidrug-resistant Gram-negative bacterial infections. Lancet Infect Dis 6:589–601. doi: 10.1016/S1473-3099(06)70580-1. [DOI] [PubMed] [Google Scholar]
- 7.Falagas ME, Rafailidis PI, Matthaiou DK. 2010. Resistance to polymyxins: mechanisms, frequency and treatment options. Drug Resist Updat 13:132–138. doi: 10.1016/j.drup.2010.05.002. [DOI] [PubMed] [Google Scholar]
- 8.Humphries RM. 2015. Susceptibility testing of the polymyxins: where are we now? Pharmacotherapy 35:22–27. doi: 10.1002/phar.1505. [DOI] [PubMed] [Google Scholar]
- 9.Cannatelli A, Di Pilato V, Giani T, Arena F, Ambretti S, Gaibani P, D'Andrea MM, Rossolini GM. 2014. In vivo evolution to colistin resistance by PmrB sensor kinase mutation in KPC-producing Klebsiella pneumoniae is associated with low-dosage colistin treatment. Antimicrob Agents Chemother 58:4399–4403. doi: 10.1128/AAC.02555-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cheng HY, Chen YF, Peng HL. 2010. Molecular characterization of the PhoPQ-PmrD-PmrAB mediated pathway regulating polymyxin B resistance in Klebsiella pneumoniae CG43. J Biomed Sci 17:60. doi: 10.1186/1423-0127-17-60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Choi MJ, Ko KS. 2014. Mutant prevention concentrations of colistin for Acinetobacter baumannii, Pseudomonas aeruginosa and Klebsiella pneumoniae clinical isolates. J Antimicrob Chemother 69:275–277. doi: 10.1093/jac/dkt315. [DOI] [PubMed] [Google Scholar]
- 12.Jayol A, Poirel L, Brink A, Villegas MV, Yilmaz M, Nordmann P. 2014. Resistance to colistin associated with a single amino acid change in protein PmrB among Klebsiella pneumoniae isolates of worldwide origin. Antimicrob Agents Chemother 58:4762–4766. doi: 10.1128/AAC.00084-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cannatelli A, D'Andrea MM, Giani T, Di Pilato V, Arena F, Ambretti S, Gaibani P, Rossolini GM. 2013. In vivo emergence of colistin resistance in Klebsiella pneumoniae producing KPC-type carbapenemase mediated by insertional inactivation of the PhoQ/PhoP mgrB regulator. Antimicrob Agents Chemother 57:5521–5526. doi: 10.1128/AAC.01480-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cannatelli A, Giani T, D'Andrea MM, Di Pilato V, Arena F, Conte V, Tryfinopoulou K, Vatopoulos A, Rossolini GM. 2014. MgrB inactivation is a common mechanism of colistin resistance in KPC-producing Klebsiella pneumoniae of clinical origin. Antimicrob Agents Chemother 58:5696–5703. doi: 10.1128/AAC.03110-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Poirel L, Jayol A, Bontron S, Villegas MV, Özdamar M, Turkoglü S, Nordmann P. 2015. The mgrB gene as a key target for acquired resistance to colistin in Klebsiella pneumoniae. J Antimicrob Chemother 70:75–80. doi: 10.1093/jac/dku323. [DOI] [PubMed] [Google Scholar]
- 16.Wright MS, Suzuki Y, Jones MB, Marshall SH, Rudin SD, van Duin D, Kaye K, Jacobs MR, Bonomo RA, Adams MD. 2015. Genomic and transcriptomic analyses of colistin-resistant clinical isolates of Klebsiella pneumoniae reveal multiple pathways of resistance. Antimicrob Agents Chemother 59:536–543. doi: 10.1128/AAC.04037-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gunn JS. 2008. The Salmonella PmrAB regulon: lipopolysaccharide modifications, antimicrobial peptide resistance and more. Trends Microbiol 16:284–290. doi: 10.1016/j.tim.2008.03.007. [DOI] [PubMed] [Google Scholar]
- 18.Groisman EA. 2001. The pleiotropic two-component regulatory system PhoP-PhoQ. J Bacteriol 183:1835–1842. doi: 10.1128/JB.183.6.1835-1842.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mitrophanov AY, Jewett MW, Hadley TJ, Groisman EA. 2008. Evolution and dynamics of regulatory architectures controlling polymyxin B resistance in enteric bacteria. PLoS Genet 4:e1000233. doi: 10.1371/journal.pgen.1000233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Potron A, Poirel L, Rondinaud E, Nordmann P. 2013. Intercontinental spread of OXA-48 β-lactamase-producing Enterobacteriaceae over a 11-year period, 2001 to 2011. Euro Surveill 18(31):pii=20549 http://www.eurosurveillance.org/ViewArticle.aspx?ArticleId=20549. [DOI] [PubMed] [Google Scholar]
- 21.Clinical and Laboratory Standards Institute. 2014. Performance standards for antimicrobial susceptibility testing; 24th informational supplement. CLSI document M100-S24. Clinical and Laboratory Standards Institute, Wayne, PA. [Google Scholar]
- 22.Hindler JA, Humphries RM. 2013. Colistin MIC variability by method for contemporary clinical isolates of multidrug-resistant Gram-negative bacilli. J Clin Microbiol 51:1678–1684. doi: 10.1128/JCM.03385-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sader HS, Rhomberg PR, Flamm RK, Jones RN. 2012. Use of a surfactant (polysorbate 80) to improve MIC susceptibility testing results for polymyxin B and colistin. Diagn Microbiol Infect Dis 74:412–414. doi: 10.1016/j.diagmicrobio.2012.08.025. [DOI] [PubMed] [Google Scholar]
- 24.European Committee on Antimicrobial Susceptibility Testing. 2015. Breakpoint tables for interpretation of MICs and zone diameters. Version 2.0 http://www.eucast.org/clinical_breakpoints/.
- 25.Nordmann P, Naas T, Poirel L. 2011. Global spread of carbapenemase-producing Enterobacteriaceae. Emerg Infect Dis 17:1791–1798. doi: 10.3201/eid1710.110655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Diancourt L, Passet V, Verhoef J, Grimont PA, Brisse S. 2005. Multilocus sequence typing of Klebsiella pneumoniae nosocomial isolates. J Clin Microbiol 43:4178–4182. doi: 10.1128/JCM.43.8.4178-4182.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Adams MD, Nickel GC, Bajaksouzian S, Lavender H, Murthy AR, Jacobs MR, Bonomo RA. 2009. Resistance to colistin in Acinetobacter baumannii associated with mutations in the PmrAB two-component system. Antimicrob Agents Chemother 53:3628–3634. doi: 10.1128/AAC.00284-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.