

Abstract
Increasing evidence demonstrates induction of proinflammatory Toll-like receptor (TLR) 2 and TLR4 signaling by morphine and, TLR4 signaling by alcohol; thus indicating a common site of drug action and a potential novel innate immune-dependent hypothesis for opioid and alcohol drug interactions. Hence, the current study aimed to assess the role of TLR2, TLR4, MyD88 (as a critical TLR-signalling participant), NF-κB, Interleukin-1β (IL-1β; as a downstream proinflammatory effector molecule) and the µ opioid receptor (MOR; as a classical site for morphine action) in acute alcohol-induced sedation (4.5 g/kg) and alcohol (2.5 g/kg) interaction with morphine (5 mg/kg) by assessing the loss of righting reflex (LORR) as a measure of sedation. Wild-type male Balb/c mice and matched genetically-deficient TLR2, TLR4, and MyD88 strains were utilized, together with pharmacological manipulation of MOR, NF-κB, TLR4 and Interleukin-1β. Alcohol induced significant LORR in wild-type mice; this was halved by MyD88 and TLR4 deficiency, and surprisingly nearly completely eliminated by TLR2 deficiency. In contrast, the interaction between morphine and alcohol was found to be MOR-, NF-κB-, TLR2- and MyD88-dependent, but did not involve TLR4 or Interleukin-1β. Morphine-alcohol interactions caused acute elevations in microglial cell counts and NF-κB-p65 positive cells in the motor cortex in concordance with wild-type and TLR2 deficient mouse behavioral data, implicating neuroimmunopharmacological signaling as a pivotal mechanism in this clinically problematic drug-drug interaction.
Keywords: Drug Interactions, Toll Like Receptors, Myeloid Differentiation Factor 88, Cytokines, Naloxone, Drug overdose
Introduction
Alcohol increases the risk of heroin-related deaths (Hickman et al., 2008; Levine et al., 1995), not due to any pharmacokinetic interaction (Darke et al., 1997; Fugelstad et al., 2003), but due to a pharmacodynamic interaction. Knowledge of the mechanism of such a synergistic interaction between alcohol and opioids in the central nervous system (CNS) is surprisingly limited (White and Irvine, 1999), but is generally considered to be alcohol-induced potentiation of sedation by morphine. The molecular mechanisms implicated in the sedative effects of alcohol include neuronal gamma-aminobutyric acid (GABA) receptors, the N-methyl-D-aspartate (NMDA) receptor, and cyclic adenosine monophosphate-protein kinase A (cAMP-PKA) signalling (Boyce-Rustay and Holmes, 2005; Sharko and Hodge, 2008; Wand et al., 2001). In contrast, morphine can rapidly and differentially modulate the binding affinity of GABA for its receptors in various brain regions (Sivam et al., 1982; Ticku and Huffman, 1980), or alternatively directly cause sedation (Craft and Leitl, 2006; Kissin et al., 1991) due to activation of the mu (µ) opioid receptor (MOR) (Craft and Leitl, 2006; Kissin et al., 1991). However, other non-neuronal mechanisms of alcohol- and opioid-induced sedation may be involved.
Recent appreciation of the non-neuronal CNS actions of opioids and alcohol has led to the discovery of xenobiotic-induced proinflammatory reactivity of glia, through Toll-like receptors (TLRs) and myeloid differentiation primary response gene 88 (MyD88)-dependent pathways. Opioid-induced TLR4 activation was first identified as a modifier of opioid analgesia (Hutchinson et al., 2008a; Hutchinson et al., 2010b; Wang et al., 2012) and reward (Hutchinson et al., 2012), with TLR2 subsequently shown to play a similar role (Li et al., 2010; Li et al., 2009; Zhang et al., 2011). An intriguing parallel research track developed for alcohol where alcohol-induced sedation and motor dysfunction occurred in a TLR4-, MyD88-, and IL-1β-dependent fashion (Wu et al., 2012; Wu et al., 2011). Therefore, this common TLR-dependent neuroimmune convergence by alcohol and opioids may shed light on a new mechanism that contributes to the pharmacodynamic interaction between alcohol and opioids.
Hence, we hypothesized that: a) TLR2 is involved in the sedative effects of alcohol; and b) that TLR2, TLR4, MyD88, NF-κB, Interleukin-1 receptor, and MOR signaling are collectively involved in the synergistic sedative effects induced by alcohol plus morphine.
Thus, the aim of this study was, firstly, to examine if mice genetically deficient in TLR2 had decreased sedation to alcohol alone, since such an effect was found in all the other TLR-related genetic deficient mouse strains in our previous experiments (Wu et al., 2012). Secondly, we aimed to characterize the effect of genetic deficiency of TLR2, TLR4 or MyD88 on the interaction between alcohol and morphine. Thirdly, we aimed to determine the effect of (+)-naloxone (the MOR-inactive isomer but TLR4 signaling inhibitor (Hutchinson et al., 2010a; Hutchinson et al., 2008b)), (−)-naloxone (the MOR antagonist and TLR4 signaling inhibitor), IL-1ra (Interleukin-1 receptor antagonist) and IKK-NBD (a NF-κB inhibitor (Ghosh et al., 2007a)) on combined alcohol- and morphine-induced sedation. Examination of the impact of these genetic and pharmacological manipulations on acute microglial response within the motor cortex was also examined to provide a mechanistic cellular link to the altered behavioral responses.
Materials and Methods
Animals
Pathogen-free male Balb/c wild-type mice, and mice with null mutations in Tlr2 (Tlr2−/−), Tlr4 (Tlr4−/−), and Myd88 (Myd88−/−) (all 8–14 weeks old; weighing 18–26 g; n = 4–8) were used in the experiments. These genetically-deficient mice, back crossed onto Balb/c for more than ten generations, were sourced from Prof. Akira (Osaka University, Osaka, Japan), and the Tlr4, and Myd88 null mutant mice were kindly supplied by Dr. Simon Phipps (University of Queensland, QLD, Australia) and Prof. Paul Foster (University of Newcastle, NSW, Australia). Mice were housed under SPF conditions in temperature (23 ± 3 °C) and light/dark cycle (12/12 h) controlled rooms with standard rodent food and water available ad libitum. All animal studies were approved by the University of Adelaide Animal Ethics Committee. Every effort was taken to Replace, Refine and Reduce animal usage in the design and conduct of these experiments.
