

Abstract
18β-Glycyrrhetinic acid (GRA), a natural immunomodulator, greatly reduced the parasite load in experimental visceral leishmaniasis through nitric oxide (NO) upregulation, proinflammatory cytokine expression, and NF-κB activation. For the GRA-mediated effect, the primary kinase responsible was found to be p38, and analysis of phosphorylation kinetics as well as studies with dominant-negative (DN) constructs revealed mitogen-activated protein kinase kinase 3 (MKK3) and MKK6 as the immediate upstream regulators of p38. However, detection of remnant p38 kinase activity in the presence of both DN MKK3 and MKK6 suggested alternative pathways of p38 activation. That residual p38 activity was attributed to an autophosphorylation event ensured by the transforming growth factor β-activated kinase 1 (TAK1)-binding protein 1 (TAB1)-p38 interaction and was completely abolished upon pretreatment with SB203580 in DN MKK3/6 double-transfected macrophage cells. Further upstream signaling evaluation by way of phosphorylation kinetics and transfection studies with DN constructs identified TAK1, myeloid differentiation factor 88 (MyD88), interleukin 1 receptor (IL-1R)-activated kinase 1 (IRAK1), and tumor necrosis factor (TNF) receptor-associated factor 6 (TRAF6) as important contributors to GRA-mediated macrophage activation. Finally, gene knockdown studies revealed Toll-like receptor 2 (TLR2) and TLR4 as the membrane receptors associated with GRA-mediated antileishmanial activity. Together, the results of this study brought mechanistic insight into the antileishmanial activity of GRA, which is dependent on the TLR2/4-MyD88 signaling axis, leading to MKK3/6-mediated canonical and TAB1-mediated noncanonical p38 activation.
INTRODUCTION
The term leishmaniasis encompasses a spectrum of vector-borne protozoan parasitic diseases affecting 12 million people worldwide with 0.5 million new cases per annum. With no available vaccines for treatment of leishmaniasis, chemotherapy remains the major tool to combat infection (1). The primary treatment for leishmaniasis includes pentavalent antimonials, amphotericin B, pentamidine, miltefosine, and aminosidine, which often pose problems of high toxicity and several adverse effects. In addition, the combination of lengthy treatment schedules and the high cost of the compounds makes the treatment unsuitable in many cases (2). Evidence for a drug that specifically eliminates the infection and is safe enough to be used in the clinical field is still lacking. We have previously shown that 18β-glycyrrhetinic acid (GRA), a pentacyclic triterpene derivative of the β-amyrin type, extracted from the root of the medicinal plant licorice (Glycyrrhizza glabra L.), might completely cure experimental visceral leishmaniasis (3). The pharmacological properties of GRA include antitumorigenic (4), hepatoprotective (5), antiulcerative (6), and immunomodulatory effects (7). Apart from immunomodulation, GRA plays a role in shifting the cellular kinase/phosphatase balance toward kinases during infection (8). This host-favorable action of GRA was achieved by downregulating the expression and activity of mitogen-activated protein kinase (MAPK)-directed phosphatases (MKPs) with concomitant activation of the MAPKs, p38, and extracellular signal-regulated kinase (ERK) (8). However, the further upstream signaling pathway that leads to activation of these MAPKs upon GRA treatment is yet to be explored.
The MAPKs are an important group of serine and threonine kinases responsible for signal transduction of a variety of extracellular stimuli through a cascade of protein activation via phosphorylation. The downstream transcription factors of MAPKs are Elk-1, NF-κB, CCAAT/enhancer-binding protein β (C/EBPβ), activating transcription factor 2 (ATF-2), and c-Jun (9–11). There are in principal three MAPK family members, p46 and p54 c-Jun N-terminal kinase (JNK), p38 MAPK, and p42 and p44 ERK. MAPKs are activated by specific upstream MAPK kinases (MKKs): MKK4 and MKK7 activate JNK (12–14), MAPK ERK kinase 1 (MEK1) and MEK2 activate the ERKs (15), and MKK3, MKK6, and MKK4 activate p38 MAPK (16). Inhibition studies with p38 MAPK and mice lacking MKK3, a major upstream kinase of p38, have indicated the importance of the p38 signaling cascade in macrophage tumor necrosis factor alpha (TNF-α) and interleukin 12 (IL-12) production during lipopolysaccharide (LPS) stimulation (17, 18); these are also the major antileishmanial cytokines (19, 20). The MKKs are again activated by MKK kinases (M3Ks), including transforming growth factor β (TGF-β)-activated protein kinase 1 (TAK1), which has been shown to activate p38, and stress-activated protein kinase (SAPK)/JNK, as well as the IκB kinase complex leading to NF-κB activation (21, 22). TAK1 associates with TAK1-binding protein 1 (TAB1) and TAB2 and also relies upon ubiquitination of TNF receptor-associated factor 6 (TRAF6) for its activation (23). TRAF6 itself is linked to the Toll-like receptor (TLR) system, and this occurs through interactions with IL-1R-associated kinase 1 (IRAK1) and IRAK4 and adaptor protein myeloid differentiation factor 88 (MyD88) (24–26).
TLR signaling plays an important role in the immune response against Leishmania parasites, as evident from gene knockout studies (27). For instance, MyD88-deficient mice develop progressive lesions with a dominant Th2 response (28). In vivo studies revealed important roles of TLR4 in the control of infection via early induction of inducible nitric oxide synthase (iNOS) (29). TLR9 mediates NK cell activation and inflammatory cytokine synthesis in animal models of visceral and cutaneous leishmaniasis (30, 31). Leishmania lipophosphoglycan (LPG), an abundant molecule on the surfaces of promastigotes, is known to signal via TLR2. Our recent work suggests important roles of TLRs in both the recognition and modulation of immune responses against Leishmania parasites (32, 33). Moreover, the silencing of IRAK1, MyD88, TLR2, or TLR3 attenuated the release of TNF-α and nitric oxide following infection of gamma interferon (IFN-γ)-primed macrophages by Leishmania donovani promastigotes (34). On the basis of this information, we hypothesized that GRA-mediated MAPK activation might be associated with TLRs, and in order to define the mechanism of MAPK activation by GRA, we directed our findings to understanding the upstream signaling events that ultimately lead to proinflammatory cytokine production and elimination of intramacrophage pathogens.
MATERIALS AND METHODS
Ethics statement.
This study was carried out in accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. The protocol used was approved by the committee on the Ethics of Animal Experiments of Indian Institute of Chemical Biology (permit number 147-1999).
Cell culture, parasites, and infection.
The promastigotes of L. donovani strain MHOM/IN/1983/AG83 were maintained in medium 199 (Invitrogen Life Technologies) supplemented with 10% fetal calf serum (FCS) (Invitrogen), 50 μg/ml streptomycin, and 50 U/ml penicillin. The murine macrophage cell line RAW 264.7 was maintained at 37°C and 5% CO2 in RPMI 1640 (Invitrogen Life Technologies) supplemented with 10% FCS, streptomycin (100 μg/ml), and penicillin (100 U/ml). In vitro infection was done in the RAW 264.7 cell line using stationary-phase promastigotes at a 10:1 parasite-to-macrophage ratio. For the in vivo experiments, female BALB/c mice were injected with 107 L. donovani promastigotes via the tail vein. GRA (Sigma-Aldrich) (50 mg/kg of body weight/day) was administered intraperitoneally 3 times, 5 days apart, starting at 10 days postinfection. In certain experiments, infected mice were treated with the p38 inhibitor SB203580 HCl (10 mg/kg, twice weekly) or the ERK inhibitor PD0325901 (20 mg/kg, twice weekly) along with GRA. Infection was assessed by removing the spleens and livers from infected mice at the indicated times and determining the parasite burdens from Giemsa-stained impression smears. Data are presented as Leishman-Donovan units (LDU) as described earlier (35).
