

Abstract
The recently approved HIV-1 integrase strand transfer inhibitor (INSTI) dolutegravir (DTG) (S/GSK1349572) has overall advantageous activity when tested in vitro against HIV-1 with raltegravir (RAL) and elvitegravir (EVG) resistance signature mutations. We conducted an in vitro resistance selection study using wild-type HIV-1 and mutants with the E92Q, Y143C, Y143R, Q148H, Q148K, Q148R, and N155H substitutions to assess the DTG in vitro barrier to resistance. No viral replication was observed at concentrations of ≥32 nM DTG, whereas viral replication was observed at 160 nM RAL or EVG in the mutants. In the Q148H, Q148K, or Q148R mutants, G140S/Q148H, E138K/Q148K, E138K/Q148R, and G140S/Q148R secondary mutations were identified with each INSTI and showed high resistance to RAL or EVG but limited resistance to DTG. E138K and G140S, as secondary substitutions to Q148H, Q148K, or Q148R, were associated with partial recovery in viral infectivity and/or INSTI resistance. In the E92Q, Y143C, Y143R, and N155H mutants, no secondary substitutions were associated with DTG. These in vitro results suggest that DTG has a high barrier to the development of resistance in the presence of RAL or EVG signature mutations other than Q148. One explanation for this high barrier to resistance is that no additional secondary substitution of E92Q, Y143C, Y143R, or N155H simultaneously increased the fold change in 50% effective concentration (EC50) to DTG and infectivity. Although increased DTG resistance via the Q148 pathway and secondary substitutions occurs at low concentrations, a higher starting concentration may reduce or eliminate the development of DTG resistance in this pathway in vitro.
INTRODUCTION
Remarkable progress has been made in the global effort to overcome HIV-1 infection with the introduction and widespread use of antiretroviral (ARV) treatment and prevention measures (1). Combination antiretroviral therapy has significantly improved AIDS-related morbidity and mortality, extending the expected life span of patients with HIV-1. However, several factors contribute to the continuing issue of treatment failure and drug resistance, including suboptimal drug efficacy and/or variable pharmacokinetics, inadequate compliance to lifelong therapy, transmitted drug resistance, and acute or chronic drug toxicity. In particular, treatment with an ARV that has a high barrier to resistance and good safety and tolerability profiles might further improve long-term treatment efficacy for patients, especially those with multidrug-resistant HIV-1 infections (2).
Integrase strand transfer inhibitors (INSTIs) are the latest drug class developed for the treatment of HIV-1 infections. Raltegravir (RAL) and elvitegravir (EVG) are previously approved INSTIs that are safe and effective in both treatment-naive and treatment-experienced patients (3–6). Although these INSTIs strongly inhibit HIV-1 replication, they possess only a modest barrier to resistance, and the resulting INSTI resistance mutations are associated with resistance to both RAL and EVG (7, 8).
The most recently approved INSTI dolutegravir (DTG) is for once-daily dosing without a pharmacokinetic booster and shows efficacy for treatment-naive subjects and treatment-experienced but INSTI-naive subjects, as well as subjects who failed RAL and EVG treatment (9–13). DTG was approved by the U.S. FDA in August 2013 for treatment-naive and treatment-experienced INSTI-naive adults, as well as for INSTI-resistant adults. The pediatric indication is for treatment-naive and treatment-experienced INSTI-naive subjects age ≥12 years and weighing ≥40 kg. The INSTI class is now recognized as one of the safest and most effective anti-HIV-1 drug classes (14), and the U.S. Department of Health and Human Services (DHHS) guidelines recommend INSTI-based regimens, including DTG, as the preferred regimens for antiretroviral therapy (ART)-naive patients (15).
During the course of DTG clinical development, we expected that the attributes of DTG, i.e., low cross-resistance to RAL or EVG, should be suitable for all HIV-1-positive patients, including the critical unmet need of INSTI salvage therapy because of clinical resistance to RAL and EVG (16, 17). The INSTI-resistant patient VIKING pilot study and phase 3 (VIKING-3) study have shown the clinical utility of DTG in patient populations who have failed to respond to INSTI therapy (10, 18, 19).
Previously, we reported from our in vitro DTG resistance selection study that a mutant virus with >3-fold change as site-directed mutants (SDM) did not emerge during >100 days of culture, indicating a high barrier to resistance of DTG to wild-type HIV-1 in vitro (20). In this report, we investigated the effect of RAL and EVG signature mutations on the barrier to resistance to DTG in vitro.
MATERIALS AND METHODS
Compounds.
DTG was synthesized at Shionogi Research Laboratories (Osaka, Japan). EVG was synthesized at GSK (Research Triangle Park, NC). RAL, efavirenz (EFV), and lamivudine (3TC) were purchased from Sequoia Research Products (Pangbourne, United Kingdom).
Cells and viruses.
HeLa-CD4 cells carrying a reporter β-galactosidase gene driven by the HIV-1 long terminal repeat (LTR) were established by transfecting HeLa cells with CD4 and β-galactosidase expression vector (21). Human T-cell lines (MT-2, MT-4, and M8166) were obtained from the Institute for Virus Research, Kyoto University, Kyoto, Japan (22). HeLa-CD4 cells were grown in Dulbecco's modified minimal essential medium (DMEM) containing 10% fetal bovine serum (FBS) and 60 μg/ml kanamycin sulfate. Human T-cell lines were maintained in RPMI 1640 medium supplemented with 10% FBS and 60 μg/ml kanamycin sulfate. Molt-4 cells persistently infected with the HIV-1 IIIB strain were obtained from S. Harada (Kumamoto University, Kumamoto, Japan), and the cell culture supernatant of Molt-4/IIIB was used as an HIV-1 IIIB virus solution. HIV-1 strain NL432 was obtained from A. Adachi (Tokushima University, Tokushima, Japan) (23).
Construction of HIV-1 site-directed molecular clones.
The recombinant HIV-1 molecular clones were constructed as follows. The XbaI-EcoR I fragment from pNL-IN301 (pNL432 with the XbaI site inserted into the 5′ end of the IN region) was cloned at the XbaI-EcoR I site of cloning vector pUC18. In vitro mutagenesis was performed with the QuikChange site-directed mutagenesis kit (Stratagene, La Jolla, CA) using a pUC18 plasmid containing the IN-encoding region as a template. The amplified mutated XbaI-EcoR I fragment was ligated into pNL-IN301 to construct recombinant HIV-1 molecular clones. The plasmids were subsequently transfected into 293T cells by Lipofectamine 2000 (Life Technologies/Invitrogen, Carlsbad, CA) to generate infectious viruses. The supernatants were harvested 2 days after transfection and stored as cell-free viral stocks at −80°C.
Selection of drug-resistant viruses.
INSTI-resistant HIV-1 mutants were expanded in fresh MT-2 cells, and the viral titers were normalized with reverse transcriptase (RT) activity. For the initial infection, medium containing dilutions of the test compound was distributed into the plate. An MT-2 cell suspension (3 × 105 cells) was dispensed into each well of a 24-well tissue culture plate, and then MT-2 cells infected with INSTI-resistant HIV-1, prepared as described above, were added to each well. The passage study was performed in duplicate.
Every 3 or 4 days, the cells were passaged with or without the addition of fresh MT-2 cells. If a cytopathic effect (CPE) was observed, the culture medium, including the infected cells, was dispensed into a new plate with fresh MT-2 cell suspensions, and the concentration of the compound was held constant and/or increased 5-fold in medium containing the test compounds. If a CPE was not observed, the cells were cultivated without adding fresh MT-2 cells. When the replication of viruses was ascertained by an observed CPE, the infected cells were collected and used for genotypic and phenotypic analyses. We confirmed that all mutant viruses were able to replicate continuously with 6.4 nM DTG (data not shown) and thus selected initial drug concentrations of 6.4, 32, or 160 nM for use in this study. These concentrations are in good agreement with those used in our previous study.
