

Abstract
During the past few decades, sleep curtailment has become a very common behavior in industrialized countries. This trend for shorter sleep duration has developed over the same time period as the dramatic increase in the prevalence of obesity and diabetes. There is rapidly accumulating evidence to indicate that chronic partial sleep loss may increase the risk of obesity and diabetes. Laboratory studies in healthy volunteers have shown that experimental sleep restriction is associated with an adverse impact on glucose homeostasis. Insulin sensitivity decreases rapidly and markedly without adequate compensation in beta cell function, resulting in an elevated risk of diabetes. Prospective epidemiologic studies in both children and adults are consistent with a causative role of short sleep in the increased risk of diabetes. Sleep curtailment is also associated with a dysregulation of the neuroendocrine control of appetite, with a reduction of the satiety factor leptin and an increase in the hunger-promoting hormone ghrelin. Thus, sleep loss may alter the ability of leptin and ghrelin to accurately signal caloric need, acting in concert to produce an internal misperception of insufficient energy availability. The adverse impact of sleep deprivation on appetite regulation is likely to be driven by increased activity in neuronal populations expressing the excitatory peptides orexins that promote both waling and feeding. Consistent with the laboratory evidence, multiple epidemiologic studies have shown an association between short sleep and higher body mass index after controlling for a variety of possible confounders.
Keywords: Sleep Deprivation, Diabetes, Obesity, Glucose Tolerance, Energy Expenditure, Epidemiology, leptin, ghrelin, appetite, orexins
Introduction
The prevalence of obesity and type 2 diabetes is increasing worldwide but particularly in the US.1 Obesity and diabetes are both associated with increased age-adjusted mortality risk as well as a substantial economic burden.2 The causes of this pandemic are not fully explained by changes in traditional lifestyle factors such as diet and physical activity. One behavior that seems to have developed during the past few decades and has become highly prevalent, particularly amongst Americans, is sleep curtailment. In 1960, a survey study conducted by the American Cancer Society found modal sleep duration to be 8.0 to 8.9 hours,3 while in 1995 the modal category of the survey conducted by the National Sleep Foundation poll had dropped to 7 hours.4 Recent analyses of national data indicate that a greater percentage of adult Americans report sleeping 6 hours or less in 2004 than in 1985.5 Today, more than 30% of adult men and women between the ages of 30 and 64 years report sleeping less than 6 hours per night. 5 The decrease in average sleep duration in the U.S. has occurred over the same time period as the increase in the prevalence of obesity and diabetes.
The present review examines the existing evidence for a link between short sleep and increased risk of obesity and diabetes and explores putative causal mechanisms. By “short sleep”, we mean sleep durations under 7 hours per night. There is substantial evidence in support of an association between long sleep (>8 h) and increased morbidity and mortality,6–8 but the mechanisms linking long sleep and poor health are likely to be distinct from those mediating the adverse effects of short sleep.
Figure 1 provides a schematic representation of some of the pathways that could mediate an adverse effect of sleep loss on the risk of obesity and diabetes. Several of these pathways interact with one another. An upregulation of the activity of orexin neurons may be a primary mechanism linking sleep deprivation and adverse metabolic effects. Total or partial sleep deprivation results in increased sympathetic nervous activity, increased levels of cortisol in the evening, and increased levels of growth hormone (GH) in the daytime. All of these can, in turn, lead to increased insulin resistance and reduced glucose tolerance, and thus increase the risk of developing of diabetes. Sleep loss also impacts hormones involved in appetite regulation. After sleep restriction, the levels of leptin, a satiety factor, are lower and the levels of ghrelin, an appetite stimulant, are higher. Less time sleeping also allows for more opportunity to eat. Thus, through these pathways, sleep loss could lead to increased appetite and increased food intake, which could lead to obesity. Finally, sleep loss and the associated sleepiness and fatigue may be result in reduced energy expenditure, in particular through decreased physical exercise but also through decreases in non-exercise activity thermogenesis (NEAT). Reduced energy expenditure is to date an unexplored pathway that could link short sleep and the risk of overweight and obesity. Obesity is in itself a major risk factor for type 2 diabetes. This cascade of negative events is likely to be accelerated in many overweight and obese individuals by sleep-disordered breathing (SDB), a reported independent risk factor for insulin resistance.9, 10 The present chapter will only focus on sleep loss resulting from behavioral sleep restriction rather than from the presence of a sleep disorder.
Figure 1.
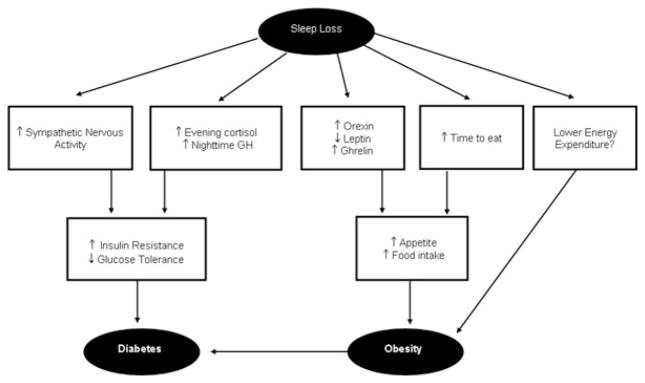
Schematic of the putative pathways leading from sleep loss to diabetes and obesity risk.
We will first review the experimental and epidemiologic evidence for an association between short sleep, alterations in glucose metabolism and increased diabetes risk. As a cautionary note, translating the effects of experimental sleep restriction in the laboratory to the real world is not straightforward. Furthermore, laboratory studies of sleep restriction cannot be conducted for periods of time extending beyond 1–2 weeks. Epidemiologic studies that involve population-based samples do not provide evidence for causal direction. In many epidemiologic studies, the reductions in sleep duration cannot be distinguished from reductions in sleep quality. A presentation of the evidence linking short sleep, upregulation of appetite and increased body mass index (BMI) will follow. Finally, we will address the possibility that individuals exposed to insufficient sleep and the resulting sleepiness and fatigue may also have lower levels of energy expenditure than well-rested adults, particularly in an environment that promotes physical inactivity.
Sleep Loss & Glucose Metabolism
Normal Physiology
Blood levels of glucose are tightly regulated within in a narrow range to avoid hypoglycemia and hyperglycemia as both conditions have adverse life threatening consequences. Glucose tolerance refers to the ability to metabolize exogenous glucose and return to baseline normoglycemia. In clinical settings, glucose tolerance is assessed by the oral glucose tolerance test, which consists of ingesting a glucose solution and measuring glucose levels at frequent intervals during the next 2 hours. Glucose tolerance may also be examined after ingestion of a carbohydrate-rich meal or after intravenous injection or infusion of glucose. Glucose tolerance is dependent on the balance between glucose production by the liver and glucose utilization by insulin-dependent tissues, such as muscle and fat, and non-insulin dependent tissues, such as the brain. Thus, glucose tolerance is critically dependent on the ability of pancreatic beta cells to release insulin both acutely (i.e. acute insulin response to glucose or beta cell responsiveness) and in a sustained fashion and on the ability of insulin to inhibit glucose production by the liver and promote glucose utilization by peripheral tissues (i.e. insulin sensitivity). Reduced insulin sensitivity, or insulin resistance, occurs when higher amounts of insulin are needed to reduce blood glucose levels following the administration of the same amount of exogenous glucose.