Drugs
Human IL-1ra (Kineret®, Amgen Inc, Thousand Oaks, CA, USA) was purchased from the Queen Elizabeth Hospital Pharmacy (Woodville, SA, Australia). (+)-Naloxone was supplied by Dr. Kenner Rice (Chemical Biology Research Branch, National Institute on Drug Abuse and National Institute on Alcohol Abuse and Alcoholism, National Institutes of Health, Rockville, MD, USA). (−)-Naloxone was purchased from Sigma (St. Louis, MO, USA). Morphine was obtained from Fauldings Australia (Adelaide, SA, Australia). Alcohol (ethanol) was obtained from Chem-Supply (99.5%; Gillman, SA, Australia). Sodium chloride was obtained from Merck (99.5%, Darmstadt, Germany). The IKK-NBD peptide was obtained from Enzo LifeSciences (Exeter, United Kingdom).
Morphine (5 mg/kg), (+)-naloxone (60 mg/kg) (Wu et al., 2012), (−)-naloxone (60 mg/kg), IL-1ra (100 mg/kg) (Wu et al., 2012) and IKK-NBD (0.75 mg/kg) (Ghosh et al., 2007a) were injected intraperitoneally (i.p.) at 0.1 mL/kg. Alcohol was injected at a dose of 4.5 g/kg and 2.5 g/kg to assess the sedative effect induced by alcohol alone and the co-administration of alcohol and morphine, respectively. The volume for injection of alcohol (20%, v/v, i.p.) varied as it was based on animal weights and was 25 g on average, (range: 22–30 g); thus the volume for injection of alcohol was 0.40 mL (range: 0.35–0.48 mL) at 2.5 g/kg and 0.71 mL (range: 0.63–0.86 mL) at 4.5 g/kg of alcohol. Saline (0.9% sodium chloride) was used as the vehicle control.
Measurement of the sedative effect of drugs (the LORR test)
After drug treatment, mice were placed into separate cages with bedding. The duration of LORR was measured as the time from LORR to the time of righting themselves 3 times in 30 s (waking up) (Wu et al., 2012; Wu et al., 2011). Animals were continually monitored for LORR. This behavioral response is considered a measure of the sedation induced by the treatments.
Experimental procedures
A. Acute Dose of Alcohol Alone on LORR
To determine LORR induced by alcohol alone in wild-type, Tlr2−/−, Tlr4−/− and MyD88−/− mice, saline was administered at −30 min followed by alcohol (4.5 g/kg) at 0 min. Assessment of the impact of pharmacological TLR4 blockade on alcohol-induced LORR in these four mice strains was conducted by administering (+)-naloxone (60 mg/kg i.p.) at −30 min followed by the alcohol at 0 min with LORR monitored until righting reflex was recovered.
B. Alcohol and morphine-induced LORR: Pharmacological antagonist assessment
To examine in wild-type mice the effects of acute morphine on alcohol-induced LORR, mice were injected with morphine (5 mg/kg, −35 min) then saline at −30 min then alcohol (2.5 g/kg) or saline at 0 min. The reduction in alcohol dosage from the acute alcohol alone study was to accommodate for the synergistic effects of alcohol-morphine administration and thus avoid a ceiling pharmacological effect and prevent overdose deaths in these mice.
To assess the influence of IL-1ra, (+)-naloxone, (−)-naloxone and IKK-NBD on alcohol-induced LORR following morphine co-administration, wild-type mice were injected with morphine (−35 min), followed by IL-1ra, (+)-naloxone, (−)-naloxone or IKK-NBD administration (−30 min). Alcohol (2.5 g/kg) being administered at 0 min.
C. Alcohol and morphine-induced LORR: Genetic knockout assessment
To examine the effects of morphine on alcohol-induced sedation across different genetically deficient mouse strains, Tlr2−/−, Tlr4−/− and Myd88−/− mice were dosed with morphine (5 mg/kg, −35 min) then saline (−30 min) and alcohol (2.5 g/kg) at 0 min. These data were compared to their wild-type counterpart data from study B.
D. Immunohistochemical assessment of the molecular alterations induced by alcohol and morphine
To further elucidate the cellular mechanisms underlying the behavioral observations presented here, immunohistochemistry of Iba1 and NF-κB expression in the motor cortex 30 min after alcohol exposure was examined in wild-type, Tlr2−/− and Tlr4−/− mice. Motor cortex was chosen as a region of interest owing to these and other studies examining alcohol-induced impairment of gross motor function (see Figure 1). The same morphine and alcohol dosing protocol that induced LORR in studies B and C was employed. Thirty minutes after alcohol administration, animals (n = 3 group, [morphine vs saline] by [alcohol vs saline]) were humanely sacrificed and transcardially perfused with 10% formalin. Brains were sectioned coronally (2 mm) using a mouse brain blocker (Kopf, Tujunga, CA, USA) and embedded in paraffin wax. Using a microtome (Microm, Walldorf, Germany), 5 µm sections were cut from each brain at Bregma −1.5 to allow examination of the motor cortex. Three sections per animal were labelled with either rabbit anti-NF-κB-p65 (1:1000, Santa Cruz, Santa Cruz, CA, USA) or goat anti-ionised calcium-binding adaptor molecule-1 (Iba1) (1:1000, Sigma, St Louis, Missouri, USA). Following overnight incubation, slides were then incubated with the appropriate secondary antibody (Dako 1:250) for 30 min followed by incubation with SPC (1:1000) for 1 h. For negative controls primary antibodies were omitted. Bound antibody was then detected with 3,3-diaminobenzidine tetrahydrochloride (DAB) in the presence of hydrogen peroxide and sections counterstained with haematoxylin. Slides were digitally scanned (Nanozoomer, Hanamatsu) and viewed with the associated software (NDP view). Four serial 40x images were taken from each section and digitally reconstructed into a montage to allow manual counting of Iba1 and NFκB-p65 positive cells using the cell count software associated with ImageJ. Morphology of microglial cells was also evaluated qualitatively, and representative images of typical appearance collated.
Figure 1.

Representative image of the region of interest for immunohistochemical cell counts. Scale bar = 1mm
To determine differences in the intensity of NF-κB-p65 staining, images were processed using Photoshop, converted to grayscale and the intensity of staining determined. All NF-κB-p65 positive cells within the motor cortex of the section were analysed for integrated density with background staining then subtracted and an average of each of these cells then taken for each animal, to negate the effects of variability of number of NF-κB positive cells between groups. Backgrounds were defined as the intensity of grey in areas without cells. Investigators who were blinded to treatment assignments conducted all quantifications.