Genotypic analyses of HIV-1 proviral DNA.
To analyze the amino acid substitutions, DNA was extracted from infected cells using a DNeasy blood and tissue kit (Qiagen, Valencia, CA), and the IN region of HIV-1 proviral DNA was amplified by PCR using a TaKaRa Taq DNA polymerase kit (TaKaRa, Shiga, Japan) and specific primers. The sequencing of the products was entrusted to the Operon Biotechnologies sequencing service (Tokyo, Japan). The sequence of the IN region derived from isolated viruses was compared with that of the wild-type NL432 IN region, and amino acid substitutions were identified.
Phenotypic analyses of viral isolates and drug-resistant molecular clones.
The drug sensitivities of the viruses isolated during the passage study and the drug-resistant molecular clones were assessed by a reporter assay with HeLa-CD4 cells, as described previously (24, 25). Briefly, viral isolates from the passage study were expanded in fresh M8166 cells. The test compounds were diluted to appropriate concentrations with culture medium, and HeLa-CD4 cell suspensions (2.5 × 104 cells/well) were dispensed into each plate. After 3 days of incubation, the cells were lysed, and the supernatant of each well was used for measuring luminescent activity using the Reporter assay kit-β-gal (Toyobo, Osaka, Japan). The luminescent activity (in relative luminescence units [RLU]) was measured using a MicroBeta TriLux instrument (PerkinElmer, Waltham, MA). The effective drug concentration reducing HIV-1 replication to 50% (EC50) was calculated.
Determination of relative infectivity of the integrase mutant molecular clones.
The relative infectivities of the IN mutant molecular clones were measured using HeLa-CD4 cells, as described previously (24, 25). Wild-type or mutant viruses were diluted serially with culture medium, and HeLa-CD4 cell suspensions (2.5 × 104 cells/well) were dispensed into each plate. After 3 days of incubation, virus-induced β-galactosidase activity was measured as described above. To evaluate the relative infectivity, virus-induced β-galactosidase activity was normalized by the RT activity of the input virus. The RT-normalized infectivity of the wild-type virus was defined as 100%, and the relative infectivity of each resistant virus was calculated in proportion to its induced β-galactosidase activity.
RESULTS
Resistance selection with dolutegravir and other INSTIs.
The in vitro passage experiments were assayed with DTG, RAL, or EVG in wild-type NL432 and RAL- or EVG-resistant HIV-1 molecular clones with the E92Q, Y143C, Y143R, Q148H, Q148K, Q148R or N155H substitution. The dose escalation curves for each resistance selection study are shown in Fig. 1, and the results of genotype assay at each time point are shown in Table 1.
FIG 1.



In vitro selection of INSTI-resistant HIV-1 mutants. HIV-1 NL432 wild type or mutants with the E92Q, N155H, Q148K, Q148R, Q148H, Y143C, or Y143R substitution were propagated in MT-2 cells in the presence of increasing concentrations of INSTIs. The highest concentrations of INSTIs at which the virus could replicate in cultured wells were plotted. The y axis shows the drug concentration (nM), and the x axis shows the days of culture. The genotypes of virus selected the first time at defined time points are indicated in the balloons. Detailed genotype analyses are shown in Table 1. (a) Resistant selection with DTG at initial concentrations of 160 nM (open circles), 32 nM (gray circles), and 6.4 nM (black circles). (b) Resistant selection with RAL at initial concentrations of 160 nM (white triangles), 32 nM (gray triangles), and 6.4 nM (black triangles). (c) Resistant selection with EVG at initial concentrations of 160 nM (white squares), 32 nM (gray squares), and 6.4 nM (black squares).
TABLE 1.
Resistance selection study with INSTIs from wild-type and RAL- or EVG-resistant signature mutants
| Initial virus | Drug | Initial concn (nM) | Change(s) at culture day: |
FC at day 56a | Final concn (nM) | |||
|---|---|---|---|---|---|---|---|---|
| 14 | 28 | 42 | 56 | |||||
| NL432 (wild type) | DTG | 6.4 | No substitution | No substitution | No substitution | E92Q | 3.1 | 6.4 |
| G193E | 3.2 | 6.4 | ||||||
| 32 | No replication | No replication | No replication | No replication | ||||
| 160 | No replication | No replication | No replication | No replication | ||||
| RAL | 6.4 | No substitution | No substitution | Q148R | N155S/D232N | ND | 32 | |
| Q148R | 300 | 800 | ||||||
| 32 | No substitution | N155H | F121Y | F121Y/D232N | 9.2 | 32 | ||
| N155H | N155H | 14 to 22 | 160 | |||||
| 160 | No replication | No replication | No replication | No replication | ||||
| EVG | 6.4 | No substitution | Q148R | Q148R | Q148R | 31 | 32 | |
| E138K/Q148R | ND | 160 | ||||||
| N155H | N155H | N155H | 1.4 to 18 | 32 | ||||
| E92Q/N155H | E92Q/N155H | 2,800 | 160 | |||||
| E92Q/N155H/R263K | ND | 800 | ||||||
| 32 | Q148R | Q148R | Q148R | Q148R | 250 | 160 | ||
| T66A | T66A | T66A | 14 to 100 | 160 | ||||
| 160 | No replication | No replication | No replication | No replication | ||||
| E92Q mutant | DTG | 6.4 | E92Q | E92Q | E92Q | E92Q | 2.9 to 4.1 | 6.4 |
| 32 | No replication | No replication | No replication | No replication | ||||
| 160 | No replication | No replication | No replication | No replication | ||||
| RAL | 6.4 | E92Q | E92Q | E92Q | E92Q | 5.7 to 13 | 32 | |
| L74M/E92Q | L74M/E92Q | 26 to 120 | 4,000 | |||||
| 32 | E92Q | E92Q | E92Q | E92Q | 4.8 to 12 | 32 | ||
| L74M/E92Q | L74M/E92Q | 21 to 110 | 4,000 | |||||
| 160 | E92Q | E92Q | E92Q | E92Q | 14 | 160 | ||
| L74M/E92Q | L74M/E92Q | 28 to 38 | 4,000 | |||||
| EVG | 6.4 | E92Q | E92Q | E92Q | E92Q | 3.2 to 82 | 160 | |
| 32 | E92Q | E92Q | E92Q | E92Q | 21 to 102 | 160 | ||
| 160 | E92Q | E92Q | E92Q | E92Q | 29 to 93 | 160 | ||
| N155H mutant | DTG | 6.4 | N155H | N155H | N155H | N155H | 2.0 to 3.9 | 32 |
| 32 | No replication | No replication | No replication | No replication | ||||