Reagents, antibodies, and constructs.
All antibodies were purchased from Santa Cruz Biotechnology and Cell Signaling Technology. All chemicals were from Sigma-Aldrich, unless indicated otherwise. Dominant-negative (DN) MyD88 (152–296) and IRAK1 (1–217) were kindly provided by M. Muzio (Department of Immunology and Cell Biology, Mario Negri Institute, Milan, Italy) (36, 37). Wild-type (WT) and DN TRAF6 were gifts from J. Inoue (Institute of Medical Science, University of Tokyo, Tokyo, Japan) (38). DN (K63W) TAK1 was a gift from K. Matsumoto (Nagoya University, Nagoya, Japan) (39). MKK3 and MKK6 were obtained from D. R. Alessi (Medical Research Council, Protein Phosphorylation Unit, Department of Biochemistry, University of Dundee, Scotland).
Immunoprecipitation and immunoblotting.
Cells were lysed in lysis buffer (Cell Signaling Technology), and the protein concentrations were estimated using the Bio-Rad protein assay (Bio-Rad). For immunoprecipitation experiments, precleared cell lysates (500 μg) were incubated overnight with the specific primary antibody at 4°C. Thereafter, 25 μl of protein A/G plus agarose beads (Santa Cruz Biotechnology) was added to the mixture and incubated for 4 h at 4°C. Immune complexes were washed three times with ice-cold lysis buffer. The immunoprecipitated samples and whole-cell lysates were resolved on 10% SDS-PAGE and then transferred to a nitrocellulose membrane (Millipore). The membranes were blocked with 5% bovine serum albumin (BSA) in wash buffer (Tris-buffered saline [TBS] with 0.1% Tween 20) for 1 h at room temperature and probed with the primary antibody overnight at the dilution recommended by the suppliers. Membranes were washed three times with wash buffer and then incubated with alkaline phosphatase-conjugated secondary antibody, washed, and detected by hydrolysis of 5-bromo-4-chloro-3-indolylphosphate (BCIP) chromogenic substrate according to the manufacturer's instructions.
Cytokine analysis by ELISA.
Enzyme-linked immunosorbent assays (ELISA) were done using a sandwich ELISA kit (Quantikine M; R&D Systems, Minneapolis, MN, USA). The detection limits of these assays were >5.1, >2.5, and >4 pg/ml for TNF-α, IL-12p70, and IL-10, respectively.
Transient transfection.
RAW 264.7 cells (2 × 106) were transfected with 1 μg of small interfering RNA (siRNA) according to the manufacturer's instructions (Santa Cruz Biotechnology). Scrambled siRNA was used as the control.
Densitometric analysis.
Densitometric analyses for all experiments were carried out using Quantity One software (Bio-Rad, Hercules, CA, USA). Band intensities were quantified, and the values were normalized to the endogenous control and are expressed in arbitrary units indicated as fold change values below the individual blots.
Statistical analysis.
The data shown are representative of three independent experiments unless otherwise stated as n values given in the legend. Results are expressed as means ± standard deviations (SD). Student's t test was employed to assess the statistical significances of differences among pairs of data sets. A P value of <0.05 was considered significant.
RESULTS
Analysis of MAPK pathway activation effect upon infection and GRA treatment.
Our earlier studies documented that GRA exerts its antileishmanial effect by upregulating production of iNOS and proinflammatory cytokines (3, 8). We next investigated the MAPK signaling pathways which are important for regulation of these downstream effector molecules. Analysis of all of the three major MAP kinases in GRA (20 μM)-treated infected RAW 264.7 cells revealed maximum phosphorylation of p38 (3.9-fold over that for infected cells, P = 0.0005) (Fig. 1A) with much lower activation of ERK (2.2-fold over that for infected, P = 0.032) at 4 h posttreatment (Fig. 1B). However, the levels were lower than only those for GRA-treated cells for p38 (4.8-fold over that for infected cells, P = 0.0005) (Fig. 1A) and for ERK (2.8-fold over infected cells, P = 0.0091) (Fig. 1B). No phosphorylation was observed for JNK in infected and/or GRA-treated cells (Fig. 1C).
FIG 1.
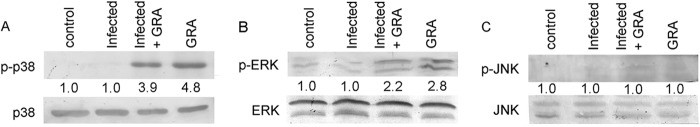
Analysis of MAPK pathway activation effect on infection and GRA treatment. (A, B, and C) RAW264.7 cells (2 × 106/ml) were infected with L. donovani promastigotes (macrophage/parasite ratio, 1:10) and then were either treated with 20 μM GRA for 4 h or left untreated. The levels of p38 and phosphorylated p38 (p-p38) (A), ERK and p-ERK (B), and JNK and p-JNK were detected by Western blotting. The bands were analyzed densitometrically, and fold changes are indicated below each blot. All experiments were repeated at least three times each, and one set of representative data is shown.
Roles of p38 and ERK in in vivo antileshmanial activity of GRA.
To ascertain the roles of p38 and ERK in GRA-mediated modulation of disease progression, the in vivo antileishmanial activity of GRA was evaluated in the presence of either the p38 inhibitor SB203580 HCl (10 mg/kg/day) or the ERK inhibitor PD0325901 (20 mg/kg/day), and the liver and spleen parasite burdens were measured. The infected control mice received only phosphate-buffered saline (PBS). In contrast to the GRA-treated mice in which at 6 weeks postinfection a marked decrease was observed in the spleen and liver parasite burdens (98.1% and 94.9% reductions for the spleen and liver, respectively, compared to those for the infected control, P = 0.0001) (Fig. 2A and B), the effect was significantly less in SB203580-treated mice (54.4% and 43.9% suppressions were found for spleen and liver, respectively, at 6 weeks compared to that for the infected control, P = 0.0007 and P = 0.0018, respectively). On the contrary, a much less pronounced effect was found in PD0325901-treated mice (Fig. 2A and B). The significant difference in LDU between 4 and 6 weeks emerged only when the p38 inhibitor was used along with GRA, not when the ERK inhibitor was used. This is in agreement with the Western blot observation and confirmed that the GRA-mediated antileishmanial effect is primarily mediated by p38, not ERK. p38 MAPK-mediated downstream signaling is known to be associated with upregulation of antileishmanial proinflammatory cytokines like TNF-α and downregulation of a disease-favoring anti-inflammatory cytokine like IL-10 (8). Therefore, to assess further the relative importance of p38 and ERK1/2 MAP kinases in the GRA-mediated antileishmanial effect, splenocytes were isolated from the control, GRA-administered, and inhibitor-treated mice, and the levels of TNF-α and IL-10 were measured (Table 1). GRA treatment resulted in a significant increase in TNF-α (8.1-fold increase over that of the untreated infected control at 4 weeks postinfection, P = 0.0001) with a concomitant decrease in IL-10 (69.5% reduction compared to that of the untreated infected control at 4 weeks postinfection, P < 0.0001) (Table 1). Selective blockade of the p38 pathway resulted in a reduction of the GRA-induced TNF-α production (63.5% reduction compared to that of the GRA-treated infected mice at 4 weeks postinfection, P = 0.0004) as well as suppression of IL-10 synthesis (24.4% compared to 69.5% in GRA-treated infected mice at 4 weeks postinfection, P < 0.0001) (Table 1). Inhibition of the ERK pathway had a much less prominent effect. All of these results collectively documented that p38 MAPK plays a more predominant role than ERK in the antileishmanial effect of GRA.
FIG 2.