Statistical analysis
Analysis of the alcohol-induced LORR, morphine-alcohol-induced LORR, Iba1 positive cells, NF-κB-p65 and the average integrated density of NF-κB-p65 per positive cell data were conducted in RStudio (Version 0.97.551; R version 3.0.1 (Team, 2013)). Linear models of the multi-way relationships were created in RStudio. Post hoc contrasts were conducted using the multcomp R package and the general linear hypothesis (glht) analysis which allows for simultaneous tests for general linear hypotheses, and multiple comparisons of means with Tukey contrasts and single-step adjusted p-values (Hothorn et al., 2008). GraphPad Prism 6.0.1 (GraphPad Software Inc., San Diego, CA, USA) was used for graphing. P-values (adjusted for repeated sampling where appropriate) of 0.05 or less were considered statistically significant. Graphically, all data are presented as mean ± SEM.
Results
A. High dose alcohol LORR: TLR2 and TLR4 signaling contribute to high dose alcohol-induced LORR
Alcohol (4.5 g/kg) produced profound loss of righting reflex in wild-type animals with statistically significant main effects of strain (F3,46 = 17.0; P < 0.0001), and a strain:(+)-naloxone interaction (F3,47 = 10.7; P < 0.0001) on alcohol-induced LORR time (Figure 2A). As expected mice given saline at −30 min and saline at 0 min did not lose righting reflex (data not shown). Post hoc analysis revealed significant reductions in alcohol-induced LORR in saline+alcohol treated Tlr2−/− (mean = 88%), Tlr4−/− (55%) and Myd88−/− (46%) animals when compared with saline+alcohol treated wild-type mice (P < 0.001; Figure 2A). Alcohol-induced LORR in Tlr2−/− mice was reduced significantly greater compared to Tlr4−/− and Myd88−/− mice (P < 0.02, Figure 2A). Pharmacological TLR4 blockade with (+)-naloxone significantly reduced alcohol-induced LORR (45%) in wild-type animals (P = 0.002), but failed to alter the alcohol-induced LORR in Tlr4−/− and Myd88−/− animals (P > 0.74). However, (+)-naloxone treatment in Tlr2−/− mice caused a significant increase (P = 0.036) in alcohol-induced LORR to times not statistically significantly different to saline treated Tlr4−/− or Myd88−/− animals (P > 0.97; Figure 2A).
Figure 2.

(A) Decreased duration of loss of righting reflex (LORR) induced by alcohol (4.5 g/kg) in Tlr2−/− Tlr4−/− and Myd88−/− mice compared to wild-type mice and the protective effect of (+)-naloxone (60 mg/kg) (n = 6 – 8). The effects of treatments with IL-1ra, (+)-naloxone, or (−)-naloxone and the genetic deficiency of innate immune genes in TLR signaling cascades on the increased duration of LORR interaction between morphine and alcohol. (B) In wild-type mice, (−)-naloxone and NK-κB inhibitor IKK-NBD blocked the potentiated duration of LORR effect of alcohol by morphine. (C) In Tlr2−/− and Myd88−/− mice, the enhancement of alcohol-induced sedation by morphine was significantly decreased, but in Tlr4−/− mice the LORR response was enhanced (n = 4 – 6). Data are presented as mean ± SEM. * P < 0.05; ** P < 0.01; *** P < 0.001.
B. Morphine:Alcohol synergistic interactions: Potentiation of the duration of alcohol-induced LORR by morphine is inhibited by (−)-naloxone and IKK-NBD in wild-type mice
A low dose of morphine (5 mg/kg) in wild-type mice significantly increased the duration of alcohol-induced LORR when compared to alcohol alone by 313% (t = 3.7; P = 0.01) or morphine alone (t = 4.9; P < 0.001) (Figure 2B) (multi-way ANOVA with post hoc analysis). This synergistic effect of morphine on alcohol-induced LORR was blocked by MOR antagonism using (−)-naloxone (t = 4.9; P < 0.001) and by NF-κB inhibition (IKK-NBD; t = 3.4; P = 0.02) but not by pharmacological TLR4 blockade with (+)-naloxone (t = 0.38; P = 0.99) or with the recombinant human IL-1ra (t = 0.61; P = 0.99) (Figure 2B).
C. Morphine:Alcohol synergistic interactions: Key roles of TLR2 and MyD88 signaling in synergistic Morphine+Alcohol induced LORR
To further examine the influence of innate immune receptor signaling on morphine+alcohol interactions, strains of mice deficient in TLR2, TLR4 and MyD88 were administered morphine or saline followed by alcohol. Multi-way ANOVA with post hoc analysis of alcohol- and morphine-alcohol-induced LORR revealed that morphine significantly potentiated alcohol-induced LORR in wild-type mice (t = 5.4, P < 0.001) and Tlr4−/− mice (P < 0.019) but not in Tlr2−/− or Myd88−/− mice (P < 0.001; Figure 2C).
D. Morphine:Alcohol synergistic interactions: Morphine+Alcohol acutely increase Iba1 microglial counts in the motor cortex
Morphine and alcohol treatments caused significant alterations in Iba1 microglia counts in the motor cortex, that were dependent on strain (F2,24 = 4.0; P = 0.03) and morphine-alcohol interactions (F1,24 = 5.3; P = 0.03; multi-way ANOVA [alcohol by morphine by strain]) (Figure 3A). Morphine+alcohol caused significant increases in wild-type Iba1 cell counts compared to saline+alcohol (t = 3.7, P = 0.04) and saline+saline (t = 3.9, P = 0.03) treated wild-type; and compared to morphine+alcohol treated Tlr2−/− animals (t = 3.7, P = 0.04) (Figure 3C). In addition to microglial number, microglial morphology was evaluated, with the typical appearance of microglia within layer 5 of the motor cortex (Figure 4). Morphine+alcohol treatment in WT and Tlr4−/− mice led to a typical `bushy’ appearance of the microglia suggestive of heightened reactivity, which was not seen in the Tlr2−/− mice, or in mice treated with morphine or alcohol alone (Figure 4).
Figure 3.