| 160 | No replication | No replication | No replication | No replication | ||||
| RAL | 6.4 | N155H | N155H | N155H | N155H | 22 to 79 | 160 | |
| N155H/D232N | G70R/N155H | ND | 800 | |||||
| N155H/G163R/D232N | 38 to 54 | 4,000 | ||||||
| 32 | N155H | N155H | N155H | N155H | 26 to 250 | 160 | ||
| S119R/N155H | S119R/N155H | 88 to 180 | 4,000 | |||||
| 160 | N155H | N155H | N155H | N155H | 25 to 52 | 4,000 | ||
| P142T/N155H/G163R | P142T/N155H/G163R | 270 | 800 | |||||
| EVG | 6.4 | N155H | N155H | N155H | N155H | 41 to 130 | 160 | |
| N155H/S230K | N155H/S230K | ND | 160 | |||||
| 32 | N155H | N155H | N155H | N155H | 38 to 41 | 160 | ||
| N155H/D232N | N155H/D232N | 77 | 800 | |||||
| 160 | N155H | N155H | N155H | N155H | ND | 800 | ||
| G70R/V75I/N155H | N155K/E170K | N155K/E170K | ND | 160 | ||||
| G70R/V75I/N155H | G70R/V75I/N155H | ND | 160 | |||||
| Y143C mutant | DTG | 6.4 | Y143C | No replication | No replication | No replication | ||
| 32 | No replication | No replication | No replication | No replication | ||||
| 160 | No replication | No replication | No replication | No replication | ||||
| RAL | 6.4 | Y143C | Y143R | Y143R | Y143R | 11 to 28 | 800 | |
| Y143R/G163R | 21 | |||||||
| 32 | Y143C | Y143R | Y143R | Y143R | 16 to 51 | 4,000 | ||
| E92Q/Y143R | 45 | |||||||
| 160 | Y143C | Y143C | G163R | G163R/E170A | ND | 160 | ||
| Y143R mutant | DTG | 6.4 | Y143R | Y143R | Y143R | Y143R | 1.7 | 6.4 |
| 32 | No replication | No replication | No replication | No replication | ||||
| 160 | No replication | No replication | No replication | No replication | ||||
| RAL | 6.4 | Y143R | Y143R | Y143R | Y143R | ND | 800 | |
| 32 | Y143R | Y143R | Y143R | Y143R | ND | 800 | ||
| 160 | Y143R | Y143R | Y143R | L74M/Y143R | 78 | 4,000 | ||
| Y143R/N155H | >1,400 | |||||||
| Q148K mutant | DTG | 6.4 | E138K/Q148K | E138K/Q148K | E138K/Q148K | E138K/Q148K | 47 to 190 | 32 |
| 32 | No replication | No replication | No replication | No replication | ||||
| 160 | No replication | No replication | No replication | No replication | ||||
| RAL | 6.4 | Q148K | Q148K | Q148K | Q148K | ND | 160 | |
| E138K/Q148K | E138K/Q148K | ND | 4,000 | |||||
| 32 | Q148K | Q148K | Q148K | Q148K | >510 | 160 | ||
| E138K/Q148K | E138K/Q148K | E138K/Q148K | ND | 800 | ||||
| 160 | Q148K | Q148K | Q148K | Q148K | ND | 160 | ||
| E138K | E138K/Q148K | E138K/Q148K | >510 | 800 | ||||
| E138K/Q148K | E138K/Q148K | E138K/Q148K | >510 | 4,000 | ||||
| EVG | 6.4 | Q148K | Q148K | Q148K | Q148K | 950 to 2,500 | 32 | |
| E138K/Q148K | E138K/Q148K | 4,900 to >5,400 | 4,000 | |||||
| 32 | Q148K | Q148K | Q148K | Q148K | 1,500 to 2,500 | 32 | ||
| E138K/Q148K | E138K/Q148K | E138K/Q148K | 2,800 to 4,900 | 4,000 | ||||
| 160 | Q148K | E138K/Q148K | E138K/Q148K | E138K/Q148K | 3,100 to >5,400 | 800 | ||
| Q148R mutant | DTG | 6.4 | Q148R | G140S/Q148R | G140S/Q148R | G140S/Q148R | 16 | 6.4 |
| G140S/Q148R/V201I | 39 | 32 | ||||||
| E138K/Q148R | E138K/Q148R | E138K/Q148R | E138K/G140S/Q148R | 13 | 6.4 | |||
| 32 | No replication | No replication | No replication | No replication | ||||
| 160 | No replication | No replication | No replication | No replication | ||||
| RAL | 6.4 | Q148R | Q148R | Q148R | Q148R | 84 to 190 | 160 | |
| G140S/Q148R | >510 | 800 | ||||||
| G140S/Q148R/V259I | ND | 4,000 | ||||||
| 32 | Q148R | Q148R | Q148R | Q148R | 190 to 250 | 160 | ||
| G140S/Q148R | G140S/Q148R | ND | 4,000 | |||||
| 160 | Q148R | Q148R | Q148R | Q148R | 220 to 370 | 160 | ||
| G140S/Q148R | G140S/Q148R | G140S/Q148R | ND | 4,000 | ||||
| L74M/Q148R | L74M/G140S/Q148R | L74M/G140S/Q148R | ND | 4,000 | ||||
| EVG | 6.4 | Q148R | Q148R | Q148R | Q148R | 170 to 710 | 160 | |
| E138K/Q148R | 720 to 1,500 | 800 | ||||||
| 32 | Q148R | Q148R | Q148R | Q148R | 470 | 32 | ||
| E138K/Q148R | 670 | 160 | ||||||
| 160 | Q148R | Q148R | Q148R | E138K/Q148R | 1,100 to 1,200 | 4,000 | ||
| Q148H mutant | DTG | 6.4 | G140S/Q148H | G140S/Q148H | G140S/Q148H | G140S/Q148H | 4.8 to 8.0 | 6.4 |
| T97A/G140S/Q148H | 44 | 32 | ||||||
| V75I/E138K/G140S/Q148H/M154I | 46 | 32 | ||||||
| 32 | No replication | No replication | No replication | No replication | ||||
| 160 | No replication | No replication | No replication | No replication | ||||
| RAL | 6.4 | Q148H | Q148H | Q148H | G140S/Q148H | >510 | 4,000 | |
| G140S/Q148H | G140S/Q148H | |||||||
| 32 | Q148H | Q148H | G140S/Q148H | G140S/Q148H | >510 | 4,000 | ||
| G140S/Q148H | ||||||||
| 160 | Q148H | G140S/Q148H | G140S/Q148H | G140S/Q148H | >510 | 4,000 | ||
| G140S/Q148H | ||||||||
| EVG | 6.4 | Q148H | Q148H | G140S/Q148H | G140S/Q148H | 520 to >5,400 | 4,000 | |
| 32 | G140S/Q148H | G140S/Q148H | G140S/Q148H | G140S/Q148H | 2800 to >5,400 | 4,000 | ||
| 160 | G140S/Q148H | G140S/Q148H | G140S/Q148H | G140S/Q148H | 4,000 to >5,400 | 4,000 | ||
Data from a single experiment, which was performed in duplicate. ND, not determined, because the low viral titer of the culture supernatant did not provide enough signal to evaluate the EC50.
The resistance selection studies of each drug were started with concentrations of 6.4, 32, or 160 nM each, and the compound concentrations were increased in a stepwise manner. Table 1 and Fig. 1 show the viral replication for each drug concentration. Viral replication was detected at all concentrations of RAL and EVG and in every strain, except for wild-type NL432 in both drugs at a concentration of 160 nM. The concentrations of RAL or EVG were able to be increased and emerging IN-resistant mutants allowed to replicate at up to 800 or 4,000 nM, respectively. In contrast, no viral replication was detected with an initial exposure to 32 nM or 160 nM DTG in the wild-type NL432 virus and all mutant viruses. When initially exposed to 6.4 nM DTG, the viruses were able to replicate, but if the DTG concentration was increased during the passage at or beyond 160 nM, complete viral suppression occurred.