In normal, healthy individuals, glucose tolerance varies across the day such that plasma glucose responses to exogenous glucose are markedly higher in the evening than in the morning, and glucose tolerance is at its minimum in the middle of the night.11 The reduced glucose tolerance in the evening is at least partly due to a reduction in insulin sensitivity concomitant with a reduction in the insulin secretory response to elevated glucose levels. The further decrease in glucose tolerance during the night is dependent on the occurrence of sleep. Indeed, a variety of mechanisms intervene to maintain stable glucose levels during the extended overnight fast associated with sleep.11 Overall glucose utilization is greatest during wake and lowest during non-REM (Stages 2, 3 & 4) sleep with intermediate levels during REM sleep.12 In the first half of the night, glucose metabolism is slower, partly because of the predominance of slow wave sleep that is associated with a marked reduction in cerebral glucose uptake,13, 14 and may be also because of a reduction in peripheral glucose utilization. These effects are reversed during the second half of the night, when light non-REM sleep and REM sleep are dominant and awakenings are more likely to occur. These major modulatory effects of sleep on glucose regulation can also be observed when the sleep period occurs during the daytime.15
Laboratory studies of glucose metabolism and sleep loss
Nearly 40 years ago, a study examining the impact of 72–126 hours of total sleep deprivation on oral glucose tolerance found that levels of glucose were markedly higher throughout the test when subjects were sleep-deprived.16 Since then, a large body of evidence has accumulated to indicate that sleep has major modulatory effects on glucose regulation15. It may therefore be somewhat surprising that the possibility that recurrent sleep loss may be associated with adverse metabolic effects has only been considered in recent years. An explanation to this paradox may be that nearly all early studies used the paradigm of acute total sleep deprivation, a condition that is necessarily of short duration in humans and invariably followed by sleep recovery. Alterations evidenced during acute total sleep deprivation are readily corrected following sleep recovery and therefore the possibility that sleep loss may result in long-term adverse effects on glucose tolerance appeared unlikely. However, as pointed out elsewhere, there are differences in the EEG and hormonal effects of acute total as compared to recurrent partial sleep deprivation.17 For example, after recovery sleep following total sleep deprivation, slow-wave sleep and growth hormone levels rebound whereas during recurrent sleep restriction, slow-wave sleep and growth hormone levels are not higher than at baseline.17, 18 During total sleep deprivation, thyroid stimulating hormone (TSH) levels are more than doubled whereas after 3–5 days of partial sleep deprivation, TSH levels are markedly depressed.19–21
The first detailed laboratory study that examined the effects of recurrent partial sleep deprivation on glucose metabolism involved healthy young men who were subjected to 6 nights of 4 hours in bed (“sleep debt”) followed by 7 nights of 12 hours in bed (“sleep recovery”).21 The subjects ate identical carbohydrate-rich meals and were at continuous bed rest on the last two days of each condition. During each bedtime condition, the subjects underwent an intravenous glucose tolerance test (ivGTT) on the morning of the 5th or 6th day that was followed by 24 hours of blood sampling at 10–30 minutes intervals.21 The results of the ivGTT may be analyzed using a mathematical model of glucose homeostasis, referred at as the “minimal model”.22 Fitting the minimal model to the measured values of plasma glucose and insulin levels allows for the derivation of critical parameters contributing to glucose metabolism. The results obtained during the sleep debt condition are shown in Table 1. The rate of glucose clearance during the initial phase of the test (glucose tolerance; KG) was 40% lower, glucose effectiveness (SG), a measure of non-insulin dependent glucose disposal, was 30% lower, and the acute insulin response to glucose (AIRG) was also 30% lower. A trend for reduced insulin sensitivity (SI) was evident but failed to reach statistical significance. The disposition index (DI) is the product of AIRG x SI, and is a marker of diabetes risk that is used in genetic studies.23 In individuals with normal glucose tolerance, the product AIRg x SI remains constant because beta cell function is able to compensate for insulin resistance with increased insulin release.24 Type 2 diabetes occurs when beta-cell function is unable to be sufficiently upregulated to compensate for insulin resistance, resulting in hyperglycemia. Thus low DI values represent a higher risk of type 2 diabetes. DI values of 2,000 and above are typical of subjects with normal glucose tolerance while DI values under 1,000 have been reported in populations at high risk for type 2 diabetes, such as Hispanic women with prior gestational diabetes.25 In the sleep debt condition, the DI was 40% lower than after sleep recovery and 3 of the 11 subjects had DI values under 1,000. Table 2 compares the glucose tolerance (KG) values observed in these healthy young men at the end of the sleep debt condition and when fully rested to values obtained in different subject populations using the same ivGTT protocol and mathematical analysis. The values observed after 5 days of sleep restriction were similar to those observed in older adults with impaired glucose tolerance,26 while the values observed after sleep recovery were, as expected, in the range typical of healthy young subjects.27 When glucose responses to a high carbohydrate breakfast presented on the 6th day of each sleep condition were examined, glucose levels were higher after sleep restriction than after sleep recovery despite levels of insulin secretion that were slightly, but not significantly, higher.21 Calculations of the homeostatic model assessment (HOMA) levels, an index of insulin resistance directly proportional to the product insulin x glucose, revealed that the area under the HOMA curve for the breakfast meal was more than 50% higher after 6 days of sleep restriction than when the subjects were fully rested.28, 29 Although the HOMA has only been validated as a measure of insulin resistance under fasting conditions, these results suggest that insulin sensitivity was lower on the 6th than on the 5th day of sleep restriction and thus that insulin resistance may develop progressively with increasing exposure to partial sleep loss. Preliminary findings from ongoing studies in our laboratory are supporting this hypothesis although reduced insulin sensitivity to oral glucose administration was already observed after one night of total sleep deprivation in an earlier study.30 In any case, insulin resistance is a well-recognized risk factor for type 2 diabetes. It is possible that insulin resistance could also promote increased adiposity and weight gain.
Table 1.
Results from intravenous glucose tolerance tests including glucose tolerance (KG), acute insulin response to glucose (AIRg), glucose effectiveness (SG), insulin sensitivity (SI) and disposition index (DI) in healthy males while fully rested and after 5 days of sleep restriction.
| Fully rested | After 5 days of sleep restriction | p level | |
|---|---|---|---|
| KG (% per minute) | 2.40±0.41 | 1.45 ±0.31 | <0.04 |
| AIRg (μU.ml-1.min) | 548 ± 158 | 378 ± 136 | 0.05 |
| SG (%/min) | 2.6±0.2 | 1.7±0.2 | p<0.0005 |
| SI (104.min−1.(μU/ml) −1) | 6.73 ± 1.24 | 5.41 ± 0.60 | 0.28 |
| DI | 2897 ± 404 | 1726 ± 395 | 0.0006 |
Table 2.