Representative images of Iba1 (A) and NF-κB-p65 (B) staining in layer 5 of the motor cortex taken at 30 min following pharmacological treatments in each of the wild-type and innate immune genetic deficient mouse strains. (Images are representative of n = 3 – 4 per group) (Scale bar = 500 µm (A) or 100µm (B)). Immunohistochemical analysis of sections of the motor cortex and: (C) quantitative counts of the number of Iba1 positive cells expressed as cells/mm2; (D) quantitative counts of the number of NF-κB-p65 positive cells expressed as cells/mm2; and (E) calculation of the average integrated density of NF-κB-p65 positive cells. (n = 3 per group). * P < 0.05; ** P < 0.01; *** P < 0.001.
Figure 4.
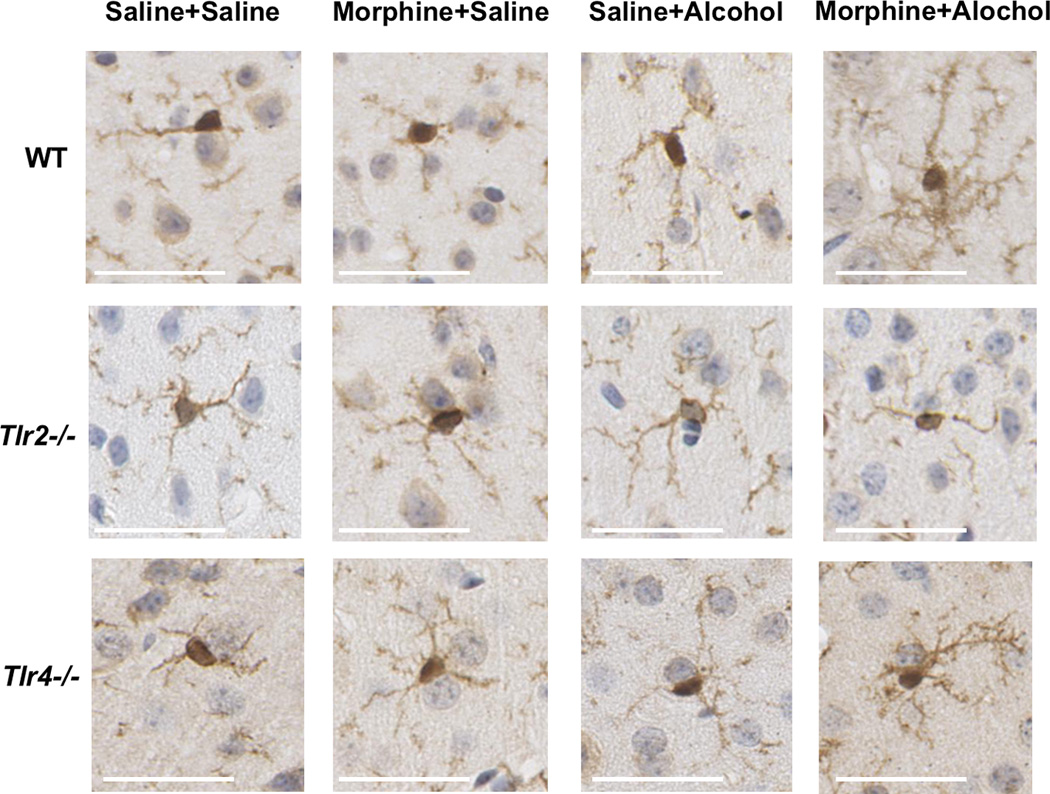
Phenotypic changes of Iba1 positive microglia in layer 5 of the motor cortex taken at 30 min following pharmacological treatments in each of the wild-type and innate immune genetic deficient mouse strains (scale bar = 10µm).
E. Morphine:Alcohol synergistic interactions: Alcohol +Morphine acutely increases NF-κB-p65 in Iba1 microglia in the motor cortex
Activation of NF-κB-p65 signaling in the motor cortex in wild-type, Tlr2−/− and Tlr4−/− mice is shown in Figure 3B. Once again, morphine and alcohol treatments caused significant changes in NF-κB-p65 cell counts that were dependent on drug (morphine [F1,24 = 6.6, P = 0.02], alcohol [F1,24 = 4.8, P = 0.04]) and drug strain interactions (morphine:strain [F2,24 = 6.6, P = 0.005] and alcohol:strain [F2,24 = 4.0, P = 0.03]). Morphine+alcohol caused significant increases in wild-type in NF-κB-p65 positive cell counts compared to wild-type saline+alcohol (t = 4.0, P = 0.02) and saline+saline treated wild-type mice (t = 5.6, P < 0.001) (Figure 3D). Moreover, this occurred in a TLR2- and TLR4-dependent fashion as no significant changes in NF-κB-p65 positive cells were observed between saline+saline and morphine+alcohol treated Tlr2−/− (t = 0.5, P = 1.0) or Tlr4−/− mice (t = 0.8, P = 1.0; Figure 3D). Interestingly, significantly lower cell counts were seen when comparing morphine+alcohol between wild-type and Tlr2−/− (t = 3.7, P = 0.04) and Tlr4−/− (t = 3.8, P = 0.03) animals.
Further examination of the integrated density of the NF-κB-p65 positive cells demonstrated that morphine (F1,24 = 5.0, P = 0.04), strain (F2,24 = 36.6, P < 0.0001), and an alcohol:strain (F2,24 = 8.2, P = 0.002) interactions significantly influenced the straining (Figure 3E). There was a significant increase in the integrated density of staining induced by morphine+alcohol in wild-type animals compared to saline+saline treatments (t = 5.2, P < 0.01), a response not observed in Tlr2−/− (t = 0.5, P = 1.0) or Tlr4−/− (t = 0.7, P = 1.0) mice. Tlr2−/− mice displayed significantly reduced NF-κB-p65 staining density under all four treatments compared with wild-type (morphine+saline (t = 6.2, P = 0.01); saline+alcohol (t = 5.2, P = 0.01) and Tlr4−/− mice (morphine+alcohol [t = 3.9, P = 0.02]; saline+saline [t = 4.6, P < 0.01]; Figure 3E).