Genotypic analyses of wild-type NL432 exposed to RAL showed that the N155H substitution was observed on day 28, the F121Y and Q148R substitutions were observed on day 42, and the F121Y/D232N and N155S/D232N double substitutions were observed on day 56. When wild-type viruses were exposed to EVG, the Q148R substitution was observed on day 14, the T66A and N155H substitutions were observed on day 28, the E92Q/N155H double substitution was observed on day 42, and the E138K/Q148R and E92Q/N155H/R263K substitutions were observed at day 56. When the wild-type viruses were exposed to DTG, the E92Q and G193E substitutions were observed on day 56.
Genotypic analyses of mutations with the E92Q substitution showed that the only changes detected were the combined secondary substitutions L74M/E92Q on day 42 of exposure to RAL; no secondary substitution was detected with EVG or DTG.
Genotypic analyses of the N155H mutant exposed to RAL detected secondary substitutions S119R/N155H, N155H/D232N, and P142T/N155H/G163R observed on day 42 and G70R/N155H and N155H/G163R/D232N on day 56. When exposed to EVG, G70R/V75I/N155H was observed on day 28, and N155H/S230K, N155H/D232N, and N155H/E170K were observed on day 42. No additional substitution was detected when the N155H mutants were exposed to DTG.
With Y143C mutants exposed to RAL starting at a concentration of 6.4 or 32 nM, Y143C was replaced by Y143R on day 28 and persisted throughout the remaining passages. Additionally, the secondary substitutions E92Q/Y143R and Y143R/G163R were observed on day 56. Starting from 160 nM RAL, revertant Y143 virus harboring G163R from the Y143C initial mutant virus was also detected on day 42, and the additional G163R/E170A substitutions were observed on day 56 during passage with RAL starting at a concentration of 160 nM. In the case of DTG, no additional substitutions were detected, and no viral replication was observed after day 28 with all concentrations of DTG.
With Y143R mutants exposed to RAL, L74M/Y143R and Y143R/N155H were detected on day 56, whereas no additional substitution was detected with DTG.
With the strains carrying the Q148K substitution that were exposed to RAL, EVG, or DTG, E138K/Q148K was observed on day 14 or 28 and persisted throughout the remaining passages.
With Q148R exposed to RAL, L74M/Q148R and G140S/Q148R were observed on day 28, L74M/G140S/Q148R was observed on day 42, and G140S/Q148R/V259I was observed on day 56. With EVG, E138K/Q148R was observed on day 56. With DTG, E138K/Q148R and G140S/Q148R were observed on days 14 and 28, respectively, and E138K/G140S/Q148R and G140S/Q148R/V201I were observed on day 56.
With the strains carrying the Q148H substitution that were exposed to RAL or EVG, G140S/Q148H was observed on day 14 and persisted throughout the remaining passages. In the case of DTG, G140S/Q148H was observed on day 14, and T97A/G140S/Q148H and V75I/E138K/G140S/Q148H/M154I were observed on day 56.
Sensitivities of site-directed molecular clones identified in DTG resistance selection study to INSTIs.
The in vitro selection of DTG resistance in wild-type or Q148H, Q148K, or Q148R mutants resulted in emerging amino acid substitutions in the IN region. Several INSTI-resistant SDMs were constructed and evaluated for their susceptibilities to DTG, RAL, and EVG, and the fold change in EC50 (FC) was compared (Table 2), using EFV as a reference compound. From wild-type NL432, E92Q and G193E were isolated in 6.4 nM DTG but did not reduce the antiviral activity of DTG. In the Q148H, Q148K, or Q148R mutants, RAL and EVG had reduced activities against all mutants with the secondary mutations identified in this study (FC, >130). The mutants with the G140S/Q148R, E138K/G140S/Q148R, E138K/G140S/Q148H, and E138K/G140S/Q148H/M154I substitutions showed moderate resistance to DTG (FCs, 8.4, 8.3, 4.5, and 8.4, respectively), and the mutants with the E138K/Q148K, G140S/Q148R/V201I, T97A/G140S/Q148H, and V75I/E138K/G140S/Q148H/M154I substitutions showed high resistance to DTG (FCs, 19, 10, 13, and 21, respectively).
TABLE 2.
Fold changes in EC50 against molecular clones with IN substitutions identified in resistance selection study with DTG
| Virus substitution(s) | Mean fold change in EC50a: |
|||
|---|---|---|---|---|
| Dolutegravir | Raltegravir | Elvitegravir | Efavirenz | |
| NL432 (wild type) | 1.0 | 1.0 | 1.0 | 1.0 |
| E92Q | 1.6 (0.12) | 3.5 (1.4) | 19 (8.1) | 1.2 (0.22) |
| G193E | 1.3 (0.25) | 1.3 (0.35) | 1.3 (0.40) | 1.1 (0.032) |
| Q148K | 1.1 (0.19) | 83 (6.6) | >1,700 | 2.1 (0.26) |
| E138K/Q148K | 19 (8.0) | 330 (75) | 370b | 1.2b |
| Q148R | 1.2 (0.21) | 47 (9.3) | 240 (91) | 1.9 (0.21) |
| E138K/Q148R | 4.0 (1.1) | 110 (37) | 460 (230) | 1.0 (0.31) |
| G140S/Q148R | 8.4 (4.0) | 200 (5.3) | 270b | 1.5b |
| E138K/G140S/Q148R | 8.3 (1.1) | >660 | 190 (43) | 1.0 (0.10) |
| G140S/Q148R/V201I | 10 (3.0) | >660 | 420 (80) | 1.9 (0.28) |
| Q148H | 0.97 (0.67) | 13 (5.0) | 7.3 (2.3) | 1.4 (0.83) |
| G140S/Q148H | 2.6 (1.4) | >130 | >890 | 1.7 (0.99) |
| T97A/G140S/Q148H | 13 (2.1) | >660 | 3,900 (630) | 1.3 (0.046) |
| E138K/G140S/Q148H | 4.5 (1.1) | 500 (210) | 1,600 (370) | 0.92 (0.026) |
| E138K/G140S/Q148H/M154I | 8.4 (1.5) | >660 | 2,400 (670) | 0.87 (0.099) |
| V75I/E138K/G140S/Q148H/M154I | 21 (6.6) | >660 | 2,600 (450) | 1.2 (0.16) |
Each value represents the mean FC. The standard deviations from 3 to 5 independent experiments are shown in parentheses. Each data point was calculated from duplicate experiments. FC values of >10 are shown in bold type.
Data from one or two experiments.
Effect on viral infectivity of integrase mutants.
The relative infectivities of the mutant viruses in the absence of INSTIs were also measured using the single-cycle reporter assay with HeLa-CD4 cells (Fig. 2). Q148K reduced infectivity to 11%, although adding the E138K substitution partially restored its infectivity to 36% (Fig. 2a). Similarly, the relative infectivity of the Q148R mutant virus was reduced to 19%, whereas the subsequent addition of the E138K substitution partially restored its infectivity to 40% (Fig. 2a). Also, the relative infectivity of the Q148H mutant was reduced to 27%, whereas the subsequent addition of the G140S substitution partially restored its infectivity to 72%. However, the differences in the viral infectivity of Q148R versus those of the mutants with the G140S/Q148R, E138K/G140S/Q148R, or G140S/Q148R/V201I substitutions were not statistically significant. On the contrary, the relative infectivity of the mutant with the V75I/E138K/G140S/Q148H/M154I substitutions (49%) was reduced from that of the mutant with the E138K/G140S/Q148H/M154I substitutions (76%) (Fig. 2b). Therefore, further additional substitutions to G140S/Q148H, which were introduced only while selecting for DTG resistance, did not restore viral infectivity but exhibited higher INSTI resistance.