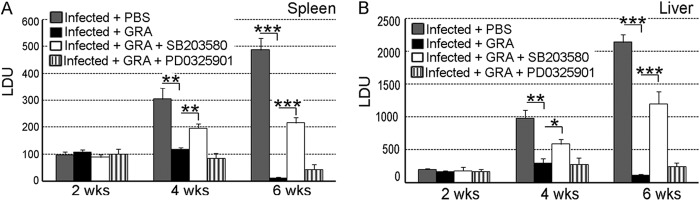
Determination of p38-mediated antileishmanial activity of GRA in vivo. Spleen (A) and liver (B) parasite burdens were determined every 2 weeks in each group and are expressed as LDU ± SD. Animal experiments were done with five animals per group. Error bars represent means ± SD; n = 5. Statistical significance was determined by Student’s t test and indicated by asterisks as follows: *P < 0.05; **P < 0.01; ***P < 0.001.
TABLE 1.
Cytokine levels in 4-week L. donovani-infected BALB/c mice after various treatments
| Treatment | TNF-α (pg/ml)a | IL-10 (pg/ml)a |
|---|---|---|
| Infected + PBS | 102.4 ± 13.1 A | 1,043.3 ± 69.1 A |
| Infected + GRA | 824.9 ± 82.7 ABC | 318.3 ± 24.1 ABD |
| Infected + GRA | ||
| SB203580 | 301.4 ± 12.8 B | 789.1 ± 43.6 B |
| PD00325901 | 606.2 ± 69.8 C | 584.2 ± 51.6 D |
Values are means ± SD; n = 5. Letters indicate combinations whose values were significantly different by Student's t test: A and B, P < 0.001; C, P < 0.05; D, P < 0.01.
MKK3/6 is involved in GRA-mediated p38 activation.
The identification of p38 as the major MAP kinase responsible for the GRA-mediated antileishmanial effect prompted us to investigate the status of the immediate upstream kinases of p38, MKK3, and MKK6. Similar to that of p38, the maximum phosphorylation of MKK3/6 was found at 4 h in GRA-treated infected macrophages (3.8-fold, P = 0.0001) (Fig. 3A). On the other hand, infected macrophages did not show any MKK3/6 phosphorylation (Fig. 3A). Other MAP kinase kinase (MAPKK) activation (MKK4/7) was also not observed in either GRA-treated or untreated Leishmania-infected macrophages, indicating the absence of any role of this pathway in GRA-mediated p38 activation (data not shown). To further understand the role of MKK3/6 in GRA-mediated p38 activation, macrophages were transiently transfected with the dominant-negative constructs of MKK3 or MKK6. The activation of p38 was significantly diminished in DN MKK3-transfected (47.4% reduction from that for WT MKK3, P = 0.01) (Fig. 3B) or DN MKK6-transfected (46.9% reduction from that for WT MKK6, P = 0.017) (Fig. 3C) macrophages, suggesting the importance of these MAP kinase kinases in GRA-mediated p38 activation. To further ascertain whether double transfection of MKK3 and MKK6 dominant-negative constructs might completely inhibit p38 activation, we simultaneously transfected GRA-treated infected macrophages with both the constructs. Surprisingly, some p38 activation was still found to be present in the presence of both the dominant-negative constructs (54.2% reduction from that for WT MKK3/6, P = 0.007) (Fig. 3D), thus hinting that some other mode of upstream activation is responsible for the residual activity of p38 in GRA-treated cells.
FIG 3.
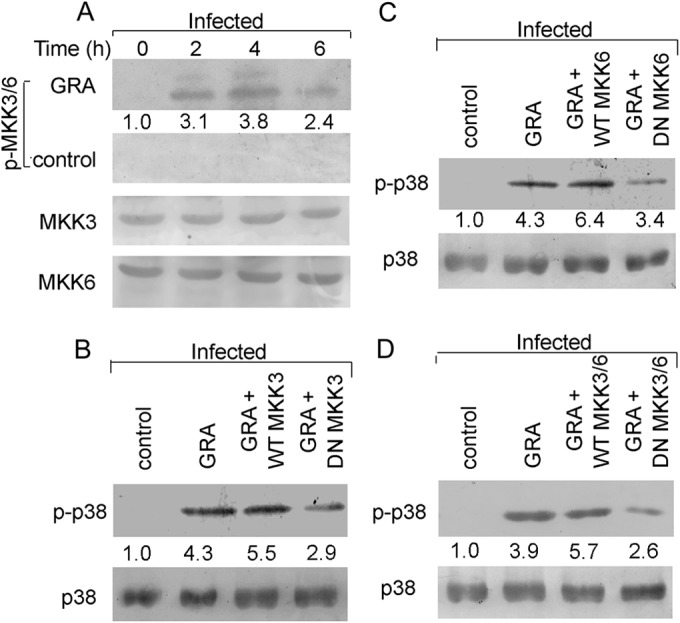
Role of MKK3/6 in GRA-mediated p38 activation. (A and B) Infected cells as stated in the legend to Fig. 1 were either treated with GRA (20 μM) (A) or left untreated (B) for different times, and the total and phosphorylated levels of various MAP kinases were detected by Western blotting using the respective anti-MAP kinase antibodies. (B, C, and D) Cells were transiently transfected with the wild-type or dominant-negative constructs of MKK3 (B), the wild-type or dominant-negative constructs of MKK6 (C), and the wild-type or dominant-negative constructs of both MKK3 and MKK6 (D) for 24 h. Thereafter, they were infected and treated with GRA for 4 h, and in all the samples, the levels of total and phosphorylated p38 were determined by Western blot analysis. The bands were analyzed densitometrically, and the fold changes are indicated below each blot. All experiments were repeated at least three times each, and one set of representative data is shown.
Residual phosphorylation of p38 is a result of an autophosphorylation event.
Incomplete inhibition of p38 phosphorylation in DN MKK3/6-transfected cells indicated an alternative p38 activation mechanism. It was earlier reported that p38 activation can be achieved in an MKK-independent pathway where autophosphorylation of p38 takes place upon association with TAB1 (40). We thus thought it worthwhile to check if p38 autophosphorylation also occurs in GRA-stimulated cells. To this end, we used the pharmacological inhibitor SB203580, which does not prevent MKK-mediated p38 transphosphorylation but instead occupies the ATP-binding pocket within the p38 kinase cleft, thereby preventing its downstream kinase activity (41). Accordingly, cells were transfected with both DN MKK3 and DN MKK6 and treated with SB203580 prior to GRA stimulation and infection. Whereas WT MKK3/6-transfected SB203580-pretreated cells showed 25.8% reduced p38 phosphorylation compared to GRA-treated infected cells (Fig. 4A), DN MKK3/6-transfected SB203580-treated cells showed almost complete inhibition of p38 activation (77.6% reduction from that of WT MKK3/6-transfected SB203580-treated cells, P = 0.0005) (Fig. 4A), indicating both autophosphorylation and MKK3/6-mediated transphosphorylation for GRA-mediated p38 activation. To confirm whether GRA stimulation in infected cells might induce an interaction between p38 and TAB1 to stimulate the residual p38 activation in the absence of MKK3/6, coimmunoprecipitation studies were performed by Western blotting with immunoprecipitated phosphorylated p38 (p-p38) using anti-TAB1 antibody. This revealed a significantly higher association between p-p38 and TAB1 in GRA-treated infected cells compared to that in the infected control (4.3-fold, P = 0.0013) (Fig. 4B). Transfection with DN MKK3/6 significantly reduced the level of phosphorylated p38, but a strong association with TAB1 was still seen, suggesting that the TAB1-mediated p38 autophosphorylation is independent of MKK3/6. On the other hand, SB203580-mediated inhibition resulted in a marked decrease in this association (74.5%, P = 0.0017) (Fig. 4B) in spite of a higher p-p38 level. In order to validate these findings, coimmunoprecipitation studies were performed with immunoprecipitated TAB1 and immunoblotting with anti-p-p38 antibody, and similar results were observed (84.2% reduction in cells pretreated with SB203580 from that in GRA-treated infected cells, P = 0.0004) (Fig. 4C). No p-p38 was detected in the infected control (Fig. 4B and C), documenting that both pathways remained inactive during infection. Next, to assess the roles of these pathways in GRA-mediated proinflammatory cytokine production, macrophage cells were either transfected with DN MKK3/6 alone and/or preincubated for 1 h with the p38 inhibitor SB203580 before infection and GRA stimulation. Selective blockade of the MKK3/6 pathway resulted in reductions of 57.4% for IL-12 and 57.5% for TNF-α compared to those for WT MKK3/6 (P = 0.0005), whereas simultaneous transfection with DN MKK3/6 and pretreatment with SB203580 led to reductions of 67.8% for IL-12 and 66.7% for TNF-α (P = 0.0002) (Table 2). Interestingly, pretreatment of cells with SB203580 led only to decreases in IL-12 and TNF-α similar to those for simultaneous transfection with DN MKK3/6 and pretreatment with SB203580 (Table 2). These results indicate that although SB203580 only selectively blocks p38 phosphorylation, it inhibits all of the downstream activities of p38 kinase. This also indicates that both trans- and autophosphorylation of p38 play important roles in GRA-treated infected cells.