Glucose tolerance (KG) derived from glucose disappearance curve during IVGTT from the Sleep Debt Study 21 and reference populations 26, 27.
| Sleep Debt Study | ||
| 18–27 year old men in sleep debt condition | 18–27 year old men fully rested | |
| KG (% per minute) | 1.45 ± 0.31 | 2.40 ± 0.41 |
| Reference Populations | ||
| 61–80 yr old adults with impaired glucose tolerance | 21–30 yr old fit subjects | |
| KG (% per minute) | Range: 1.30 – 2.10 | Range: 2.20 – 2.90 |
A criticism of this initial “sleep debt study” was that there may be an order effect,31 since the fully rested condition always followed the sleep debt condition.21 Partly to address this concern, a second study that examined the impact of sleep restriction (4 hours per night for 2 nights) as compared to sleep extension (10 hours per night for 2 nights) used a randomized cross-over design and confirmed the findings of the “sleep debt” study. In both bedtime conditions, plasma levels of glucose and insulin were examined during constant glucose infusion in healthy lean young subjects. Morning glucose levels were higher, and insulin levels tended to be lower, after 2 nights with 4 hours in bed as compared to 2 nights with 10 hours in bed.28 A further critique of both these studies is that they used a severe restriction of sleep (i.e. only 4 hours in bed), and thus that it is not clear if more moderate amounts of sleep restriction would have similar effects.31 Ongoing studies in our laboratory examine the effects of more extended periods of less severe sleep curtailment (e.g. 8 days of 5 hours in bed, and 2 weeks of bedtimes restricted by only 1.5 hours per night) and both have provided preliminary evidence of impaired glucose homeostasis, consistent with our previous studies.
The mechanisms underlying alterations in glucose metabolism following recurrent partial sleep restriction are likely to be multifactorial. The reduction in SG, a measure of non-insulin mediated glucose disposal, is suggestive of a decrease in cerebral glucose metabolism, consistent with the findings of positron emission tomography (PET) studies of subjects submitted to total sleep deprivation.32 The acute reduction in insulin release could be due to increased sympathetic nervous activity at the level of the pancreatic beta-cell. While changes in sympatho-vagal balance at the level of the pancreas have not yet been assessed in any study, in both laboratory studies of sleep restriction described above, cardiac sympatho-vagal balance, derived from estimations of heart rate variability, was elevated (likely reflecting an increased influence of sympathetic tone) when sleep was restricted.21, 29 Disturbances in the secretory profiles of the counter-regulatory hormones, GH and cortisol, may also contribute to the alterations in glucose regulation observed during sleep loss. Indeed, 6 days of sleep restriction were associated with an extended duration of elevated nighttime GH concentrations 18 and with an increase in evening cortisol levels.21 An extended exposure of peripheral tissues to higher GH levels may induce a rapid decrease in muscular glucose uptake adversely affecting glucose regulation. Also, elevated evening cortisol concentrations are likely to result in reduced insulin sensitivity on the following morning.11 Finally, acute total sleep loss or even a 2-hour reduction of sleep/night for one week is associated with increased levels of proinflammatory cytokines and low grade inflammation, a condition known to predispose to insulin resistance and diabetes.33, 34 There is recent evidence that decreased sleep quality without changes in sleep duration also results in impaired glucose metabolism.35
Epidemiologic studies of sleep and diabetes
There have been a few studies that examined cross-sectional associations between sleep and markers of diabetes risk or control. An overnight polysomnographic recording was conducted in obese children aged 3 to 19 years and sleep measures were compared to the results of an oral glucose tolerance test.36 The results indicated that obese children who slept less than 6 hours during the PSG had higher fasting insulin and higher insulin resistance based on the HOMA method.36 A cross-sectional study among adults in Canada found an increased risk of prevalent diabetes for those reporting sleeping less than 7 hours (OR 1.58, 95% CI 1.13, 2.31).37 Furthermore, fasting plasma glucose, fasting plasma insulin, and the HOMA insulin resistance index were higher in the short sleepers.37 A survey study among patients with type 2 diabetes found that subjective poor sleep quality and subjective insufficient sleep were associated with higher levels of hemoglobin A1c, which is a marker of worse glycemic control.38 Finally, a cross-sectional study found that subjective sleep quality was associated with higher glucose and insulin levels, higher estimated insulin resistance, and higher BMI, body fat percentage and waist circumference.39 These cross-sectional analyses suggest a significant relationship between glucose metabolism and sleep duration and quality.
Several prospective studies have examined the association between sleep duration or disturbance and the development of diabetes. Results from the Nurses Health Study, which included only women, found an increased risk of incident symptomatic diabetes over 10 years among those reporting sleep durations of 5 hours or less relative to 7–8 hours, even after controlling for many covariates such as BMI, shiftwork, hypertension, exercise and depression.7 A study in Japan followed adult men for 8 years from 1984 to 1992, and high frequency of difficulty initiating sleep or difficulty maintaining sleep, which are both likely to result in shorter sleep duration, had an increased age-adjusted risk of developing type 2 diabetes.40 One Swedish study examined men aged 35–51 years once between 1974 and 1984 and again 7–22 years later, and found an increased risk of incident diabetes among those who reported difficulty falling asleep or use of sleeping pills (OR 1.52, 95% CI: 1.05, 2.20) after controlling for numerous covariates.41 Another prospective study conducted in Sweden followed 1,187 men and women free of diabetes at baseline for 12-years.42 Men who reported difficulty maintaining sleep or who reported sleep duration of 5 hours or less had a significantly greater risk of developing diabetes, but no significant associations between sleep and diabetes risk was observed in women.42 A third prospective study from Sweden followed over 600 women for 32 years beginning in 1968–69, but the incidence of diabetes over a 32-year period was not associated with the self-reported sleep problems, sleep medication use or sleep duration at baseline.43 A prospective study from Germany interviewed 8,269 non-diabetic men and women aged 25–74 years and followed them for an average of 7.5 years.44 The results demonstrated a significant increased risk of incident type 2 diabetes for those who reported difficulty maintaining sleep at baseline, even after adjustment for numerous covariates.44 The Massachusetts Male Aging Study observed that among men without diabetes at baseline, a sleep duration of 6 hours or less per night was associated with twice the risk of developing diabetes after adjustment for covariates such as age, hypertension, smoking, self-rated health, waist circumference and education.45 Finally, recent analysis of data from the First National Health and Nutrition Examination Survey found that people reporting sleeping five hours or less and those reporting sleeping 9 or more hours were at an increased risk of developing diabetes.46
The majority of these studies, which involved different subject populations and originated from different geographical locations and cultures, were consistent in indicating that short or poor sleep may increase the risk of developing type 2 diabetes. One important limitation of all these epidemiologic studies is that nearly all of them relied on self-reported measures of sleep. Additional studies that use objective measures of sleep and preferably an interventional design are required to determine if sleep loss could indeed be part of the causal mechanisms leading to the development of diabetes.
Sleep Loss & Appetite Regulation
Normal Conditions
Appetite is regulated by the interaction between metabolic and hormonal signals and neural mechanisms. The arcuate nucleus of the hypothalamus has two opposing sets of neuronal circuitry, appetite simulating and appetite-inhibiting, and several peripheral hormonal signals have been identified that affect these neuronal regions.47 Among these peripheral signals are leptin, an appetite-inhibiting hormone, and ghrelin, an appetite stimulating hormone. Leptin is primarily secreted by adipose tissue and appears to promote satiety.47 Ghrelin is a peptide released primarily from the stomach. In rodents, ghrelin generates a positive energy balance and increased adiposity through increased food intake and reduced fat oxidation.48 Studies in humans also indicate that ghrelin increases appetite and food intake.48 Plasma ghrelin levels are rapidly suppressed by food intake and then rebound after 1.5–2 hours, paralleling the resurgence in hunger. Thus, leptin and ghrelin exert opposing effects on appetite. Evidence from animal studies suggests that leptin and ghrelin also have opposing effects on energy expenditure (see following section).