Discussion
Here we show that TLRs and their associated signaling pathways are novel contributors to acute alcohol- and morphine+alcohol-induced cellular and behavioral responses. Firstly, a novel role for TLR2 was established in the acute sedative effects of alcohol, with near complete protection from alcohol-induced LORR in Tlr2−/− mice. However, an intriguing and complex TLR2-TLR4 interaction was uncovered as the Tlr2−/− mice LORR protection was halved by combining pharmacological TLR4 blockade in Tlr2−/− mice. Secondly, a key role for innate immune TLR signaling in the synergistic drug-drug interaction elicited by combining morphine and alcohol was also established. Specifically, TLR2-MyD88-dependent NF-κB signaling was found to contribute to the synergistic drug-drug interaction of alcohol- and morphine-induced LORR, as demonstrated pharmacologically and through immunohistochemical staining. Finally the role of microglia were assessed in wild-type and TLR deficient strains, with an increase in microglial number associated with a change in morphology suggestive of activation in morphine+alcohol treated wild-type mice, that was not seen in the Tlr2−/− mice. However, discordant results were observed in Tlr4−/− mice, which did not have an increase in microglial number, although microglia appeared to adopt an activated phenotype, despite a behavioural response to morphine+alcohol administration (Table 2 results summary). Nonetheless, these results uncover a new potential non-neuronal cellular target of drug-drug interactions and reaffirm our previous hypothesis that xenobiotics such as morphine and alcohol (or their metabolites (Lewis et al., 2010; Lewis et al., 2013), act as xenobiotic-associated molecular patterns (XAMPs) to activate TLRs causing altered pharmacodynamic responses (Hutchinson et al., 2012).
Table 2.
Summary of the main findings from this study investigating the effects of alcohol and morphine administration and the current study.
| Morphine+Alcohol LORR |
Iba1 count |
Iba1 Morphology |
NF-κB count |
NF-κb Intensity |
|
|---|---|---|---|---|---|
| Wild-type | ++++ | M+A ++++ | M+A Activated | M+A ++++ | M+A ++ |
| Tlr2−/− | −−− | −−− | M+A Resting | −−− | −−− |
| Tlr4−/− | ++++++ | −−− | M+A Activated | −−− | −−−* |
| Myd88−/− | −−− | ||||
| IL-1ra | ++++ | ||||
| (+)-Naloxone | ++++ | ||||
| (−)-Naloxone | −−− | ||||
| NF-κB-NBD | ++ |
LORR = Loss of righting reflex. M = morphine, A= Alcohol. Number of plus signs signifies approximate increase in the magnitude of response, whilst the minus signs signify blockade of the response.
Elevated levels of staining intensity basally compared to other groups, but no effect of drug treatment.
Firstly, this study has demonstrated that the acute neurological effects of alcohol are dependent on both TLR4 and TLR2 signaling. This builds on our previous work indicating that treatment with IL-1ra and minocycline reduced alcohol-induced sedation in mice, as measured by the LORR (Wu et al., 2012; Wu et al., 2011). As both the TLR2 and TLR4 signalling pathways leads to the production of IL-1β (Wu et al., 2012; Wu et al., 2011), it can be hypothesized that activation of TLR2 and TLR4 signalling pathways, promoting IL-1β production by microglia, are key participants in the pharmacodynamic actions of alcohol. Alcohol has been shown to recruit TLR2 and TLR4 to lipid rafts in microglial cells, with an associated release of pro-inflammatory cytokines (Fernandez-Lizarbe et al., 2013) TLR4 signalling has previously been linked to other behavioural changes following alcohol exposure including acute changes in motor function (Wu et al., 2012) and development of conditional learning and memory deficits in the object recognition task following chronic alcohol consumption (Pascual et al., 2011).
It is intriguing that Tlr2−/− mice display significantly less acute alcohol-induced LORR than the other strains tested, indicating TLR2 is a vital participant in this acute alcohol pharmacodynamic response. However, complex TLR2/4 and MyD88 interactions are also evident in this acute alcohol response, as pharmacological TLR4 blockade with (+)-naloxone in Tlr2−/− mice increases their LORR, although this still remains less than that in untreated wild-type mice. This complexity is further emphasized by the increase in LORR in response to alcohol in Myd88−/− mice compared to Tlr2−/− mice, despite TLR2 signalling being MyD88 dependent.
This highlights the underlying complexity of the relationship between alcohol and the innate immune system. Complex interactions in innate immune signaling have been hypothesized recently (Nish and Medzhitov, 2011), with opportunities for cooperation, complementation and compensation in the signaling cascade. Indeed, it has been demonstrated that ethanol promotes interaction between TLR4 and TLR2 following their recruitment into lipid rafts which act to stabilise TLRs and form signalling platforms (Fernandez-Lizarbe et al., 2013). Our results suggest that in the genetic absence of TLR2, TLR4 signaling by alcohol is ineffective, whilst in the absence of TLR4 (genetic or pharmacological), TLR2 signaling can continue to produce some, but reduced acute alcohol effects. However, in the case of the Myd88−/− or Tlr2−/− mice given (+)-naloxone, we are presented with a unique situation where the action of a third unknown signaling factor, biased signaling or an acutely activated compensatory mechanism appears to be contributing to the alcohol response. Further investigations of these acute proinflammatory TLR-dependent contributions to the acute actions of alcohol are warranted, as how these signaling interactions occur and the mechanism(s) remains to be determined. Moreover, the developmental consequences of the genetic absence of innate immune genes needs to be considered as contributing to the different responses observed here.
Nonetheless, having demonstrated that the behavioural response to alcohol is immune dependent, we sought to determine whether innate immune signalling was promoting the interaction between alcohol and morphine. We were able to demonstrate that this interaction appears to be dependent on MOR, TLR2 and MyD88-NFκB-signaling, as Tlr2−/− and Myd88−/− mice and MOR or IKK-NBD blockade protects against morphine+alcohol LORR. In contrast targeting IL-1β and TLR4 are without effect (see Table 2 for a summary).
To address the possible underlying cellular mechanisms at play in morphine+alcohol LORR, alterations in microglial cell number (Iba1 positive cells) and NF-κB-p65 expression were examined in the motor cortex 30 min after drug administration. In wild-type mice significant morphine+alcohol-induced increases in Iba1 positive microglia were observed in the motor cortex. This rapid and substantial elevation in Iba1 positive cells may reflect both recruitment of Iba1 positive cells as well as possible elevations in Iba1 expression by resident populations. However, this is a very immediate effect and will require further research to elucidate the precise mechanism.