FIG 4.
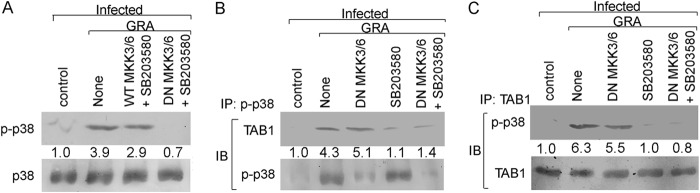
p38 autophosphorylation in GRA-stimulated cells. (A) RAW 264.7 cells were transiently transfected with either WT or DN MKK3/6 expression plasmids (24 h) and treated with SB203580 (30 μM) for 1 h before infection and stimulation with GRA (4 h). The levels of total and phosphorylated p38 were determined by Western blotting. (B) Cells were treated as described for panel A. Cell lysates were then subjected to immunoprecipitation by anti-p-p38 antibody and Western blotting using anti-TAB1 antibody. (C) Cells were treated as stated for panel A. Cell lysates were then subjected to immunoprecipitation by anti-TAB1 antibody and Western blotting using anti-p-p38 antibody. The bands were analyzed densitometrically, and the fold changes are indicated below each blot. All experiments were repeated at least three times each, and one set of representative data is shown. IP, immunoprecipitation using the indicated antibody (Ab); IB, immunoblot analysis using the indicated Ab.
TABLE 2.
Cytokine levels in infected RAW 264.7 macrophages after various treatments
| Treatment | IL-12 (pg/ml)a | TNF-α (pg/ml)a |
|---|---|---|
| Infected | 79.8 ± 8.1 | 34.2 ± 3.6 |
| Infected + GRA | 282.3 ± 13.2 | 207.6 ± 21.6 |
| Infected + GRA | ||
| WT MKK3/6 | 328.3 ± 28.2 ABC | 231.3 ± 20.1 ABC |
| DN MKK3/6 | 139.7 ± 13.8 A | 98.3 ± 10.2 A |
| SB203580 | 113.7 ± 10.4 B | 78.3 ± 7.8 B |
| DN MKK3/6 + SB203580 | 105.7 ± 9.5 C | 77.2 ± 7.3 C |
Values are means ± SD; n = 3. Letters indicate combinations whose values were significantly different by Student's t test at P < 0.001.
Importance of TAK1 in GRA-mediated p38 activation.
Upstream of MKK3/6 is the MAP kinase kinase kinase (MAPKKK), TAK1, which is responsible for MKK3/6 phosphorylation and activation leading to p38 activation (42). Similar to p38 and MKK3/6, TAK1 maximum activation was found at 4 h in GRA-treated infected cells (3.4-fold over that of the infected control, P = 0.0007) (Fig. 5A). To further assess the role of TAK1 in GRA-mediated p38 activation during infection, we transiently transfected cells with DN constructs of TAK1 and checked for the activation of MKK3/6 and p38. TAK1 inactivation led to a significant reduction in GRA-mediated phosphorylation of p38 (58.9% reduction from that for WT TAK1, P = 0.0032) (Fig. 5B) and MKK3/6 (78.9% reduction from that for WT TAK1, P = 0.0002) (Fig. 5C) during infection, indicating the importance of TAK1 in activation of downstream MAP kinases. We also wanted to assess the direct role of TAK1 in TAB1-mediated p38 autoactivation. Again, residual p38 phosphorylation (2.1-fold over that of the infected control, P = 0.012) (Fig. 5B) was detected in DN TAK1-transfected cells, suggesting that p38 autophosphorylation may not depend on TAK1. SB203580 treatment led to a further decline in the p38 phosphorylation status in DN TAK1-transfected cells, suggesting thereby that TAB1-mediated p38 autophosphorylation is independent of TAK1 (80.1% decrease from that in WT TAK1-transfected cells, P = 0.0007) (Fig. 5D). To further ascertain the role of TAK1 in GRA-mediated antileishmanial activity, DN TAK1-transfected and infected cells were treated with GRA, and parasite survival was measured. GRA-mediated parasite killing was markedly attenuated in DN TAK1-transfected cells (63.1% decrease in parasite killing from that in WT TAK1-transfected cells, P = 0.0007) (Table 3), indicating the importance of TAK1 in the antileishmanial effect of GRA. Interestingly, when DN TAK1-transfected cells were subjected to SB203580 treatment, a 72.9% reduction in parasite killing was observed compared to that in WT TAK1-transfected cells (P = 0.0003) (Table 3). Therefore, both autophosphorylation and transphosphorylation of p38 take place in GRA-treated infected cells, and as for cytokine levels, parasite killing is also regulated by TAK1-dependent and TAB-mediated signaling.
FIG 5.
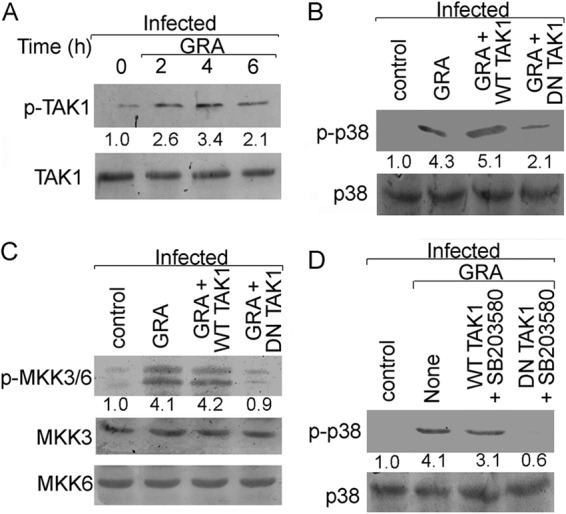
Role of TAK1 in GRA-mediated antileishmanial activity. (A) RAW 264.7 cells were infected with L. donovani and treated with 20 μM GRA for various time periods, and the levels of total and phosphorylated TAK1 were determined by Western blotting. (B and C) Cells were transiently transfected with WT or DN TAK1 expression plasmid (24 h) and then infected with L. donovani and treated with GRA (4 h). The levels of total and phosphorylated p38 (B) and MKK3/6 (C) were determined by Western blotting. (D) WT or DN TAK1-transfected cells were treated with SB203580 for 1 h before infection, and GRA stimulation and total and phosphorylated p38 levels were determined by Western blotting. The bands were analyzed densitometrically, and fold changes are indicated below each blot. All experiments were repeated at least three times each and one set of representative data is shown.