Under normal conditions, the 24-hour profile of human plasma leptin levels shows a marked nocturnal rise, which is partly dependent on meal intake.49 Nevertheless, a study using continuous enteral nutrition to eliminate the impact of meal intake showed the persistence of a sleep-related leptin elevation, although the amplitude was lower than during normal feeding conditions.50 The 24-hour profile of ghrelin levels also shows a nocturnal rise, which may partly reflect the post-dinner rebound. However, ghrelin levels spontaneously decrease in the second half of the sleep period, despite the maintenance of the fasting condition.51
In rodents, food shortage or starvation results in decreased sleep,52 and, conversely, total sleep deprivation leads to marked hyperphagia.53 The identification in the lateral hypothalamus and perifornical area of a population of neurons that express two excitatory neuropeptides (orexin A and orexin B, also referred to as hypocretin A and hypocretin B) derived from the same precursor (pre-pro-orexin) that have potent wake promoting effects and stimulate food intake, has provided a molecular basis for the interactions between feeding and sleeping.54, 55 Figure 2 presents a simplified schematic representation of the orexin system. Orexins activate all the components of the ascending arousal system, including noradrenergic, cholinergic and serotonergic cell groups in the brain stem and histaminergic cells in the tuberomammillary nucleus (TMN) of the hypothalamus. Orexin neurons also project diffusely to the entire cerebral cortex.56 Thus, orexinergic neurons promote wake. Orexin exhibits excitatory effects on neurons of the nucleus tractus solitaries (NTS) and of the hypothalamic paraventricular nucleus (PVN),57 resulting in increased sympathetic nervous activity. The orexin system also activates the appetite-promoting neuropeptide Y neurons in the arcuate nucleus of the hypothalamus. Furthermore, orexin neurons in the lateral hypothalamus have dense projections to the dopaminergic ventro-tegmental area (VTA) and nucleus accumbens (NA), which are important in the hedonic control of food intake.58, 59 Thus, the activation of the orexin system would lead to increased hedonic and homeostatic feeding. Orexinergic activity is in turn influenced by both central and peripheral signals, with glucose and leptin exerting inhibitory effects while ghrelin promotes further activation.55 In animal models, experimental sleep deprivation – which invariably involves increased activity and/or stress – results in increased orexinergic activity.60 It is not known whether sleep deprivation in humans under comfortable sedentary conditions, e.g. in an armchair in front of a television set, is similarly associated with an upregulation of the orexin system. Rodent studies have provided evidence suggesting that a decrease in leptin levels may be involved in the hyperphagia associated with sleep deprivation. Indeed, in rats submitted to prolonged total sleep deprivation as well as in their yoked controls who experienced partial sleep deprivation, leptin levels declined in the first four days of the deprivation period.61 Rats submitted to selective REM sleep deprivation for 2 weeks also showed reduced levels of leptin as well as increased NPY expression in the arcuate nucleus.62, 63 Finally, a brief period of sleep deprivation (5 hours) did not alter the levels of leptin but did acutely increase the levels of ghrelin.64 The results from animal studies are consistent with findings from human studies as will be discussed below. In general both animal and human studies have observed that sleep loss is associated with increased food intake and appetite, decreased leptin and increased ghrelin, altered glucose utilization and increased sympathetic nervous activity.65 (For a more detailed review, see 65)
Figure 2.

Schematic of the Orexin System. TMN: tuberomammillary nucleus; NTS: nucleus tractus solitaries; PVN: paraventricular nucleus; VTA: ventro-tegmental area; NA: nucleus accumbens (NA). Grey arrows represented stimulatory (+) pathways and black dashed lines reflect inhibitory (−) pathways.
Recent rodent data indicate that orexins also play a role in the control of reward and motivation58 and raise the possibility that insufficient sleep may affect the amount and composition of non-homeostatic food intake (i.e. food intake that is not needed to fulfill a caloric need) related to emotional and psychosocial factors in humans. Consistent with this hypothesis, epidemiological data show an association between short sleep duration and irregular eating habits, snacking between meals, excessive food seasoning, and reduced consumption of vegetables.66, 67 Evidence for a role of sleep loss in the dysregulation of mechanisms involved in the non-homeostatic control of food intake, i.e. food intake that is not in response to a caloric need, but rather to social, hedonic or other factors, is currently lacking in humans.
Laboratory studies of leptin and ghrelin during sleep loss
Only a couple of studies have examined human leptin or ghrelin levels during acute total sleep deprivation. In subjects receiving continuous enteral nutrition, plasma leptin levels rose slightly during nocturnal sleep deprivation and the maximum level occurred later in night compared to during nocturnal sleep 50. Eighty-eight hours of total sleep deprivation with scheduled meals designed to maintain body weight resulted in a decrease in the amplitude of the leptin diurnal variation without change in body weight.68 The nocturnal rise of ghrelin is modestly, but significantly, reduced during acute total sleep deprivation as compared to normal nocturnal sleep.51
In the laboratory study that subjected healthy men to 4-hour bedtimes for 6 nights followed by 6 nights of 12-hour recovery sleep, mean leptin levels were 19% lower, the nocturnal acrophase was 2 hours earlier and 26% lower and the amplitude of the diurnal variation was 20% lower during sleep restriction.29 These changes occurred despite identical caloric intake and physical activity with no change in BMI.29 Maximal leptin levels between the state of sleep debt and the fully rested state differed on average by 1.7 ng/ml, which is somewhat larger than the decrease reported in young adults after three days of dietary intake restricted to 70% of energy requirements (a caloric deficit of approximately 900 Kcal per day).29 Ghrelin levels were not measured and subjective feelings of hunger and appetite were not assessed. Another laboratory study, which used a randomized cross-over design to compare the impact of restricted versus extended sleep, observed similar effects of sleep restriction on leptin. This study involved 2 days of 4-hour bedtimes and 2 days of 10-hour bedtimes in subjects receiving a constant glucose infusion as their only source of caloric intake. Daytime levels of leptin, ghrelin, hunger and appetite were measured at 20 (for leptin) to 60 minutes intervals following the 2nd night of sleep restriction or extension.69 Figure 3 illustrates the levels of hunger, appetite and the ghrelin-to-leptin ratio in the 4-hour and 10-hour condition in a representative individual. Importantly, the change in the ratio of ghrelin-to-leptin between the two conditions was strongly correlated to the change in hunger ratings, suggesting that the changes observed in these appetite hormones was partially responsible for the increase in appetite and hunger. These observed changes would suggest that these subjects, if allowed ad libidum food, may have increased their food intake.
Figure 3.