The importance of NF-κB signalling in this interaction was also investigated, both pharmacologically through the administration of the IKK-NBD peptide, and immunohistochemically through evaluation of NF-κB-p65 staining. Treatment with the IKK-NBD peptide significantly reduced LORR. It should be noted that previous studies utilizing IP injection of the NDD-IKK peptide demonstrate that it is able to enter into the brain where it inhibits activation of NF-κB signaling (Ghosh et al., 2007b). The importance of this component of the signaling cascade was highlighted by the NF-κB-p65 changes seen in wild-type mice, which corresponded to a synergistic increase in LORR in wild-type animals. Moreover, in Tlr2−/− mice, where there was complete blockade of LORR, no such elevations in microglial Iba1 cell number, NF-κB-p65 positive cells or staining intensity occurred, thus agreeing with the behavioral responses, compared to wild-type mice. However, in Tlr4−/− mice that displayed a significant morphine+alcohol response there was no change in the number of NF-κB-p65 positive cells nor staining intensity, although the latter may have been masked by an elevation in basal NF-κB-p65 staining intensity.
Thus, whilst a simple NF-κB-p65 activation can potentially explain the synergistic morphine+alcohol interaction in wild-type versus Tlr2−/− mice, the same cannot be said for the Tlr4−/− strain. This suggests that non-immune signaling may play a greater role in Tlr4−/− mice than in wild-type mice. This may relate to activation of MOR, as (−)-naloxone was found to be effective at reducing LORR in WT mice or it is possible that the genetic deletion of TLR4 has allowed other novel pathways to become involved in the promotion of LORR in response to morphine+alcohol, which would require further investigation. Nevertheless, in contrast to the effects of morphine or alcohol alone involving both TLR2 and TLR4, the current results suggest that the co-administration of morphine and alcohol in wild-type mice elicits the sedative effect via the TLR2-MyD88-NF-κB signaling cascade, highlighting a new XAMP-induced innate immune signal that alters drug responses.
The exact mechanisms behind how morphine+alcohol act synergistically via activation of TLR2 to promote sedation is yet to be fully elucidated. In glial cells the net result of the TLR2 signaling cascades is Myd88 dependent NF-κB activation leading to the robust and transient transcription of a myriad of pro-inflammatory factors including cytokines (TNF-α, IL-6), chemokines and immune receptors, promoting glial reactivity (Kumar et al., 2009). Apart from cytokines, reactive glial cells also release a variety of other substances such as cytokines, prostaglandins, brain-derived neurotrophic factor, adenosine triphosphate (ATP) and glutamate (Kettenmann et al., 2013), which can all modulate neuronal activity, although the mechanisms remain poorly understood. Of note inhibitory GABA neurotransmission is enhanced by the chemokine (C-X-C motif) ligand 12 (CXCL12) (Heinisch and Kirby, 2010) and IL-6 (De Laurentiis et al., 2000), providing a potential mechanism for enhanced neuronal inhibition. The prior exposure to morphine may prime microglia to have an exaggerated proinflammatory cytokine response to the subsequent effects of alcohol, as seen by the appearance of activated microglia and upregulation of NF-κB. This leads to an enhanced release of cytokines and chemokines from microglia, potentially exaggerating the behavioral response. Several mechanisms have been proposed as the basis for this exaggerated response by primed microglia including enhanced NF-κB activation and chromatin remodelling, where histone acetylation and phosphorylation make cytokine gene promoters more readily accessible (Pestka and Zhou, 2006).
It should also be noted that apart from modulating neuronal signaling via microglial cross-talk, stimulation of TLRs on neurons themselves may also directly affect neuronal function. An increase in TLR4 expression has been noted within hippocampal neurons in a model of kainate induced seizures, with TLR4 blockade reducing seizure intensity, suggesting a role in modulating excitatory neurotransmission (Maroso et al., 2010). Activation of MOR also plays an important role in the synergistic effects of morphine+alcohol, given the reversal of LORR following treatment with (–)-naloxone. Both alcohol and morphine interact with MOR, with the acute anxiolytic and locomotor effects of alcohol prevented in µ-opioid receptor knockout mice (Ghozland et al., 2005), whilst morphine induced sedation has been linked to activation of the MOR (Craft et al, 2006; Kissen et al, 1991). This suggests that there is complex interplay of microglia-neuronal crosstalk that can be triggered by activation of both TLR2 and MOR signaling pathways promoted by release of a host of factors including cytokines, chemokines and neurotransmitters that ultimately leads to the change of behavior noted in these mice.
In conclusion, it is apparent that TLR2-MyD88-NF-κB signaling is important in the acute sedative effects of alcohol and the interaction between alcohol and morphine in mice. Given the novel XAMP sensitivity of, at minimum the TLR2 pathway, this may prove crucial in explaining novel xenobiotic-alcohol synergistic interactions. As such, a unique opportunity has presented itself whereby novel pharmacological targeting of the TLR2-MyD88-NF-κB signaling pathway may beneficially avoid these predominantly unwanted synergistic drug-drug interactions.
Table 1.
Summary of the main findings from past research (as indicated by #) (Wu et al., 2012; Wu et al., 2011) and the current study investigating the acute neurological effects of alcohol alone.
| Alcohol LORR |
|
|---|---|
| Wild-type | ++++ |
| Tlr2−/− | −−− |
| Tlr4−/− | ++ |
| Myd88−/− | ++ |
| IL-1ra# | ++ |
| (+)-Naloxone | ++ |
| Minocycline# | ++ |
LORR = Loss of righting reflex. M = morphine, A= Alcohol. Number of plus signs signifies approximate increase in the magnitude of response, whilst the minus signs signify blockade of the response.
Acknowledgements
Yue Wu was the recipient of China Scholarship Council (CSC)-University of Adelaide Joint Postgraduate Scholarship. Mark R. Hutchinson is a NHMRC CJ Martin Fellow (ID 465423; 2007–2010) and an Australian Research Council Research Fellow (DP110100297). Janet K. Coller is a FTT Fricker Research Fellow (Medical Endowment Funds), Faculty of Health Sciences, University of Adelaide. Kerrilyn R. Diener is supported by a NHMRC ECR Fellowship. Linda Watkins is supported by a NIDA K05 award (DA024044).