TABLE 3.
Parasite loads in infected macrophages during various treatments
| Treatment | No. of amastigotes/100 macrophagesa |
|---|---|
| Infected | 356.3 ± 29.8 |
| Infected + GRA | 79.1 ± 8.5 |
| Infected + GRA | |
| WT TAK1 | 88.0 ± 9.1 AB |
| DN TAK1 | 238.3 ± 26.5 A |
| DN TAK1 + SB203580 | 324.7 ± 33.9 B |
| WT MyD88 | 64.3 ± 7.2 C |
| DN MyD88 | 193.0 ± 18.0 C |
| WT IRAK1 | 56.7 ± 4.9 D |
| DN IRAK1 | 186.3 ± 19.5 D |
| WT TRAF6 | 83.1 ± 9.9 E |
| DN TRAF6 | 241.1 ± 23.8 E |
| Scrambled TLR2 siRNA | 78.3 ± 8.1 F |
| TLR2 siRNA | 219.0 ± 22.3 F |
| Scrambled TLR4 siRNA | 86.3 ± 9.2 G |
| TLR4 siRNA | 231.3 ± 23.5 G |
Values are means ± SD; n = 3. Letters indicate combinations whose values were significantly different by Student's t test at P < 0.001.
MyD88, IRAK1, and TRAF6 play important roles in antileishmanial activity of GRA.
Involvement of the TAK1-MKK3/6-p38 pathway in the GRA-mediated antileishmanial response led us to check the further upstream molecules TRAF6, IRAK1, and MyD88, which are known for TAK1-mediated NF-κB and MAPK activation (25, 26, 43). We thus transiently transfected cells with DN constructs of MyD88, IRAK1, and TRAF6, which severely impaired the parasite-killing ability (66.7%, 69.6%, and 65.6% reduced parasite killing, respectively, compared to those for the WT constructs of MyD88, IRAK1, and TRAF6, P = 0.0003, P = 0.0004, and P = 0.0004, respectively), indicating that the antileishmanial activity of GRA is dependent upon the upstream signaling molecules MyD88, IRAK1, and TRAF6 (Table 3). The GRA-induced p38 activation was also severely impaired in DN MyD88-, DN IRAK1-, and DN TRAF6-transfected cells (55.1%, 51.9%, and 53.1% decreases, respectively, compared to those for WT construct-transfected GRA-treated infected cells, P = 0.0016, P = 0.0055, and P = 0.0024, respectively), suggesting thereby the involvement of MyD88 signaling in the GRA-mediated activation of p38 (Fig. 6A). Next we wanted to study the status of TLR2 and TLR4, the ultimate upstream receptors primarily implicated in the proinflammatory response against Leishmania (32, 33). For this, we used an in vitro siRNA knockdown system for TLR2 and TLR4. As seen in Fig. 6B and C, TLR2 and TLR4 were effectively downregulated by the respective siRNAs (82.5% and 83.3% reductions, respectively, compared with those for the control siRNA-treated cells, P = 0.0002 and P = 0.0004, respectively). The siRNA-mediated inhibition of either TLR2 or TLR4 significantly reduced the NF-κB reporter activity (57.1% and 54.6% reductions, respectively, from those for the scrambled siRNA control, P = 0.0009 and P = 0.0007, respectively) (Fig. 6D) and parasite killing (64.3% and 62.8% reductions from those for scrambled siRNA control, P = 0.0005 and P = 0.0006, respectively) (Table 3) in GRA-administered infected cells. All of these results collectively demonstrate that GRA exerts its antileishmanial effect by activating the TLR2- and TLR4-mediated MyD88-dependent TAK1–MKK–p38–NF-κB pathway.
FIG 6.
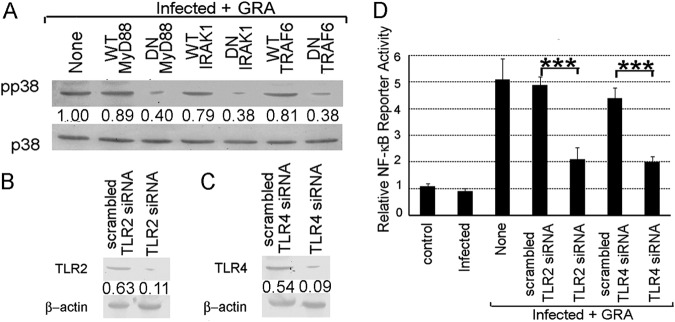
Importance of TLR-MyD88-IRAK1-TRAF6 axis in GRA-mediated antileishmanial response. (A) Cells were transiently transfected with the indicated constructs, infected with L. donovani, and stimulated with GRA for 4 h. Cells were lysed and processed for p38 expression and phosphorylation by Western blotting. (B to D) To determine the effect of TLR inhibition, macrophages were transfected (24 h) with either TLR2 siRNA (B) or TLR4 siRNA (C), and TLR expression was determined by Western blotting. Cells were transiently transfected with TLR2 or TLR4 siRNA, respectively, infected with L. donovani, stimulated with GRA for 4 h, and processed for determination of NF-κB luciferase activity (D). The bands were analyzed densitometrically, and fold changes are indicated below each blot. All experiments were repeated at least three times each, and one set of representative data is shown. Error bars represent means ± SD; n = 3. ***, P < 0.001; Student's t test.
DISCUSSION
GRA is a natural compound which decreases parasite burden to a great extent (8, 44). Recently, GRA has been shown to induce Th1 immunological adjuvant activity in Candida albicans infection (45, 46). GRA treatment showed enhanced expression of iNOS, NF-κB, and IL-12 (47, 48) and shifted the kinase/phosphatase balance toward kinases, where p38 plays a pivotal role (8). But in order for GRA to be used as a drug, it is of utmost importance to know its precise mode of action, which called for a deeper understanding of the activation of p38 MAPK by GRA. The results of the present study demonstrated that TLR2/4-MyD88-dependent TAK1-MKK3/6-mediated canonical activation and TAB-mediated noncanonical activation are crucial for GRA-mediated p38−NF-κB activation and its antiparasitic effects.
p38 belongs to the MAPK family, the members of which are known to regulate a variety of stress responses in a wide range of organisms (49–51). p38 pathway activation has been shown to play an important role in Leishmania elimination (52). Moreover, p38 activation by sodium antimony gluconate treatment has been shown to induce NO production, thereby inhibiting parasite survival within the macrophages (53). Our previous study revealed the importance of p38 in GRA-mediated antileishmanial activity (8). Activation of the p38 pathway is a result of a series of phosphorylation events involving proximal upstream MAPKKs such as MKK3, MKK6, and MKK4/MKK7 (54, 55). In our study, we observed that MKK3/6 activation was indeed dampened in infection, and GRA treatment increased both the kinetics and extent of induction of MKK3/6, indicating the importance of these kinase kinases in p38 activation. On the other hand, no induction of MKK4/7 was observed in GRA-stimulated infected macrophage cells, which is in line with the earlier observation that JNK phosphorylation, downstream of MKK4/7, was not seen. As MKK3/6 specifically phosphorylates p38, downregulation of these kinases should be able to completely abrogate p38 activation in GRA-stimulated cells. Instead, we observed that dual transfection with the dominant-negative constructs of MKK3 and MKK6 did not completely inhibit p38 activation, and thus we looked for an additional p38 activation mechanism in GRA-stimulated cells. We then discovered the possible existence of MKK3/6-independent p38 kinase activation by GRA. Further investigation revealed that TAB1 directly binds to p38, leading to its autophosphorylation in a MKK3/6-independent but SB203580-sensitive manner. Several other reports also implicated TAB1 in p38 activation (56, 57). TAB1 is normally known to function in TAK1 activation (22, 42), but other reports suggest a TAB splice variant that may selectively activate p38 (58). Our data indicated that indeed there is a strong association between TAB1 and p38 which might have contributed to the residual p38 phosphorylation seen in DN MKK3/6-transfected cells. It has been shown that activation of p38 in the MKK3/6-independent manner leads to IL-12 production in Toxoplasma gondii infection (59). Thus, it might be suggested that noncanonical p38 activation also contributes to the proinflammatory responses generated in GRA-stimulated cells.