FIG 2.
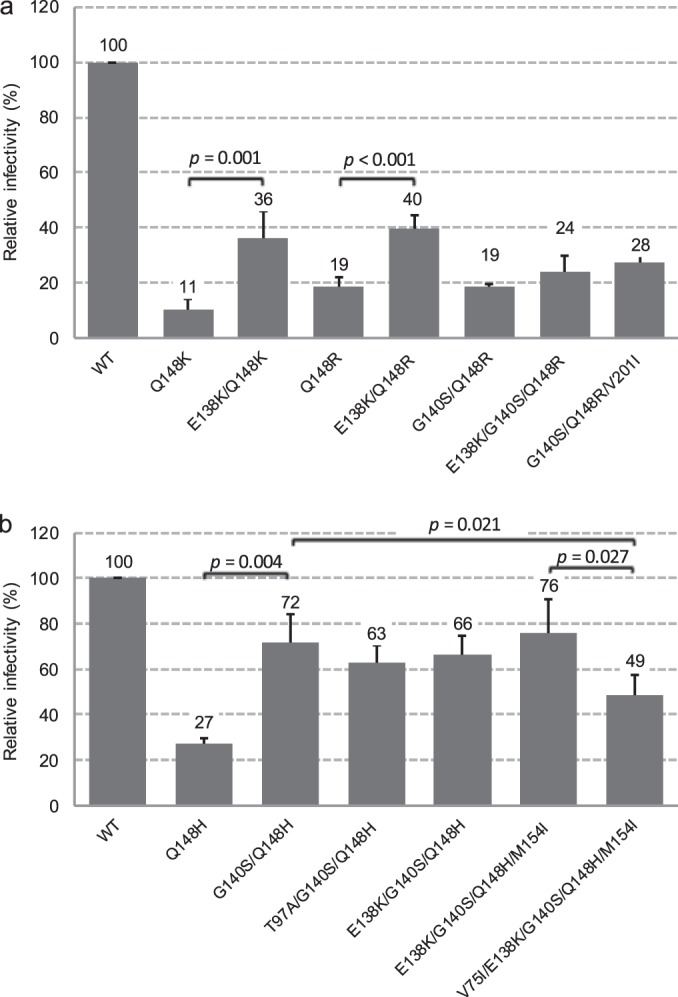
Relative infectivities in HeLa-CD4 assay of mutant viruses observed in this study. Each bar represents the mean from three to five independent experiments. The error bars represent standard deviations. Student's t test was used for statistical analysis, and the P values are shown.
DISCUSSION
DTG has low-nanomolar EC50s against both wild-type HIV-1 and many resistant mutants to previously approved INSTIs in vitro. A difficulty in generating variants with high-level phenotypic resistance to DTG from the wild-type virus was reported (20) and suggests that DTG has a high barrier to resistance against wild-type virus in vitro.
In mutant HIV-1 with the RAL and EVG resistance mutations encoded by the E92Q, Y143C, Y143R, and N155H substitutions, viral replication was not observed in medium containing ≥32 nM DTG. In the Y143C mutants, viral replication was not detected for 28 days, even at an exposure of 6.4 nM DTG. However, in the Q148H, Q148K, or Q148R mutant viruses, secondary substitutions were observed when the initial concentration was 6.4 nM DTG. In particular, the emergence of E138K during passage with the Q148K and Q148R mutant viruses and G140S for the Q148R mutant was associated with the partial restoration of viral infectivity and/or an increase in resistance to INSTIs (Fig. 2a and Table 2). We previously reported that partial restorations of infectivity of these substitutions were associated with viral replication capacity and the partial restoration of viral DNA integration efficiency, measured by replication kinetics in PBMC or Jurkat cells and by the amount of integrated viral DNA (25). However, the emergence of the E138K or V201I substitution when G140S/Q148R was present had no impact on viral infectivity and the FC values against DTG. Although the explanation for the propagation of the additional substitution was unclear, a slight advantage in viral replication in cell culture is one possibility. When the Q148H mutant was exposed to DTG, the additional emergence of G140S was observed, followed by the addition of T97A or V75I/E138K/M154I. The emergence of G140S in the Q148H mutant provided a partial restoration of viral infectivity (Fig. 2b) and also increased resistance to RAL and EVG but not to DTG (Table 2). The addition of the T97A substitution to G140S and Q148H provided no impact on viral infectivity and was selected solely on the basis of increased resistance to DTG. The addition of the V75I, E138K, and M154I substitutions was associated with an 8-fold increase in DTG resistance instead of further reduced infectivity versus that of G140S/Q148H. The FC value of the G140S/Q148R substitutions with EVG was similar to that of Q148R. In contrast, the FC value of the E138K/Q148R substitutions with EVG was higher than that of the G140S/Q148R substitutions, and partially restored infectivity was also observed (Fig. 2a and Table 2); this suggests that the E138K/Q148R substitutions apparently provide a greater advantage than do the G140S/Q148R substitutions to viral replication in the presence of EVG. The drug resistance of the G140S/Q148H substitutions to RAL or EVG was high, and the addition of a third mutation may not have been required for higher-level replication. These data indicate that the effect of secondary substitutions on restoring viral infectivity and/or increasing resistance varies with signature mutations and INSTI.
In this resistance selection study, the lowest starting dose of 6.4 nM DTG was less than the EC90 (8.4 nM) but higher than the EC50 (2.1 nM) to wild-type NL432 in MT-2 cells under our standard conditions (Shionogi Research Laboratories, unpublished data). It is important to note that the in vitro resistance selection condition was optimized, in that resistant mutants were able to replicate continuously, while under those conditions, wild-type virus or inoculated mutants need to be replicated to some extent, because a genetically mutated virus at the reverse transcription step is not yet phenotypically drug resistant. Eventually, the newly replicated resistant virus is phenotypically drug resistant and will overcome the wild-type virus under drug pressure. Nevertheless, the addition of secondary substitutions to mutants with the E92Q, Y143C, Y143R, or N155H substitution was not observed in DTG-containing culture. This suggests that the secondary substitutions added to these primary mutations would be unable to increase the FC to DTG sufficiently to allow replication with ≥6.4 nM DTG under the NL432/MT-2 selection conditions. This is consistent with our previous data regarding DTG FCs to multiple SDMs (20). Although susceptible to DTG, secondary substitutions were observed in the Q148H, Q148K, or Q148R mutants. In the present study, drug concentrations were increased stepwise by 5-fold when a CPE indicating viral replication was observed. As shown in Table 1 and Fig. 1a, the E138K/Q148R, G140S/Q148R, and G140S/Q148H secondary substitutions emerged at 6.4 nM DTG, which is 3-fold higher than the EC50; the FCs of these mutants were 4.0, 8.4, and 2.6, respectively. Therefore, these mutants should have replicated at 6.4 nM DTG, and then double or multiple substitutions should have emerged subsequently. However, Q148H, Q148K, or Q148R mutants exposed to 32 nM DTG (approximately 15 times higher than the EC50 and 4 times higher than the EC90) resulted in the complete suppression of viral replication. The FC value of the E138K/Q148K mutant was 19 but displayed reduced replication in ≥32 nM DTG. In contrast, single mutations that emerged upon exposure to RAL or EVG were associated with high resistance.