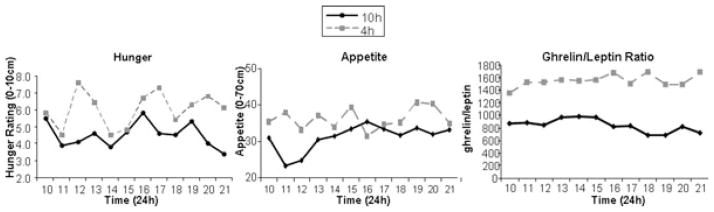
Profiles of hunger ratings, appetite ratings and the ghrelin-to-leptin ratio during the long sleep (10 hour) and short sleep (4 hour) conditions in a single representative subject.
A population-based study, The Wisconsin Sleep Cohort Study, also observed an association between sleep duration, leptin and ghrelin.70 This study collected sleep diaries from which average nightly sleep was calculated, and each subject underwent one night of polysomnography (PSG) in the laboratory. In the morning following the PSG, a single blood sample was obtained for the measurement of hormonal levels. The results indicated that total sleep time from PSG was negatively associated with ghrelin levels (beta coefficient = −0.69, p=0.008) while average usual sleep duration from the diaries was positively associated with leptin levels independently of BMI (beta coefficient = 0.11, p=0.01).70 Thus, ghrelin levels were associated with an acute, short-term measure of sleep duration while leptin levels were associated with the more chronic measure. The findings from the Wisconsin Sleep Cohort Study regarding the relationship between sleep duration and BMI, leptin and ghrelin, were not confirmed in a subsequent smaller study including only post-menopausal women.71 Cross-sectional analysis did not reveal an association between short sleep, higher BMI, lower leptin and higher ghrelin. Differences in age and gender composition of the sample between the two studies could play a role in the divergent findings, as could the fact that the smaller study included relatively fewer individuals with sleep durations under 7 hours.
The notion of “leptin resistance” has been introduced to explain the paradoxical observation that most obese individuals have high, rather, than low plasma leptin levels. While a number of putative mechanisms have been proposed to underlie leptin resistance, one suggestion that is particularly relevant to studies of sleep loss is the hypothesis that leptin binds to circulating levels of C-reactive protein (CRP), an inflammatory marker that is elevated in obesity, resulting in an attenuation of its physiological effects.72 Several studies of acute total as well as recurrent partial sleep deprivation in healthy lean adults have reported an elevation of CRP with sleep loss73 and ongoing studies from our laboratory confirm these findings. The combination of reduced leptin levels with increased CRP concentrations in sleep-deprived subjects might involve a larger negative impact on energy balance than that of the leptin reduction alone.
Taken together, these studies indicate that sleep duration may play an important role in the regulation of human leptin and ghrelin levels, hunger and appetite. In the laboratory studies, differences in energy expenditure between the two bedtime conditions were minimal because the subjects were at bed rest. The findings from the Wisconsin Sleep Cohort Study further support the hypothesis that sleep loss may alter the ability of leptin and ghrelin to accurately signal caloric need, acting in concert to produce an internal perception of insufficient energy availability. This suggests that sleep loss could lead to increased food intake, which is consistent with reports of increased food intake in human subjects and in laboratory rodents submitted to total sleep deprivation.53, 74 Study designs that examine actual food intake under different sleep duration conditions will be needed to further test this hypothesis.
Epidemiologic studies examining sleep duration and BMI
In recent years, evidence from large epidemiological studies from seven different countries has rapidly accumulated to indicate the existence of a negative association between sleep duration and BMI in both adults (Table 3) and children (Table 4). Of note, the association was observed in studies that enrolled subjects with different BMI, from lean or mildly overweight to frankly obese.
Table 3.
Summary of findings from studies examining the association between sleep and body mass index (BMI) in adults.
| Authors | Date of study | BMI range | Results | Country |
|---|---|---|---|---|
| Vioque et al, 2000 | 1994 | Mean BMI was 26.3 kg/m2 and 16.0% obese among men; mean BMI was 25.7 kg/m2 and 18.1% were obese among women. | Prevalence of obesity decreases with increasing sleep duration. Prevalence Odds Ratios adjusted for sex, age & population size: ≤6h/day: 1.00 7h/day: 0.70 (95% CI 0.48–1.01) 8h/day: 0.59 (95% CI 0.41–0.84) ≥9h/day: 0.47 (95% CI: 0.30–0.74) |
Spain |
| Shigeta et al, 2001 | 1998–1999 | Mean BMI was 23.4 (SD 3.0) kg/m2 | Odds ratio for obesity for sleeping <6 h/night: 1.98 (95% CI 1.03–3.82) versus sleeping 6 or more hours. | Japan |
| Kripke et al, 2002 | 1982 | not reported | A negative association between BMI & sleep in men; a U-shaped association in women. | US, Cancer Prevention II Study |
| Taheri et al, 2004 | 1995 | BMI Quartiles: 25th = 26.2 kg/m2, 50th = 29.7 kg/m2, 75th = 34.7 kg/m2 |
Average time in bed was associated with BMI in a U-shaped manner where lowest mean BMI was associated with 7.7 h/night. | US |
| Patel et al, 2004 | 1986–2002 | not reported | Sleep duration was associated with BMI in a U-shaped manner where the lowest mean BMI was among those sleeping 7–8 h/night. | US (Nurses Health Study) |
| Cournot et al, 2004 | 1996 | 9.8% were obese | Among women, mean BMI was higher for those reporting 6 hours or less sleep per night versus those reporting more than 6 hours (24.4 vs. 23.4 kg/m2). This difference was not observed among men. | France (VISAT study) |
| Vorona et al, 2005 | not reported | Mean BMI was 30 kg/m2 (SD 6) | Mean total sleep time was significantly shorter in the obese group relative to normal weight. The overweight and severely obese group did not differ from normal weight. | US |
| Singh et al, 2005 | not reported | Mean BMI was 27.2 kg/m2, 24.% obese. | Odds ratio for obesity was 1.7 (95% Ci 1.3–2.3) for < 5 h sleep/night and 1.4 (95% CI 1.1–1.8) for 5–6 h sleep/night relative to 7–8 h sleep/night | US |
| Gangwisch et al, 2005 | 1982–1992 | not reported | Cross-sectional analysis of sleep and obesity risk among 32–49 year olds: OR 2.35 (95% CI 1.36–4.05) for 2–4 h/night; OR 1.60 (95% CI 1.12–2.29) for 5 h/night; OR 1.27 (95% CI 1.01–1.60) for 6 h/night relative to 7 h/night. | US |
| Hasler et al, 2004 | 1978–1999 | Mean BMI was 21.2 (SD 2.5) kg/m2 in 1979 and 23.3 (SD 3.8) kg/m2 in 1999. | Prospective Study. Longitudinal analysis resulted in an odds ratio of 0.50 for sleep duration predicting obesity. | Switzerland |
| Patel et al, 2006 | 1986–2002 | Mean BMI ranged from 24.9 (SD 4.5) to 26.1 (SD 5.5) depending on sleep duration group | Prospecive Study. Those who slept ≤ 5 hours gained 1.14 kg (95% CI: 0.49, 1.79) and those sleeping 6 hours gained 0.71 kg (95% CI: 0.41, 1.00) more than those sleeping 7 hours adjusting for age and baseline BMI. | US |
| Kohatsu et al, 2006 | 1999–2004 | 29.51 kg/m2 (SD 5.79) | Cross-sectional analysis indicated sleep duration was negatively associated with BMI (Beta = −0.42; 95% CI: −0.77 to −0.07) after adjustment for covariates. | Rural US |
| Bjorvatn et al, 2007 | 1997–1999 | Mean BMI ranged from 25.05 kg/m2 (SD 3.7) to 26.34 kg/m2 (SD 4.3) depending on sleep duration group | Cross-sectional analysis indicated a u-shaped association between sleep duration and BMI after adjustment for gender and smoking. <5h: beta = 1.17 (p<.01) 5–<6 h: beta = 0.68 (p<.01) 6–<7 h: beta = 0.12 (p>.05) 7–<8: ref 8–<9: beta = 0.28 (p>.05) ≥9: beta = 1.07 (p<.01) |
Norway |
| Chaput et al, 2007 | not reported | Mean BMI was 28.6 kg/m2 (SD 5.0) | Among women ≥50 years, no significant differences between short (<7 hours) and long (≥7 hours) sleepers | Canada |
| Rontoyanni et al, 2007 | 2005 | Mean BMI was 25.7 kg/m2 (SD 3.38) | Sleep duration based on diaries was significantly associated with BMI (beta= −2.0), % body fat (beta= −2.8), and sum of four skinfolds (beta= − 15.0), in women aged 30–60 years. | Greece |
| Ko et al, 2007 | 2000–2002 | Mean BMI was 24.1 (SD 3.3) for men & 22.6 (SD 3.5) for women | In fully adjusted models, sleep duration significantly predicted BMI in men only (beta = −.051). | Hong Kong |
Table 4.