TAK1 is an important member of the MAP kinase kinase kinase family of enzymes which is upstream of MKK3/6 and is known to play a critical role in p38 and NF-κB activation (21, 22). The kinetics of TAK1 activation in GRA-stimulated cells matched that of MKK3/6 activation, which was significantly reversed in DN TAK1-transfected cells. The antileishmanial activity of GRA can be attributed to this vital MAPKKK, as DN TAK1-transfected cells also showed considerable decreases in parasite numbers after GRA treatment. The fact that the GRA-induced antileishmanial response relies on TAK1 activation and that this may in turn rely on the TLR-MyD88-TRAF6 axis prompted us to look further into this pathway. MyD88 has been implicated in the control of infection as MyD88-deficient mice showed impaired T-cell responses and protective immunity (11). Moreover, Leishmania major infection in MyD88-deficient mice showed progressive lesions with dominant Th2 responses (12). Silencing of TLR, MyD88, or IRAK1 has been shown to downregulate TNF and NO production in IFN-γ-primed macrophages infected with Leishmania promastigotes (13). Thus, for comprehensive elucidation of the role of this pathway in GRA-mediated responses, we used WT and DN constructs of TRAF6, IRAK1, and MyD88, which revealed the involvement of these signaling components in GRA-mediated p38 activation and parasite killing. Further analysis also confirmed that TLR2 and TLR4 were the potential receptors for the GRA-mediated regulation and also seemed indispensable for effective parasite killing. One other report suggests involvement of TLR4 in GRA-mediated activity (60), but inhibition studies revealed roles for both TLR2 and TLR4 in GRA-mediated antileishmanial activity.
In summary, GRA triggers phosphorylation of p38 in macrophages via two different mechanisms: (i) through an MAPKK-dependent TLR-MyD88-TAK1-mediated axis and (ii) through an MKK-independent TAB1-mediated pathway (Fig. 7). Apart from providing detailed mechanistic insight for GRA, this study also opens up new avenues for future development of additional compounds where the knowledge of the immunomodulatory pathways may help to generate new drugs effective against intracellular parasites.
FIG 7.

Schematic representation depicting mechanistic details of GRA-mediated host cell activation. Leishmania inhibits proinflammatory cytokine production and host cell activation. GRA treatment resulted in both (i) TLR2/4-MyD88-IRAK-TRAF6-TAK1-dependent MKK3/6-mediated canonical p38 activation and (ii) TAB1-dependent noncanonical p38 activation. Overall, p38 activation by GRA thus leads to proinflammatory cytokine production and host cell activation.
ACKNOWLEDGMENTS
This work was supported by the University Grants Commission, Council of Scientific and Industrial Research (CSIR), Department of Science and Technology (SB/SO/BB-00055/2013 and SERB/F/4467/2013-14) and a J. C. Bose Fellowship, Government of India.
REFERENCES
- 1.Croft SL, Coombs GH. 2003. Leishmaniasis—current chemotherapy and recent advances in the search for novel drugs. Trends Parasitol 19:502–508. doi: 10.1016/j.pt.2003.09.008. [DOI] [PubMed] [Google Scholar]
- 2.Singh S, Sivakumar R. 2004. Challenges and new discoveries in the treatment of leishmaniasis. J Infect Chemother 10:307–315. doi: 10.1007/s10156-004-0348-9. [DOI] [PubMed] [Google Scholar]
- 3.Ukil A, Biswas A, Das T, Das PK. 2005. 18 Beta-glycyrrhetinic acid triggers curative Th1 response and nitric oxide up-regulation in experimental visceral leishmaniasis associated with the activation of NF-kappa B. J Immunol 175:1161–1169. doi: 10.4049/jimmunol.175.2.1161. [DOI] [PubMed] [Google Scholar]
- 4.Rossi T, Galatulas I, Bossa R, Tampieri A, Tartoni P, Baggio G, Ruberto AI, Castelli M. 1995. Influence of glycyrrhizin on the evolution and respiration of Ehrlich ascites tumour cells. In Vivo 9:183–186. [PubMed] [Google Scholar]
- 5.Jeong HG, You HJ, Park SJ, Moon AR, Chung YC, Kang SK, Chun HK. 2002. Hepatoprotective effects of 18beta-glycyrrhetinic acid on carbon tetrachloride-induced liver injury: inhibition of cytochrome P450 2E1 expression. Pharmacol Res 46:221–227. doi: 10.1016/S1043-6618(02)00121-4. [DOI] [PubMed] [Google Scholar]
- 6.Farina C, Pinza M, Pifferi G. 1998. Synthesis and anti-ulcer activity of new derivatives of glycyrrhetic, oleanolic and ursolic acids. Farmaco 53:22–32. doi: 10.1016/S0014-827X(97)00013-X. [DOI] [PubMed] [Google Scholar]
- 7.Kroes BH, Beukelman CJ, van den Berg AJ, Wolbink GJ, van Dijk H, Labadie RP. 1997. Inhibition of human complement by beta-glycyrrhetinic acid. Immunology 90:115–120. doi: 10.1046/j.1365-2567.1997.00131.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ukil A, Kar S, Srivastav S, Ghosh K, Das PK. 2011. Curative effect of 18beta-glycyrrhetinic acid in experimental visceral leishmaniasis depends on phosphatase-dependent modulation of cellular MAP kinases. PLoS One 6:e29062. doi: 10.1371/journal.pone.0029062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kyriakis JM, Avruch J. 2001. Mammalian mitogen-activated protein kinase signal transduction pathways activated by stress and inflammation. Physiol Rev 81:807–869. [DOI] [PubMed] [Google Scholar]
- 10.Karin M, Liu Z, Zandi E. 1997. AP-1 function and regulation. Curr Opin Cell Biol 9:240–246. doi: 10.1016/S0955-0674(97)80068-3. [DOI] [PubMed] [Google Scholar]
- 11.Ip YT, Davis RJ. 1998. Signal transduction by the c-Jun N-terminal kinase (JNK)–from inflammation to development. Curr Opin Cell Biol 10:205–219. doi: 10.1016/S0955-0674(98)80143-9. [DOI] [PubMed] [Google Scholar]
- 12.Dérijard B, Raingeaud J, Barrett T, Wu IH, Han J, Ulevitch RJ, Davis RJ. 1995. Independent human MAP-kinase signal transduction pathways defined by MEK and MKK isoforms. Science 267:682–685. doi: 10.1126/science.7839144. [DOI] [PubMed] [Google Scholar]
- 13.Lin A, Minden A, Martinetto H, Claret FX, Lange-Carter C, Mercurio F, Johnson GL, Karin M. 1995. Identification of a dual specificity kinase that activates the Jun kinases and p38-Mpk2. Science 268:286–290. doi: 10.1126/science.7716521. [DOI] [PubMed] [Google Scholar]
- 14.Sánchez I, Hughes RT, Mayer BJ, Yee K, Woodgett JR, Avruch J, Kyriakis JM, Zon LI. 1994. Role of SAPK/ERK kinase-1 in the stress-activated pathway regulating transcription factor c-Jun. Nature 372:794–798. doi: 10.1038/372794a0. [DOI] [PubMed] [Google Scholar]