We included in vitro selection studies with the wild-type HIV-1 NL432 strain with DTG, and amino acid substitutions in the IN region, E92Q and G193E, were observed. Although the FC values did not significantly change and viral replication was inhibited by 32 nM DTG (Table 2), we supposed that viruses containing the E92Q or G193E substitution had still a little more advantage over the wild-type under drug selection pressure. E92Q is a well-established EVG resistance substitution (26, 27) in vitro and in clinical trials, and the FC value against EVG was 19 (20). However, the antiviral responses of DTG and RAL are similar (FCs, 1.6 and 3.5, respectively). G193E is a polymorphic substitution in the IN region that occurs in 2.3% of the 2,997 IN sequences available prior to 2005 (Los Alamos Database and Stanford Resistance Database), before RAL had progressed to clinical trials (28). In vitro selection studies with HIV-1 clinical isolates of subtype B, C, or A/G and DTG were reported previously, and amino acid substitutions in the IN region, G118R and R263K, were observed, which confer low-level resistance to INSTIs and are deficient in IN strand transfer activity (29, 30). We detected the emergence of R263K from the E92Q/N155H mutant during EVG resistance selection. However, G118R and R263K had not been detected during DTG exposure. One possible explanation might involve the differences in passage conditions or in the genetic backgrounds of the laboratory strains and clinical isolates. Importantly, the FC values of DTG against all mutants isolated in in vitro selection studies of wild-type HIV-1 were not significantly changed, and the selection studies did not lead to the emergence of highly resistant viruses with multiple substitutions.
On the basis of this and previous studies, DTG has been found to have a high barrier to the development of resistance to wild-type HIV-1 and to INSTI-resistant mutants with a single substitution, with the exception of Q148H, Q148K, or Q148R. DTG was designed to maintain activity against many INSTI-resistant mutants with single or multiple substitutions (20, 31–35), and this is consistent with biochemical and structural observations (36, 37). Studies showed that the dissociation half-life of DTG from IN/substrate–DNA/INSTI triple complex in the presence of the amino acid substitutions Q148H, Q148K, or Q148R and N155H significantly reduced the dissociation half-life from that of wild-type IN but was still similar to that of RAL to wild-type IN. This observation may explain why there is no significant increase in the FC observed with DTG to these mutant viruses. The INSTI-resistant double mutants had a further reduced dissociation half-life to DTG, but it was still longer than the dissociation half-lives of RAL and EVG to mutants with a single amino acid substitution in the IN (37). In vitro virology studies and the biochemical study suggest that the binding of DTG to the IN-substrate-DNA complex is less affected by the IN active site surrounding amino acid substitutions than by RAL and EVG. The HIV-1 IN/substrate–DNA/INSTI docking model suggested that the interaction of DTG with the 3′-terminal two nucleotides of substrate DNA and strong binding ability to the metal ions, along with the streamlined DTG architecture, might be a likely explanation for the distinct resistance profile of DTG (36).
Our data are consistent in various aspects with the results from the VIKING-3 clinical trial (10) regarding treatment-experienced virologic failure with either RAL or EVG. The DTG response rate can be categorized into three types with the baseline IN genotypes. The first category was non-Q148 mutants, which comprised N155H without a Q148 substitution, having an 88% response, and Y143C, Y143H, or Y143R without a Q148 substitution, having a 75% response. The second category was Q148H or Q148R plus 1 secondary INSTI resistance substitution, with a 59% response, and the third category was Q148H or Q148R plus ≥2 INSTI resistance substitutions, with a 24% response. In our in vitro selection study starting from RAL- or EVG-resistant signature mutants, the DTG resistance isolation pattern was clearly different between the E92Q, N155H, Y143C, or Y143R group, and Q148H, Q148K, or Q148R mutants. No addition of secondary substitutions to the E92Q, N155H, Y143C, or Y143R group in vitro is consistent with the high response rate and low virologic failure with additional mutations in the VIKING-3 subjects with the baseline IN genotype of this group.
In VIKING-3, most of the subjects who failed RAL treatment and had the Q148 substitution already had a G140 or E138 substitution at baseline. This suggests that once Q148 substitutions are introduced, secondary substitutions can be added quickly. In our in vitro studies, if the start was from wild-type virus, non-Q148 mutants were isolated from DTG, but if the start was from Q148H, Q148K, or Q148R, the addition of a secondary substitution occurred within 14 days.
All of the mutations in patients with protocol-defined virologic failure in the VIKING studies were detected in this study or had been identified as having common secondary substitutions (i.e., T97A or E138K), and no new secondary INSTI-resistant substitutions emerged (18). This study suggests that additional substitutions leading to virologic failure would be unlikely to emerge in patients infected with HIV-1 containing the E92Q, Y143C, or N155H substitution. However, additional substitutions previously observed during RAL or EVG therapy might emerge from the Q148H, Q148K, or Q148R background.
In conclusion, the lack of selection under DTG in the presence of RAL or EVG signature substitutions, except Q148 substitutions, is consistent with a high barrier to resistance in these in vitro studies. This finding is supported by the data showing that the secondary substitutions into the E92Q, Y143C, Y143R, or N155H substitution were not associated with a large enough increase in the FC to DTG. Increased resistance to DTG in the Q148 pathway can occur via the E138K or G140S secondary substitution, with subsequent T97A or V75I substitutions. DTG might completely suppress the replication of those mutant viruses with a starting concentration of >32 nM in vitro.
ACKNOWLEDGMENTS
We recognize that the entire Shionogi-GSK HIV-1 integrase drug discovery team has contributed to the discovery and preclinical development of dolutegravir (DTG, S/GSK1349572, or GSK1349572).
REFERENCES
- 1.UNAIDS. 2013. Joint United Nations Programme on HIV/AIDS (UNAIDS) report on the global AIDS epidemic 2013. UNAIDS, Geneva, Switzerland: http://www.unaids.org/sites/default/files/en/media/unaids/contentassets/documents/epidemiology/2013/gr2013/UNAIDS_Global_Report_2013_en.pdf. [Google Scholar]