Abbreviations
- CNS
central nervous system
- GABA
gamma-aminobutyric acid
- IL-1ra
interleukin-1 receptor antagonist
- MOR
µ opioid receptor
- MyD88
myeloid differentiation primary response gene 88
- NMDA
N-methyl-D-aspartate
- LORR
loss of righting reflex
- TLR
toll-like receptor
- NF-κB
Nuclear Factor-κB
Footnotes
Conflict of Interest
None
References
- Boyce-Rustay JM, Holmes A. Functional roles of NMDA receptor NR2A and NR2B subunits in the acute intoxicating effects of ethanol in mice. Synapse. 2005;56:222–225. doi: 10.1002/syn.20143. [DOI] [PubMed] [Google Scholar]
- Craft RM, Leitl MD. Potentiation of morphine antinociception by pentobarbital in female vs. male rats. Pain. 2006;121:115–125. doi: 10.1016/j.pain.2005.12.009. [DOI] [PubMed] [Google Scholar]
- Darke SG, Zador DA, Sunjic SD. Heroin-related deaths in south-western Sydney. Med. J. Aust. 1997;167:107. doi: 10.5694/j.1326-5377.1997.tb138794.x. [DOI] [PubMed] [Google Scholar]
- De Laurentiis A, Pisera D, Lasaga M, Diaz M, Theas S, Duvilanski B, Seilicovich A. Effect of interleukin-6 and tumor necrosis factor-alpha on GABA release from mediobasal hypothalamus and posterior pituitary. Neuroimmunomodulation. 2000;7:77–83. doi: 10.1159/000026423. [DOI] [PubMed] [Google Scholar]
- Fernandez-Lizarbe S, Montesinos J, Guerri C. Ethanol induces TLR4/TLR2 association, triggering an inflammatory response in microglial cells. J. Neurochem. 2013;126:261–273. doi: 10.1111/jnc.12276. [DOI] [PubMed] [Google Scholar]
- Fugelstad A, Ahlner J, Brandt L, Ceder G, Eksborg S, Rajs J, Beck O. Use of morphine and 6-monoacetylmorphine in blood for the evaluation of possible risk factors for sudden death in 192 heroin users. Addiction. 2003;98:463–470. doi: 10.1046/j.1360-0443.2003.00330.x. [DOI] [PubMed] [Google Scholar]
- Ghosh A, Roy A, Liu X, Kordower JH, Mufson EJ, Hartley DM, Ghosh S, Mosley RL, Gendelman HE, Pahan K. Selective inhibition of NF-kappaB activation prevents dopaminergic neuronal loss in a mouse model of Parkinson’s disease. Proc Natl Acad Sci U S A. 2007a;104:18754–18759. doi: 10.1073/pnas.0704908104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh HS, Spencer JV, Ng B, McBurney MW, Robbins PD. Sirt1 interacts with transducin-like enhancer of split-1 to inhibit nuclear factor kappaB-mediated transcription. Biochem. J. 2007b;408:105–111. doi: 10.1042/BJ20070817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghozland S, Chu K, Kieffer BL, Roberts AJ. Lack of stimulant and anxiolytic-like effects of ethanol and accelerated development of ethanol dependence in mu-opioid receptor knockout mice. Neuropharmacology. 2005;49:493–501. doi: 10.1016/j.neuropharm.2005.04.006. [DOI] [PubMed] [Google Scholar]
- Heinisch S, Kirby LG. SDF-1alpha/CXCL12 enhances GABA and glutamate synaptic activity at serotonin neurons in the rat dorsal raphe nucleus. Neuropharmacology. 2010;58:501–514. doi: 10.1016/j.neuropharm.2009.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hickman M, Lingford-Hughes A, Bailey C, Macleod J, Nutt D, Henderson G. Does alcohol increase the risk of overdose death: the need for a translational approach. Addiction. 2008;103:1060–1062. doi: 10.1111/j.1360-0443.2008.02134.x. [DOI] [PubMed] [Google Scholar]
- Hothorn T, Bretz F, Westfall P. Simultaneous inference in general parametric models. Biometrical Journal. 2008;50:346–363. doi: 10.1002/bimj.200810425. [DOI] [PubMed] [Google Scholar]
- Hutchinson MR, Coats BD, Lewis SS, Zhang Y, Sprunger DB, Rezvani N, Baker EM, Jekich BM, Wieseler JL, Somogyi AA, Martin D, Poole S, Judd CM, Maier SF, Watkins LR. Proinflammatory cytokines oppose opioid-induced acute and chronic analgesia. Brain. Behav. Immun. 2008a;22:1178–1189. doi: 10.1016/j.bbi.2008.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutchinson MR, Lewis SS, Coats BD, Rezvani N, Zhang Y, Wieseler JL, Somogyi AA, Yin H, Maier SF, Rice KC, Watkins LR. Possible involvement of toll-like receptor 4/myeloid differentiation factor-2 activity of opioid inactive isomers causes spinal proinflammation and related behavioral consequences. Neuroscience. 2010a;167:880–893. doi: 10.1016/j.neuroscience.2010.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutchinson MR, Northcutt AL, Hiranita T, Wang X, Lewis SS, Thomas J, van Steeg K, Kopajtic TA, Loram LC, Sfregola C, Galer E, Miles NE, Bland ST, Amat J, Rozeske RR, Maslanik T, Chapman TR, Strand KA, Fleshner M, Bachtell RK, Somogyi AA, Yin H, Katz JL, Rice KC, Maier SF, Watkins LR. Opioid Activation of Toll-Like Receptor 4 Contributes to Drug Reinforcement. J. Neurosci. 2012;32:11187–11200. doi: 10.1523/JNEUROSCI.0684-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutchinson MR, Zhang Y, Brown K, Coats BD, Shridhar M, Sholar PW, Patel SJ, Crysdale NY, Harrison JA, Maier SF, Rice KC, Watkins LR. Non-stereoselective reversal of neuropathic pain by naloxone and naltrexone: involvement of toll-like receptor 4 (TLR4) Eur. J. Neurosci. 2008b;28:20–29. doi: 10.1111/j.1460-9568.2008.06321.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutchinson MR, Zhang Y, Shridhar M, Evans JH, Buchanan MM, Zhao TX, Slivka PF, Coats BD, Rezvani N, Wieseler J, Hughes TS, Landgraf KE, Chan S, Fong S, Phipps S, Falke JJ, Leinwand LA, Maier SF, Yin H, Rice KC, Watkins LR. Evidence that opioids may have toll-like receptor 4 and MD-2 effects. Brain. Behav. Immun. 2010b;24:83–95. doi: 10.1016/j.bbi.2009.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kettenmann H, Kirchhoff F, Verkhratsky A. Microglia: new roles for the synaptic stripper. Neuron. 2013;77:10–18. doi: 10.1016/j.neuron.2012.12.023. [DOI] [PubMed] [Google Scholar]