Summary of findings from studies examining the association between sleep and body mass index (BMI) in children.
| Authors | Date of Study | Age (y) | % Overweight or obese | Results | Country |
|---|---|---|---|---|---|
| Locard et al, 1992 | 1988–1989 | 5 y | 4.8% obese | OR for obesity was: 4.9 (95% CI 1.9–12.7) for <10 h/night; 2.8 (95% CI 1.2– 6.3) for 10–11 h/night; reference group was > 12 h/night. | France |
| Gupta et al, 2002 | not reported | 11–16 y | 26.6% obese | Total sleep time adjusted OR was 0.20 (95% CI 0.11–0.34) predicting obesity | US |
| von Kries et al, 2002 | 1999–2000 | 5–6 y | 10% were overweight; 3% were obese. | Adjusted Odds Ratio for being overweight was 0.77 (95% CI 0.59–0.99) for sleep times 10.5–11 h/night; 0.54 (95%CI 0.40–0.73) for ≥11.5 h/night relative to ≤10 h/night. Adjusted OR for being obese was 0.53 (95% CI 0.35–0.80) for 10.5–11 h/night and 0.45 (95% CI 0.28–0.75) for ≥11.5 h/night. | Germany |
| Sekine et al, 2002 | 1989–1990 | 6–7 y | 12.9% obese | OR for obesity relative to ≥ 10 h sleep/night. < 8h: OR 3.06 (95% CI 1.72–5.36); 8–9 h: OR 2.01 (95% CI 1.43–2.91) | Japan |
| Agras et al, 2004 | not reported | Sleep measured at 3–5 y & overweight at 9.5 y | 25.3% overweight. | The difference in mean sleep at ages 3–5 y between those who became overweight and those who did not was 30 minutes, most of which was daytime sleep. | US |
| Reilly et al, 2005 | 1991–1992 | Sleep at 38 months & obesity at 7 y | 8.6% obese | OR for obesity was 1.45 (95% CI 1.10–1.89) for <10.5 h and 1.32 (95% CI 1.02–1.79) for 10.5–11.4 h relative to ≥12 h per night. | UK |
| Chaput et al, 2006 | 2002 | 5–10 y | 15.3% overweight, 6.75 obese) | OR for overweight/obesity was 3.45 (95% CI 2.61–4.67) for 8–10 h and 1.42 (95% CI 1.09–1.98) for 10.5– 11.5 h relative to 12–13 hours per night. | Canada |
| Knutson, 2005 | 1996 | Mean age 16 y | 11% overweight | OR for overweight among males was 0.90, (95% CI: 0.82–1.00). Not significant among females | US |
| Seicean et al 2007 | 2004 | 14–18 y | 20% overweight | Adjusted OR (95% CI) for overweight was <5 h: 7.65 (1.87, 31.30) 5–6h: 2.80 (1.00, 7.79) 6–7 h: 2.55 (1.02, 6.38) 7–8 h: 1.38 (0.54, 3.53) >8: reference |
US |
| Eisenmann et al, 2006 | 1985 | 7–15 y | 9% of males & 10.6% of females were overweight 1.8% of males & 1.5% of females were obese | In males, age-adjusted OR (95% CI) for overweight: ≤8 h: 3.06 (2.11, 4.46) 8–9 h: 1.83 (1.30, 2.58) 9–10 h: 1.61 (1.19, 2.17) ≥10 h: reference No association in females. |
Australia |
| Dieu et al 2007 | 2005 | 4–5 years | 20.5% were overweight & 16.3% were obese | In multivariate models, sleep duration was significantly associated with overweight/obesity (OR 0.87, 95% CI 0.78, 0.98) and obesity alone (OR 0.75, 95% CI 0.60, 0.94) | Vietnam |
| Lumeng et al 2007 | 2000–2003 | 8–13 years | 18% were overweight in 6th grade | Sleep duration in 3rd grade predicted overweight in 6th grade (OR 0.60, 95% CI 0.36, 0.99) after adjustment for gender, race, SES, BMI in 3rd grade and change in sleep duration between 3rd and 6th grade. | US |
Cross-sectional studies conducted in adults from Spain,75 Japan,76 and the U.S.77,78 have all observed a significant association between short sleep duration and being obese. Among studies conducted in the US, an analysis of data collected in 1982 by the American Cancer Society indicated a decrease in BMI with increasing sleep durations among men, but a U-shaped relationship between sleep duration and BMI among women.6 The Wisconsin Sleep Cohort Study conducted in 1995 found that the average time spent in bed based on diaries was significantly associated with BMI in a U-shaped manner after adjusting for age and sex, where the minimum BMI was observed at an average bedtime of 7.7 hours per night.70 Analysis of the Nurses Health Study data from over 80,000 women also observed a cross-sectional U-shaped association between sleep duration and BMI where the lowest mean BMI was observed among those reporting sleeping 7–8 hours per night.79 Longitudinal analysis of the same cohort observed that sleep durations less than 7 hours were at an increased risk of weight gain.80 In a study from France, mean BMI was slightly but significantly higher among women reporting sleeping 6 hours or less as compared to those sleeping more than 6 hours (24.4 vs. 23.4 kg/m2) after adjustment for age and area of residence, but no difference was observed among men.81 A study of a rural population in the US observed a negative association between sleep duration and BMI.82 A study from Norway found that sleep durations less than 6 hours were associated with higher BMI in 40–45 year olds.83 Analysis of a sample of adults from Hong Kong revealed a significant negative association between sleep duration and BMI in men.84 Among Greek women aged 30–60 years longer nocturnal sleep duration based on a week of sleep diaries was a significant predictor of lower BMI, lower percent body fat and lower sum of skinfolds.85 However, a study of women 50 years or older from Quebec, Canada, found no significant differences in adiposity indices or BMI between short (<7 hours) and long (≥7 hours) sleepers.86
All of the studies discussed above were cross-sectional in design, however, two published studies in adults have exploited a longitudinal design. First, analysis of data from the first National Health and Nutrition Examination Survey (NHANES I) indicated that among the 32–49 year age group those reporting sleeping 2–4 h, 5 h or 6 h/night in 1982–84 had a higher mean BMI in 1982–84, 1987 and 1992 relative to those reporting 7 h/night in 1982–84.87 The second longitudinal study analyzed the association between sleep and BMI over a 13-year period, and reported that the odds ratio for sleep duration predicting obesity was 0.50, which means that every extra hour increase of sleep duration was associated with a 50% reduction in risk of obesity.88
Table 4 lists the studies to date that have observed a negative association between sleep and BMI among children. Cross-sectional studies in children in Canada,89 France,90 Germany,91 Japan92, the US,93 and Vietnam94 have found increased risk of overweight and/or obesity associated with short sleep durations. A prospective study in the US followed children from birth to 9.5 years of age, and sleep duration was assessed annually between the ages of 2 to 5 years.95 Average sleep duration between the ages 3 to 5 years was negatively associated with being overweight at 9.5 years of age, however the difference in sleep between overweight and lean children was primarily due to daytime naps and not to nocturnal sleep.95 A study in the UK collected sleep duration information at 38 months of age and examined obesity at age 7 years, and observed that sleep durations of <10.5 h and 10.5–11.4 h were associated with an increased risk of obesity at age 7 relative to sleep durations of ≥ 12 h per night.96 A longitudinal study of children in the US found that short sleep duration in 3rd grade (when they were 9 years old) was significantly associated with increased risk of overweight in 6th grade (when they were 12 years old) after adjustment