- 2.Fernández-Montero JV, Vispo E, Soriano V. 2014. Emerging antiretroviral drugs. Expert Opin Pharmacother 15:211–219. doi: 10.1517/14656566.2014.863277. [DOI] [PubMed] [Google Scholar]
- 3.DeJesus E, Berger D, Markowitz M, Cohen C, Hawkins T, Ruane P, Elion R, Farthing C, Zhong L, Cheng AK, McColl D, Kearney BP, 183-0101 Study Team. 2006. Antiviral activity, pharmacokinetics, and dose response of the HIV-1 integrase inhibitor GS-9137 (JTK-303) in treatment-naive and treatment-experienced patients. J Acquir Immune Defic Syndr 43:1–5. doi: 10.1097/01.qai.0000233308.82860.2f. [DOI] [PubMed] [Google Scholar]
- 4.DeJesus E, Rockstroh JK, Henry K, Molina JM, Gathe J, Ramanathan S, Wei X, Yale K, Szwarcberg J, White K, Cheng AK, Kearney BP, GS-236-0103 Study Team. 2012. Co-formulated elvitegravir, cobicistat, emtricitabine, and tenofovir disoproxil fumarate versus ritonavir-boosted atazanavir plus co-formulated emtricitabine and tenofovir disoproxil fumarate for initial treatment of HIV-1 infection: a randomised, double-blind, phase 3, non-inferiority trial. Lancet 379:2429–2438. doi: 10.1016/S0140-6736(12)60918-0. [DOI] [PubMed] [Google Scholar]
- 5.Markowitz M, Morales-Ramirez JO, Nguyen BY, Kovacs CM, Steigbigel RT, Cooper DA, Liporace R, Schwartz R, Isaacs R, Gilde LR, Wenning L, Zhao J, Teppler H. 2006. Antiretroviral activity, pharmacokinetics, and tolerability of MK-0518, a novel inhibitor of HIV-1 integrase, dosed as monotherapy for 10 days in treatment-naive HIV-1-infected individuals. J Acquir Immune Defic Syndr 43:509–515. doi: 10.1097/QAI.0b013e31802b4956. [DOI] [PubMed] [Google Scholar]
- 6.Markowitz M, Nguyen BY, Gotuzzo E, Mendo F, Ratanasuwan W, Kovacs C, Prada G, Morales-Ramirez JO, Crumpacker CS, Isaacs RD, Gilde LR, Wan H, Miller MD, Wenning LA, Teppler H, Protocol 004 Part II Study Team. 2007. Rapid and durable antiretroviral effect of the HIV-1 integrase inhibitor raltegravir as part of combination therapy in treatment-naive patients with HIV-1 infection: results of a 48-week controlled study. J Acquir Immune Defic Syndr 46:125–133. doi: 10.1097/QAI.0b013e318157131c. [DOI] [PubMed] [Google Scholar]
- 7.Blanco JL, Varghese V, Rhee SY, Gatell JM, Shafer RW. 2011. HIV-1 integrase inhibitor resistance and its clinical implications. J Infect Dis 203:1204–1214. doi: 10.1093/infdis/jir025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Garrido C, Villacian J, Zahonero N, Pattery T, Garcia F, Gutierrez F, Caballero E, Van Houtte M, Soriano V, de Mendoza C, SINRES Group. 2012. Broad phenotypic cross-resistance to elvitegravir in HIV-infected patients failing on raltegravir-containing regimens. Antimicrob Agents Chemother 56:2873–2878. doi: 10.1128/AAC.06170-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cahn P, Pozniak AL, Mingrone H, Shuldyakov A, Brites C, Andrade-Villanueva JF, Richmond G, Buendia CB, Fourie J, Ramgopal M, Hagins D, Felizarta F, Madruga J, Reuter T, Newman T, Small CB, Lombaard J, Grinsztejn B, Dorey D, Underwood M, Griffith S, Min S, Extended SAILING Study Team. 2013. Dolutegravir versus raltegravir in antiretroviral-experienced, integrase-inhibitor-naive adults with HIV: week 48 results from the randomised, double-blind, non-inferiority SAILING study. Lancet 382:700–708. doi: 10.1016/S0140-6736(13)61221-0. [DOI] [PubMed] [Google Scholar]
- 10.Castagna A, Maggiolo F, Penco G, Wright D, Mills A, Grossberg R, Molina JM, Chas J, Durant J, Moreno S, Doroana M, Ait-Khaled M, Huang J, Min S, Song I, Vavro C, Nichols G, Yeo JM, VIKING-3 Study Group. 2014. Dolutegravir in antiretroviral-experienced patients with raltegravir- and/or elvitegravir-resistant HIV-1: 24-week results of the phase III VIKING-3 study. J Infect Dis 210:354–362. doi: 10.1093/infdis/jiu051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Clotet B, Feinberg J, van Lunzen J, Khuong-Josses MA, Antinori A, Dumitru I, Pokrovskiy V, Fehr J, Ortiz R, Saag M, Harris J, Brennan C, Fujiwara T, Min S, ING114915 Study Team. 2014. Once-daily dolutegravir versus darunavir plus ritonavir in antiretroviral-naive adults with HIV-1 infection (FLAMINGO): 48 week results from the randomised open-label phase 3b study. Lancet 383:2222–2231. doi: 10.1016/S0140-6736(14)60084-2. [DOI] [PubMed] [Google Scholar]
- 12.Raffi F, Jaeger H, Quiros-Roldan E, Albrecht H, Belonosova E, Gatell JM, Baril JG, Domingo P, Brennan C, Almond S, Min S, Extended SPRING-2 Study Group. 2013. Once-daily dolutegravir versus twice-daily raltegravir in antiretroviral-naive adults with HIV-1 infection (SPRING-2 study): 96 week results from a randomised, double-blind, non-inferiority trial. Lancet Infect Dis 13:927–935. doi: 10.1016/S1473-3099(13)70257-3. [DOI] [PubMed] [Google Scholar]
- 13.Walmsley SL, Antela A, Clumeck N, Duiculescu D, Eberhard A, Gutierrez F, Hocqueloux L, Maggiolo F, Sandkovsky U, Granier C, Pappa K, Wynne B, Min S, Nichols G, SINGLE Investigators. 2013. Dolutegravir plus abacavir-lamivudine for the treatment of HIV-1 infection. N Engl J Med 369:1807–1818. doi: 10.1056/NEJMoa1215541. [DOI] [PubMed] [Google Scholar]
- 14.Arribas JR, Eron J. 2013. Advances in antiretroviral therapy. Curr Opin HIV AIDS 8:341–349. doi: 10.1097/COH.0b013e328361fabd. [DOI] [PubMed] [Google Scholar]
- 15.U.S. Department of Health & Human Services. 2013. Recommendation on integrase inhibitor use in antiretroviral treatment-naive HIV-infected individuals from the HHS Panel on Antiretroviral Guidelines for Adults and Adolescents. AIDSinfo, U.S. Department of Health and Human Services, Rockville, MD: http://aidsinfo.nih.gov/contentfiles/AdultARV_INSTIRecommendations.pdf. [Google Scholar]
- 16.Geretti AM, Armenia D, Ceccherini-Silberstein F. 2012. Emerging patterns and implications of HIV-1 integrase inhibitor resistance. Curr Opin Infect Dis 25:677–686. doi: 10.1097/QCO.0b013e32835a1de7. [DOI] [PubMed] [Google Scholar]
- 17.Wensing AM, Calvez V, Gunthard HF, Johnson VA, Paredes R, Pillay D, Shafer RW, Richman DD. 2014. 2014 Update of the drug resistance mutations in HIV-1. Top Antivir Med 22:642–650. [PMC free article] [PubMed] [Google Scholar]
- 18.Eron JJ, Clotet B, Durant J, Katlama C, Kumar P, Lazzarin A, Poizot-Martin I, Richmond G, Soriano V, Ait-Khaled M, Fujiwara T, Huang J, Min S, Vavro C, Yeo J, VIKING Study Group. 2013. Safety and efficacy of dolutegravir in treatment-experienced subjects with raltegravir-resistant HIV type 1 infection: 24-week results of the VIKING Study. J Infect Dis 207:740–748. doi: 10.1093/infdis/jis750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Eron JJ, Kumar P, Lazzarin A, Richmond G, Soriano V, Huang J, Vavro C, Ait-Khaled M, Min S, Yeo J. 2011. DTG in subjects with HIV exhibiting RAL resistance: functional monotherapy results of VIKING study cohort II, abstr 151LB. 18th Conf Retrovir Oppor Infect, 27 February to 2 March 2011, Boston, MA. [Google Scholar]