- Kissin I, Brown PT, Robinson CA, Bradley EL., Jr Acute tolerance to the hypnotic effect of morphine in rats. Anesth. Analg. 1991;73:619–621. doi: 10.1213/00000539-199111000-00018. [DOI] [PubMed] [Google Scholar]
- Kumar H, Kawai T, Akira S. Toll-like receptors and innate immunity. Biochemical and biophysical research communications. 2009;388:621–625. doi: 10.1016/j.bbrc.2009.08.062. [DOI] [PubMed] [Google Scholar]
- Levine B, Green D, Smialek JE. The role of ethanol in heroin deaths. J. Forensic Sci. 1995;40:808–810. [PubMed] [Google Scholar]
- Lewis SS, Hutchinson MR, Rezvani N, Loram LC, Zhang Y, Maier SF, Rice KC, Watkins LR. Evidence that intrathecal morphine-3-glucuronide may cause pain enhancement via toll-like receptor 4/MD-2 and interleukin-1beta. Neuroscience. 2010;165:569–583. doi: 10.1016/j.neuroscience.2009.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis SS, Hutchinson MR, Zhang Y, Hund DK, Maier SF, Rice KC, Watkins LR. Glucuronic acid and the ethanol metabolite ethyl-glucuronide cause toll-like receptor 4 activation and enhanced pain. Brain Behav Immun. 2013;30:24–32. doi: 10.1016/j.bbi.2013.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Li H, Zhang Y, Sun X, Hanley GA, LeSage G, Sun S, Peng Y, Yin D. Toll-like receptor 2 is required for opioids-induced neuronal apoptosis. Biochem. Biophys. Res. Commun. 2010;391:426–430. doi: 10.1016/j.bbrc.2009.11.074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Sun X, Zhang Y, Huang J, Hanley G, Ferslew KE, Peng Y, Yin D. Morphine promotes apoptosis via TLR2, and this is negatively regulated by beta-arrestin 2. Biochem. Biophys. Res. Commun. 2009;378:857–861. doi: 10.1016/j.bbrc.2008.12.001. [DOI] [PubMed] [Google Scholar]
- Maroso M, Balosso S, Ravizza T, Liu J, Aronica E, Iyer AM, Rossetti C, Molteni M, Casalgrandi M, Manfredi AA, Bianchi ME, Vezzani A. Toll-like receptor 4 and high-mobility group box-1 are involved in ictogenesis and can be targeted to reduce seizures. Nature medicine. 2010;16:413–419. doi: 10.1038/nm.2127. [DOI] [PubMed] [Google Scholar]
- Nish S, Medzhitov R. Host defense pathways: role of redundancy and compensation in infectious disease phenotypes. Immunity. 2011;34:629–636. doi: 10.1016/j.immuni.2011.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pascual M, Balino P, Alfonso-Loeches S, Aragon CM, Guerri C. Impact of TLR4 on Behavioral and Cognitive Dysfunctions Associated with Alcohol-Induced Neuroinflammatory Damage. Brain. Behav. Immun. 2011;25:S80–S91. doi: 10.1016/j.bbi.2011.02.012. [DOI] [PubMed] [Google Scholar]
- Pestka J, Zhou HR. Toll-like receptor priming sensitizes macrophages to proinflammatory cytokine gene induction by deoxynivalenol and other toxicants. Toxicol Sci. 2006;92:445–455. doi: 10.1093/toxsci/kfl012. [DOI] [PubMed] [Google Scholar]
- Sharko AC, Hodge CW. Differential modulation of ethanol-induced sedation and hypnosis by metabotropic glutamate receptor antagonists in C57BL/6J mice. Alcohol. Clin. Exp. Res. 2008;32:67–76. doi: 10.1111/j.1530-0277.2007.00554.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sivam SP, Nabeshima T, Ho IK. An analysis of GABA receptor changes in the discrete regions of mouse brain after acute and chronic treatments with morphine. J. Neurochem. 1982;39:933–939. doi: 10.1111/j.1471-4159.1982.tb11479.x. [DOI] [PubMed] [Google Scholar]
- Team RC. R: A Language and Environment for Statistical Computing. Vienna, Austria: R Foundation for Statistical Computing; 2013. [Google Scholar]
- Ticku MK, Huffman RD. The effects of acute and chronic morphine administration on GABA receptor binding. Eur. J. Pharmacol. 1980;68:97–106. doi: 10.1016/0014-2999(80)90310-6. [DOI] [PubMed] [Google Scholar]
- Wand G, Levine M, Zweifel L, Schwindinger W, Abel T. The cAMP-protein kinase A signal transduction pathway modulates ethanol consumption and sedative effects of ethanol. J. Neurosci. 2001;21:5297–5303. doi: 10.1523/JNEUROSCI.21-14-05297.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang XH, Loram LC, Ramos K, de Jesus AJ, Thomas J, Cheng K, Reddy A, Somogyi AA, Hutchinson MR, Watkins LR, Yin H. Morphine activates neuroinflammation in a manner parallel to endotoxin. Proc. Natl. Acad. Sci. U. S. A. 2012;109:6325–6330. doi: 10.1073/pnas.1200130109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White JM, Irvine RJ. Mechanisms of fatal opioid overdose. Addiction. 1999;94:961–972. [PubMed] [Google Scholar]
- Wu Y, Lousberg EL, Moldenhauer LM, Hayball JD, Coller JK, Rice KC, Watkins LR, Somogyi AA, Hutchinson MR. Inhibiting the TLR4-MyD88 signalling cascade by genetic or pharmacological strategies reduces acute alcohol-induced sedation and motor impairment in mice. Br. J. Pharmacol. 2012;165:1319–1329. doi: 10.1111/j.1476-5381.2011.01572.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y, Lousberg EL, Moldenhauer LM, Hayball JD, Robertson SA, Coller JK, Watkins LR, Somogyi AA, Hutchinson MR. Attenuation of microglial and IL-1 signaling protects mice from acute alcohol-induced sedation and/or motor impairment. Brain. Behav. Immun. 2011;25:S155–S164. doi: 10.1016/j.bbi.2011.01.012. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Li H, Li Y, Sun X, Zhu M, Hanley G, Lesage G, Yin D. Essential role of toll-like receptor 2 in morphine-induced microglia activation in mice. Neurosci. Lett. 2011;489:43–47. doi: 10.1016/j.neulet.2010.11.063. [DOI] [PMC free article] [PubMed] [Google Scholar]