for gender, race, maternal education, BMI in 3rd grade and the change in sleep duration.97 An analysis of the National Longitudinal Study of Adolescent Health in the US indicated that self-reported sleep duration was weakly associated with BMI z score and risk of overweight among male adolescents but not among females.98 An Australian study of children aged 7 to 15 years also found that short sleep duration was significantly associated with overweight in males only.99 All of these studies in children used subjective measures of sleep duration either reported by the parent or by the adolescent. One study in the US, however, used a 24-hour period of actigraphy recording to measure sleep in 383 adolescents aged 11–16 years.100 This study defined obesity as a BMI above the 85th percentile for sex and age as well as having a percent body fat of 25% or above for males or 30% or above for females, as measured by bioelectrical impedance.100 After adjusting for age, sex, sexual maturity, and ethnicity, total sleep time had an odds ratio of 0.20 (95% CI 0.11–0.34) predicting obesity, which indicates that every extra hour of sleep is associated with an 80% reduction in risk of being obese.100
While the epidemiologic evidence for an association between short sleep and obesity is becoming quite impressive, it should be noted that nearly all studies, whether in adults or children, relied on subjective reports of sleep duration. Some studies in children suggest that self-reported sleep duration is fairly accurate,101 however, more studies are needed to determine the accuracy with which adults report their sleep duration and quality. Also, the majority of studies were cross-sectional in design, which means that the direction of causality cannot be inferred. Short sleep could lead to weight gain, but overweight or obesity could also lead to an inability to obtain sufficient amounts of sleep. Future studies need to overcome these limitations by employing a prospective and interventional design with objective measures of sleep and adiposity.
Sleep Loss & Energy Expenditure
Energy expenditure plays an important role in the control of body weight and adiposity. The total amount of daily energy expenditure (TEE) is divided into 3 components: a). Resting metabolic rate under basal conditions (RMR), which is measured as the energy expenditure of an individual resting in bed in the morning after sleep in the fasting state; RMR represents approximately 60% of TEE in people with sedentary occupations; b) Thermic effect of meals (TEM), which is the energy expenditure associated with the digestion, absorption, metabolism and storage of food and accounts for approximately 10% of TEE; and c) activity-related energy expenditure (AEE), which is the energy expended in all volitional and non-volitional activities. For most people, the majority of AEE is not accounted for by physical exercise but rather by low and moderate intensity activities such as sitting, standing, walking and other occupational, volitional and spontaneous activities, collectively referred to as non-exercise activity thermogenesis (NEAT).102 NEAT in humans may be considered the equivalent of spontaneous physical activity (SPA) in rodent models. AEE is the most variable component of TEE and plays a major role in the homeostatic control of body weight.103 The ability to increase NEAT when caloric intake is excessive can contribute to the maintenance of body weight. Obese individuals have lower levels of NEAT than lean subjects.104 Studies have indicated that increased physical activity is critical for maintenance of weight loss.105
Whether sleep loss in humans has an impact on TEE or its components has not been directly tested. While numerous studies have shown that recurrent sleep restriction is associated with cumulative deficits in sleepiness, vigilance and neurobehavioral function, it is not known whether these deficits are associated with reduced voluntary physical exercise and/or reduced NEAT. Subjects with sleep problems and/or excessive daytime sleepiness report a significant reduction in their levels of physical activity and energy,106, 107 which could indeed reduce AEE. Thus, it is possible sleep loss may impact human energy expenditure directly, but there is currently no direct evidence in support of this hypothesis. Discrepant effects of sleep loss on body weight have been observed in rodent as compared to human studies. In rats, studies have been consistent in indicating that total or partial sleep deprivation results in a marked increase in food intake along with weight loss, indicating a negative energy balance.61, 62, 108, 109 In humans, limited evidence suggests that hyperphagia may also occur during sleep deprivation,74 but, in contrast to rodents, this hyperphagia may be associated with weight gain rather than weight loss. Sleep depriving a laboratory animal for extended periods of time invariably involves increased physical activity and stress. While it is difficult to make meaningful comparisons of the impact of sleep deprivation between rats subjected to increased physical activity and repeated water immersions (as in studies using the disk-over-water procedure for sleep deprivation) and human volunteers studied under comfortable sedentary conditions at room temperature, it is likely that major species differences underlie the discrepant findings in rodents versus humans. Energy metabolism in brown adipose tissue, which is abundant in rodents but rarely found in humans, could be involved in differences in energy balance during sleep deprivation between humans and rodents.
Sleep loss could also affect energy expenditure via its impact on the levels of leptin and ghrelin. Indeed, in rodent models, there is good evidence to indicate that leptin and ghrelin have opposite effects on energy expenditure. Leptin appears to increase energy expenditure, possibly via increased thermogenesis in brown adipose tissue,110 while central ghrelin administration has been reported to decrease locomotor activity in rats.111 Exogenous leptin administration in lean mice prevents the decrease in energy expenditure typically associated with the reduction in food intake.112 Studies that administered leptin to obese and lean human subjects observed no effect on energy expenditure.113–116 However, one study of 4 human subjects observed that leptin reversed the decrease in energy expenditure experienced during sustained weight reduction,117 which is similar to the findings in rats. Since several human studies have demonstrated reduced levels of leptin after sleep loss,29, 69, 70 it is possible that the reduction in leptin is associated with a reduction in energy expenditure. Similarly, the increase in ghrelin after partial sleep restriction could be associated with a decrease in NEAT. Experimental evidence is currently lacking to support either hypothesis. In summary, the various putative pathways through which sleep loss might adversely affect energy balance and lead to weight gain include factors such as alterations in appetite and glucose regulation, increased food intake and reduced energy expenditure.