- 20.Kobayashi M, Yoshinaga T, Seki T, Wakasa-Morimoto C, Brown KW, Ferris R, Foster SA, Hazen RJ, Miki S, Suyama-Kagitani A, Kawauchi-Miki S, Taishi T, Kawasuji T, Johns BA, Underwood MR, Garvey EP, Sato A, Fujiwara T. 2011. In vitro antiretroviral properties of S/GSK1349572, a next-generation HIV integrase inhibitor. Antimicrob Agents Chemother 55:813–821. doi: 10.1128/AAC.01209-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Isaka Y, Sato A, Miki S, Kawauchi S, Sakaida H, Hori T, Uchiyama T, Adachi A, Hayami M, Fujiwara T, Yoshie O. 1999. Small amino acid changes in the V3 loop of human immunodeficiency virus type 2 determines the coreceptor usage for CXCR4 and CCR5. Virology 264:237–243. doi: 10.1006/viro.1999.0006. [DOI] [PubMed] [Google Scholar]
- 22.Harada S, Koyanagi Y, Yamamoto N. 1985. Infection of HTLV-III/LAV in HTLV-I-carrying cells MT-2 and MT-4 and application in a plaque assay. Science 229:563–566. doi: 10.1126/science.2992081. [DOI] [PubMed] [Google Scholar]
- 23.Adachi A, Gendelman HE, Koenig S, Folks T, Willey R, Rabson A, Martin MA. 1986. Production of acquired immunodeficiency syndrome-associated retrovirus in human and nonhuman cells transfected with an infectious molecular clone. J Virol 59:284–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Garvey EP, Johns BA, Gartland MJ, Foster SA, Miller WH, Ferris RG, Hazen RJ, Underwood MR, Boros EE, Thompson JB, Weatherhead JG, Koble CS, Allen SH, Schaller LT, Sherrill RG, Yoshinaga T, Kobayashi M, Wakasa-Morimoto C, Miki S, Nakahara K, Noshi T, Sato A, Fujiwara T. 2008. The naphthyridinone GSK364735 is a novel, potent human immunodeficiency virus type 1 integrase inhibitor and antiretroviral. Antimicrob Agents Chemother 52:901–908. doi: 10.1128/AAC.01218-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nakahara K, Wakasa-Morimoto C, Kobayashi M, Miki S, Noshi T, Seki T, Kanamori-Koyama M, Kawauchi S, Suyama A, Fujishita T, Yoshinaga T, Garvey EP, Johns BA, Foster SA, Underwood MR, Sato A, Fujiwara T. 2009. Secondary mutations in viruses resistant to HIV-1 integrase inhibitors that restore viral infectivity and replication kinetics. Antiviral Res 81:141–146. doi: 10.1016/j.antiviral.2008.10.007. [DOI] [PubMed] [Google Scholar]
- 26.Abram ME, Hluhanich RM, Goodman DD, Andreatta KN, Margot NA, Ye L, Niedziela-Majka A, Barnes TL, Novikov N, Chen X, Svarovskaia ES, McColl DJ, White KL, Miller MD. 2013. Impact of primary elvitegravir resistance-associated mutations in HIV-1 integrase on drug susceptibility and viral replication fitness. Antimicrob Agents Chemother 57:2654–2663. doi: 10.1128/AAC.02568-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.McColl DJ, Chen X. 2010. Strand transfer inhibitors of HIV-1 integrase: bringing IN a new era of antiretroviral therapy. Antiviral Res 85:101–118. doi: 10.1016/j.antiviral.2009.11.004. [DOI] [PubMed] [Google Scholar]
- 28.Vavro C, Hasan S, Madsen H, Horton J, DeAnda F, Martin-Carpenter L, Sato A, Cuffe R, Chen S, Underwood M, Nichols G. 2013. Prevalent polymorphisms in wild-type HIV-1 integrase are unlikely to engender drug resistance to dolutegravir (S/GSK1349572). Antimicrob Agents Chemother 57:1379–1384. doi: 10.1128/AAC.01791-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Quashie PK, Mesplede T, Han YS, Oliveira M, Singhroy DN, Fujiwara T, Underwood MR, Wainberg MA. 2012. Characterization of the R263K mutation in HIV-1 integrase that confers low-level resistance to the second-generation integrase strand transfer inhibitor dolutegravir. J Virol 86:2696–2705. doi: 10.1128/JVI.06591-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Quashie PK, Mesplede T, Han YS, Veres T, Osman N, Hassounah S, Sloan RD, Xu HT, Wainberg MA. 2013. Biochemical analysis of the role of G118R-linked dolutegravir drug resistance substitutions in HIV-1 integrase. Antimicrob Agents Chemother 57:6223–6235. doi: 10.1128/AAC.01835-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Johns BA, Kawasuji T, Weatherhead JG, Taishi T, Temelkoff DP, Yoshida H, Akiyama T, Taoda Y, Murai H, Kiyama R, Fuji M, Tanimoto N, Jeffrey J, Foster SA, Yoshinaga T, Seki T, Kobayashi M, Sato A, Johnson MN, Garvey EP, Fujiwara T. 2013. Carbamoyl pyridone HIV-1 integrase inhibitors 3. A diastereomeric approach to chiral nonracemic tricyclic ring systems and the discovery of dolutegravir (S/GSK1349572) and (S/GSK1265744). J Med Chem 56:5901–5916. doi: 10.1021/jm400645w. [DOI] [PubMed] [Google Scholar]
- 32.Kawasuji T, Fuji M, Yoshinaga T, Sato A, Fujiwara T, Kiyama R. 2006. A platform for designing HIV integrase inhibitors. Part 2: a two-metal binding model as a potential mechanism of HIV integrase inhibitors. Bioorg Med Chem 14:8420–8429. doi: 10.1016/j.bmc.2006.08.043. [DOI] [PubMed] [Google Scholar]
- 33.Kawasuji T, Johns BA, Yoshida H, Taishi T, Taoda Y, Murai H, Kiyama R, Fuji M, Yoshinaga T, Seki T, Kobayashi M, Sato A, Fujiwara T. 2012. Carbamoyl pyridone HIV-1 integrase inhibitors. 1. Molecular design and establishment of an advanced two-metal binding pharmacophore. J Med Chem 55:8735–8744. doi: 10.1021/jm3010459. [DOI] [PubMed] [Google Scholar]
- 34.Kawasuji T, Johns BA, Yoshida H, Weatherhead JG, Akiyama T, Taishi T, Taoda Y, Mikamiyama-Iwata M, Murai H, Kiyama R, Fuji M, Tanimoto N, Yoshinaga T, Seki T, Kobayashi M, Sato A, Garvey EP, Fujiwara T. 2013. Carbamoyl pyridone HIV-1 integrase inhibitors. 2. Bi- and tricyclic derivatives result in superior antiviral and pharmacokinetic profiles. J Med Chem 56:1124–1135. doi: 10.1021/jm301550c. [DOI] [PubMed] [Google Scholar]
- 35.Kawasuji T, Yoshinaga T, Sato A, Yodo M, Fujiwara T, Kiyama R. 2006. A platform for designing HIV integrase inhibitors. Part 1: 2-hydroxy-3-heteroaryl acrylic acid derivatives as novel HIV integrase inhibitor and modeling of hydrophilic and hydrophobic pharmacophores. Bioorg Med Chem 14:8430–8445. doi: 10.1016/j.bmc.2006.08.044. [DOI] [PubMed] [Google Scholar]
- 36.Deanda F, Hightower KE, Nolte RT, Hattori K, Yoshinaga T, Kawasuji T, Underwood MR. 2013. Dolutegravir interactions with HIV-1 integrase-DNA: structural rationale for drug resistance and dissociation kinetics. PLoS One 8:e77448. doi: 10.1371/journal.pone.0077448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hightower KE, Wang R, Deanda F, Johns BA, Weaver K, Shen Y, Tomberlin GH, Carter HL III, Broderick T, Sigethy S, Seki T, Kobayashi M, Underwood MR. 2011. Dolutegravir (S/GSK1349572) exhibits significantly slower dissociation than raltegravir and elvitegravir from wild-type and integrase inhibitor-resistant HIV-1 integrase-DNA complexes. Antimicrob Agents Chemother 55:4552–4559. doi: 10.1128/AAC.00157-11. [DOI] [PMC free article] [PubMed] [Google Scholar]