- 15.Winston BW, Remigio LK, Riches DW. 1995. Preferential involvement of MEK1 in the tumor necrosis factor-alpha-induced activation of p42mapk/erk2 in mouse macrophages. J Biol Chem 270:27391–27394. doi: 10.1074/jbc.270.46.27391. [DOI] [PubMed] [Google Scholar]
- 16.Herlaar E, Brown Z. 1999. p38 MAPK signalling cascades in inflammatory disease. Mol Med Today 5:439–447. doi: 10.1016/S1357-4310(99)01544-0. [DOI] [PubMed] [Google Scholar]
- 17.Wysk M, Yang DD, Lu HT, Flavell RA, Davis RJ. 1999. Requirement of mitogen-activated protein kinase kinase 3 (MKK3) for tumor necrosis factor-induced cytokine expression. Proc Natl Acad Sci U S A 96:3763–3768. doi: 10.1073/pnas.96.7.3763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lu HT, Yang DD, Wysk M, Gatti E, Mellman I, Davis RJ, Flavell RA. 1999. Defective IL-12 production in mitogen-activated protein (MAP) kinase kinase 3 (Mkk3)-deficient mice. EMBO J 18:1845–1857. doi: 10.1093/emboj/18.7.1845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tumang MC, Keogh C, Moldawer LL, Helfgott DC, Teitelbaum R, Hariprashad J, Murray HW. 1994. Role and effect of TNF-alpha in experimental visceral leishmaniasis. J Immunol 153:768–775. [PubMed] [Google Scholar]
- 20.Murray HW, Tsai CW, Liu J, Ma X. 2006. Responses to Leishmania donovani in mice deficient in interleukin-12 (IL-12), IL-12/IL-23, or IL-18. Infect Immun 74:4370–4374. doi: 10.1128/IAI.00422-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Davis RJ. 2000. Signal transduction by the JNK group of MAP kinases. Cell 103:239–252. doi: 10.1016/S0092-8674(00)00116-1. [DOI] [PubMed] [Google Scholar]
- 22.Wang C, Deng L, Hong M, Akkaraju GR, Inoue J, Chen ZJ. 2001. TAK1 is a ubiquitin-dependent kinase of MKK and IKK. Nature 412:346–351. doi: 10.1038/35085597. [DOI] [PubMed] [Google Scholar]
- 23.Deng L, Wang C, Spencer E, Yang L, Braun A, You J, Slaughter C, Pickart C, Chen ZJ. 2000. Activation of the IkappaB kinase complex by TRAF6 requires a dimeric ubiquitin-conjugating enzyme complex and a unique polyubiquitin chain. Cell 103:351–361. doi: 10.1016/S0092-8674(00)00126-4. [DOI] [PubMed] [Google Scholar]
- 24.Akira S. 2003. Toll-like receptor signaling. J Biol Chem 278:38105–38108. doi: 10.1074/jbc.R300028200. [DOI] [PubMed] [Google Scholar]
- 25.Kanayama A, Seth RB, Sun L, Ea CK, Hong M, Shaito A, Chiu YH, Deng L, Chen ZJ. 2004. TAB2 and TAB3 activate the NF-kappaB pathway through binding to polyubiquitin chains. Mol Cell 15:535–548. doi: 10.1016/j.molcel.2004.08.008. [DOI] [PubMed] [Google Scholar]
- 26.Jiang Z, Ninomiya-Tsuji J, Qian Y, Matsumoto K, Li X. 2002. Interleukin-1 (IL-1) receptor-associated kinase-dependent IL-1-induced signaling complexes phosphorylate TAK1 and TAB2 at the plasma membrane and activate TAK1 in the cytosol. Mol Cell Biol 22:7158–7167. doi: 10.1128/MCB.22.20.7158-7167.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tuon FF, Amato VS, Bacha HA, Almusawi T, Duarte MI, Amato Neto V. 2008. Toll-like receptors and leishmaniasis. Infect Immun 76:866–872. doi: 10.1128/IAI.01090-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.de Veer MJ, Curtis JM, Baldwin TM, DiDonato JA, Sexton A, McConville MJ, Handman E, Schofield L. 2003. MyD88 is essential for clearance of Leishmania major: possible role for lipophosphoglycan and Toll-like receptor 2 signaling. Eur J Immunol 33:2822–2831. doi: 10.1002/eji.200324128. [DOI] [PubMed] [Google Scholar]
- 29.Kropf P, Freudenberg MA, Modolell M, Price HP, Herath S, Antoniazi S, Galanos C, Smith DF, Muller I. 2004. Toll-like receptor 4 contributes to efficient control of infection with the protozoan parasite Leishmania major. Infect Immun 72:1920–1928. doi: 10.1128/IAI.72.4.1920-1928.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Liese J, Schleicher U, Bogdan C. 2007. TLR9 signaling is essential for the innate NK cell response in murine cutaneous leishmaniasis. Eur J Immunol 37:3424–3434. doi: 10.1002/eji.200737182. [DOI] [PubMed] [Google Scholar]
- 31.Schleicher U, Liese J, Knippertz I, Kurzmann C, Hesse A, Heit A, Fischer JA, Weiss S, Kalinke U, Kunz S, Bogdan C. 2007. NK cell activation in visceral leishmaniasis requires TLR9, myeloid DCs, and IL-12, but is independent of plasmacytoid DCs. J Exp Med 204:893–906. doi: 10.1084/jem.20061293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Srivastav S, Kar S, Chande AG, Mukhopadhyaya R, Das PK. 2012. Leishmania donovani exploits host deubiquitinating enzyme A20, a negative regulator of TLR signaling, to subvert host immune response. J Immunol 189:924–934. doi: 10.4049/jimmunol.1102845. [DOI] [PubMed] [Google Scholar]
- 33.Gupta P, Giri J, Srivastav S, Chande AG, Mukhopadhyaya R, Das PK, Ukil A. 2014. Leishmania donovani targets tumor necrosis factor receptor-associated factor (TRAF) 3 for impairing TLR4-mediated host response. FASEB J 28:1756–1768. doi: 10.1096/fj.13-238428. [DOI] [PubMed] [Google Scholar]
- 34.Flandin JF, Chano F, Descoteaux A. 2006. RNA interference reveals a role for TLR2 and TLR3 in the recognition of Leishmania donovani promastigotes by interferon-gamma-primed macrophages. Eur J Immunol 36:411–420. doi: 10.1002/eji.200535079. [DOI] [PubMed] [Google Scholar]
- 35.Murray HW, Miralles GD, Stoeckle MY, McDermott DF. 1993. Role and effect of IL-2 in experimental visceral leishmaniasis. J Immunol 151:929–938. [PubMed] [Google Scholar]
- 36.Jensen LE, Muzio M, Mantovani A, Whitehead AS. 2000. IL-1 signaling cascade in liver cells and the involvement of a soluble form of the IL-1 receptor accessory protein. J Immunol 164:5277–5286. doi: 10.4049/jimmunol.164.10.5277. [DOI] [PubMed] [Google Scholar]
- 37.Muzio M, Ni J, Feng P, Dixit VM. 1997. IRAK (Pelle) family member IRAK-2 and MyD88 as proximal mediators of IL-1 signaling. Science 278:1612–1615. doi: 10.1126/science.278.5343.1612. [DOI] [PubMed] [Google Scholar]
- 38.Ishida T, Mizushima S, Azuma S, Kobayashi N, Tojo T, Suzuki K, Aizawa S, Watanabe T, Mosialos G, Kieff E, Yamamoto T, Inoue J. 1996. Identification of TRAF6, a novel tumor necrosis factor receptor-associated factor protein that mediates signaling from an amino-terminal domain of the CD40 cytoplasmic region. J Biol Chem 271:28745–28748. doi: 10.1074/jbc.271.46.28745. [DOI] [PubMed] [Google Scholar]
- 39.Shibuya H, Yamaguchi K, Shirakabe K, Tonegawa A, Gotoh Y, Ueno N, Irie K, Nishida E, Matsumoto K. 1996. TAB1: an activator of the TAK1 MAPKKK in TGF-beta signal transduction. Science 272:1179–1182. doi: 10.1126/science.272.5265.1179. [DOI] [PubMed] [Google Scholar]
- 40.Ge B, Gram H, Di Padova F, Huang B, New L, Ulevitch RJ, Luo Y, Han J. 2002. MAPKK-independent activation of p38alpha mediated by TAB1-dependent autophosphorylation of p38alpha. Science 295:1291–1294. doi: 10.1126/science.1067289. [DOI] [PubMed] [Google Scholar]
- 41.Gum RJ, McLaughlin MM, Kumar S, Wang Z, Bower MJ, Lee JC, Adams JL, Livi GP, Goldsmith EJ, Young PR. 1998. Acquisition of sensitivity of stress-activated protein kinases to the p38 inhibitor, SB 203580, by alteration of one or more amino acids within the ATP binding pocket. J Biol Chem 273:15605–15610. doi: 10.1074/jbc.273.25.15605. [DOI] [PubMed] [Google Scholar]
- 42.Yamaguchi K, Shirakabe K, Shibuya H, Irie K, Oishi I, Ueno N, Taniguchi T, Nishida E, Matsumoto K. 1995. Identification of a member of the MAPKKK family as a potential mediator of TGF-beta signal transduction. Science 270:2008–2011. doi: 10.1126/science.270.5244.2008. [DOI] [PubMed] [Google Scholar]
- 43.Takaesu G, Kishida S, Hiyama A, Yamaguchi K, Shibuya H, Irie K, Ninomiya-Tsuji J, Matsumoto K. 2000. TAB2, a novel adaptor protein, mediates activation of TAK1 MAPKKK by linking TAK1 to TRAF6 in the IL-1 signal transduction pathway. Mol Cell 5:649–658. doi: 10.1016/S1097-2765(00)80244-0. [DOI] [PubMed] [Google Scholar]
- 44.Bhattacharjee S, Bhattacharjee A, Majumder S, Majumdar SB, Majumdar S. 2012. Glycyrrhizic acid suppresses Cox-2-mediated anti-inflammatory responses during Leishmania donovani infection. J Antimicrob Chemother 67:1905–1914. doi: 10.1093/jac/dks159. [DOI] [PubMed] [Google Scholar]
- 45.Kim J, Joo I, Kim H, Han Y. 2013. 18beta-glycyrrhetinic acid induces immunological adjuvant activity of Th1 against Candida albicans surface mannan extract. Phytomedicine 20:951–955. doi: 10.1016/j.phymed.2013.04.008. [DOI] [PubMed] [Google Scholar]
- 46.Utsunomiya T, Kobayashi M, Ito M, Pollard RB, Suzuki F. 2000. Glycyrrhizin improves the resistance of MAIDS mice to opportunistic infection of Candida albicans through the modulation of MAIDS-associated type 2 T cell responses. Clin Immunol 95:145–155. doi: 10.1006/clim.2000.4854. [DOI] [PubMed] [Google Scholar]
- 47.Jeong HG, Kim JY. 2002. Induction of inducible nitric oxide synthase expression by 18beta-glycyrrhetinic acid in macrophages. FEBS Lett 513:208–212. doi: 10.1016/S0014-5793(02)02311-6. [DOI] [PubMed] [Google Scholar]
- 48.Dai JH, Iwatani Y, Ishida T, Terunuma H, Kasai H, Iwakula Y, Fujiwara H, Ito M. 2001. Glycyrrhizin enhances interleukin-12 production in peritoneal macrophages. Immunology 103:235–243. doi: 10.1046/j.1365-2567.2001.01224.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kyriakis JM. 1999. Making the connection: coupling of stress-activated ERK/MAPK (extracellular-signal-regulated kinase/mitogen-activated protein kinase) core signalling modules to extracellular stimuli and biological responses. Biochem Soc Symp 64:29–48. [PubMed] [Google Scholar]
- 50.Ono K, Han J. 2000. The p38 signal transduction pathway: activation and function. Cell Signal 12:1–13. doi: 10.1016/S0898-6568(99)00071-6. [DOI] [PubMed] [Google Scholar]
- 51.Paul A, Wilson S, Belham CM, Robinson CJ, Scott PH, Gould GW, Plevin R. 1997. Stress-activated protein kinases: activation, regulation and function. Cell Signal 9:403–410. doi: 10.1016/S0898-6568(97)00042-9. [DOI] [PubMed] [Google Scholar]
- 52.Junghae M, Raynes JG. 2002. Activation of p38 mitogen-activated protein kinase attenuates Leishmania donovani infection in macrophages. Infect Immun 70:5026–5035. doi: 10.1128/IAI.70.9.5026-5035.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Mookerjee Basu J, Mookerjee A, Sen P, Bhaumik S, Banerjee S, Naskar K, Choudhuri SK, Saha B, Raha S, Roy S. 2006. Sodium antimony gluconate induces generation of reactive oxygen species and nitric oxide via phosphoinositide 3-kinase and mitogen-activated protein kinase activation in Leishmania donovani-infected macrophages. Antimicrob Agents Chemother 50:1788–1797. doi: 10.1128/AAC.50.5.1788-1797.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Inoue T, Boyle DL, Corr M, Hammaker D, Davis RJ, Flavell RA, Firestein GS. 2006. Mitogen-activated protein kinase kinase 3 is a pivotal pathway regulating p38 activation in inflammatory arthritis. Proc Natl Acad Sci U S A 103:5484–5489. doi: 10.1073/pnas.0509188103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Brancho D, Tanaka N, Jaeschke A, Ventura JJ, Kelkar N, Tanaka Y, Kyuuma M, Takeshita T, Flavell RA, Davis RJ. 2003. Mechanism of p38 MAP kinase activation in vivo. Genes Dev 17:1969–1978. doi: 10.1101/gad.1107303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Li J, Miller EJ, Ninomiya-Tsuji J, Russell RR III, Young LH. 2005. AMP-activated protein kinase activates p38 mitogen-activated protein kinase by increasing recruitment of p38 MAPK to TAB1 in the ischemic heart. Circ Res 97:872–879. doi: 10.1161/01.RES.0000187458.77026.10. [DOI] [PubMed] [Google Scholar]
- 57.Ohkusu-Tsukada K, Tominaga N, Udono H, Yui K. 2004. Regulation of the maintenance of peripheral T-cell anergy by TAB1-mediated p38 alpha activation. Mol Cell Biol 24:6957–6966. doi: 10.1128/MCB.24.16.6957-6966.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ge B, Xiong X, Jing Q, Mosley JL, Filose A, Bian D, Huang S, Han J. 2003. TAB1beta (transforming growth factor-beta-activated protein kinase 1-binding protein 1beta), a novel splicing variant of TAB1 that interacts with p38alpha but not TAK1. J Biol Chem 278:2286–2293. doi: 10.1074/jbc.M210918200. [DOI] [PubMed] [Google Scholar]
- 59.Kim L, Del Rio L, Butcher BA, Mogensen TH, Paludan SR, Flavell RA, Denkers EY. 2005. p38 MAPK autophosphorylation drives macrophage IL-12 production during intracellular infection. J Immunol 174:4178–4184. doi: 10.4049/jimmunol.174.7.4178. [DOI] [PubMed] [Google Scholar]
- 60.Peng LN, Li L, Qiu YF, Miao JH, Gao XQ, Zhou Y, Shi ZX, Xu YL, Shao DH, Wei JC, Ma ZY. 2011. Glycyrrhetinic acid extracted from Glycyrrhiza uralensis Fisch. induces the expression of Toll-like receptor 4 in Ana-1 murine macrophages J Asian Nat Prod Res 13:942–950. [DOI] [PubMed] [Google Scholar]