Conclusion
The research reviewed here suggests that chronic partial sleep loss may increase the risk of obesity and diabetes via multiple pathways, including an adverse effect on parameters of glucose regulation, a dysregulation of the neuroendocrine control of appetite leading to excessive food intake, and decreased energy expenditure. Epidemiological studies have generally supported the laboratory findings. As the causes of the dramatic increase in the prevalence of obesity and diabetes that has occurred worldwide over the past few decades remain to be fully elucidated, the possibility that chronic partial sleep curtailment, a novel behavior that appears to have developed with the advent of the 24-h society, is very intriguing, particularly because sleep habits may be amenable to behavioral intervention.
Acknowledgments
Portions of this text are excerpts from Sleep Medicine Reviews, Vol 11, Kristen L. Knutson, Karine Spiegel, Plamen Penev, Eve Van Cauter, The metabolic consequences of sleep deprivation, 163---178, Copyright (2007), with permission from Elsevier Ltd. Some of the research described in this chapter was supported by grants PO1 AG-11412, RO1 HL-075079, P60 DK-20595 and MO1 RR-00055.
References
- 1.Mokdad A, Bowman B, Ford E, Vinicor F, Marks J, Koplan J. The Continuing Epidemics of Obesity and Diabetes in the United States. JAMA. 2001;286(10):1195–1200. doi: 10.1001/jama.286.10.1195. [DOI] [PubMed] [Google Scholar]
- 2.Ettaro L, Songer TJ, Zhang P, Engelgau MM. Cost-of-illness studies in diabetes mellitus. Pharmacoeconomics. 2004;22(3):149–164. doi: 10.2165/00019053-200422030-00002. [DOI] [PubMed] [Google Scholar]
- 3.Kripke D, Simons R, Garfinkel L, Hammond E. Short and long sleep and sleeping pills. Is increased mortality associated? Archives of General Psychiatry. 1979;36(1):103–116. doi: 10.1001/archpsyc.1979.01780010109014. [DOI] [PubMed] [Google Scholar]
- 4.Gallup Organization. Sleep in America. Princeton, NJ: Gallup Organization; 1995. [Google Scholar]
- 5.National Center for Health Statistics. QuickStats: Percentage of adults who reported an average of ≤ 6 hours of sleep per 24-hour period, by sex and age group - United States, 1985 and 2004. MMWR Morb Mortal Wkly Rep. 2005;54(37):933. [Google Scholar]
- 6.Kripke DF, Garfinkel L, Wingard DL, Klauber MR, Marler MR. Mortality associated with sleep duration and insomnia. Arch Gen Psychiatry. 2002;59(2):131–136. doi: 10.1001/archpsyc.59.2.131. [DOI] [PubMed] [Google Scholar]
- 7.Ayas NT, White DP, Al-Delaimy WK, et al. A prospective study of self-reported sleep duration and incident diabetes in women. Diabetes Care. 2003 Feb;26(2):380–384. doi: 10.2337/diacare.26.2.380. [DOI] [PubMed] [Google Scholar]
- 8.Ayas NT, White DP, Manson JE, et al. A prospective study of sleep duration and coronary heart disease in women. Arch Intern Med. 2003 Jan 27;163(2):205–209. doi: 10.1001/archinte.163.2.205. [DOI] [PubMed] [Google Scholar]
- 9.Ip MS, Lam B, Ng MM, Lam WK, Tsang KW, Lam KS. Obstructive sleep apnea is independently associated with insulin resistance. Am J Respir Crit Care Med. 2002;165(5):670–676. doi: 10.1164/ajrccm.165.5.2103001. [DOI] [PubMed] [Google Scholar]
- 10.Punjabi NM, Shahar E, Redline S, Gottlieb DJ, Givelber R, Resnick HE. Sleep-disordered breathing, glucose intolerance, and insulin resistance: the Sleep Heart Health Study. Am J Epidemiol. 2004 Sep 15;160(6):521–530. doi: 10.1093/aje/kwh261. [DOI] [PubMed] [Google Scholar]
- 11.Van Cauter E, Polonsky KS, Scheen AJ. Roles of circadian rhythmicity and sleep in human glucose regulation. Endocr Rev. 1997;18:716–738. doi: 10.1210/edrv.18.5.0317. [DOI] [PubMed] [Google Scholar]
- 12.Scheen AJ, Byrne MM, Plat L, Van Cauter E. Relationships between sleep quality and glucose regulation in normal humans. Am J Physiol. 1996;271:E261–E270. doi: 10.1152/ajpendo.1996.271.2.E261. [DOI] [PubMed] [Google Scholar]
- 13.Nofzinger EA, Buysse DJ, Miewald JM, et al. Human regional cerebral glucose metabolism during non-rapid eye movement sleep in relation to waking. Brain. 2002 May;125(Pt 5):1105–1115. doi: 10.1093/brain/awf103. [DOI] [PubMed] [Google Scholar]
- 14.Maquet P. Functional neuroimaging of normal human sleep by positron emission tomography. J Sleep Res. 2000 Sep;9(3):207–231. doi: 10.1046/j.1365-2869.2000.00214.x. [DOI] [PubMed] [Google Scholar]
- 15.Van Cauter E, Blackman JD, Roland D, Spire JP, Refetoff S, Polonsky KS. Modulation of glucose regulation and insulin secretion by circadian rhythmicity and sleep. J Clin Invest. 1991;88:934–942. doi: 10.1172/JCI115396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kuhn E, Brodan V, Brodanova M, Rysanek K. Metabolic effects of sleep deprivation. Act Nerv Super (Praha) 1969 Nov 3;:165–174. [PubMed] [Google Scholar]
- 17.Spiegel K, Leproult R, Van Cauter E. Metabolic and Endocrine Changes. In: Kushida C, editor. Sleep Deprivation: Basic Science, Physiology, and Behavior. Vol. 192. New York: Marcel Dekker; 2005. pp. 293–318. [Google Scholar]
- 18.Spiegel K, Leproult R, Colecchia EF, et al. Adaptation of the 24-h growth hormone profile to a state of sleep debt. American Journal of Physiology - Regulatory Integrative & Comparative Physiology. 2000;279(3):R874–883. doi: 10.1152/ajpregu.2000.279.3.R874. [DOI] [PubMed] [Google Scholar]
- 19.Allan JS, Czeisler CA. Persistence of the circadian thyrotropin rhythm under constant conditions and after light-induced shifts of circadian phase. J Clin Endocrinol Metab. 1994;79:508–512. doi: 10.1210/jcem.79.2.8045970. [DOI] [PubMed] [Google Scholar]
- 20.Van Cauter E, Sturis J, Byrne MM, et al. Demonstration of rapid light-induced advances and delays of the human circadian clock using hormonal phase markers. Am J Physiol. 1994;266:E953–E963. doi: 10.1152/ajpendo.1994.266.6.E953. [DOI] [PubMed] [Google Scholar]
- 21.Spiegel K, Leproult R, Van Cauter E. Impact of sleep debt on metabolic and endocrine function. Lancet. 1999;354:1435–1439. doi: 10.1016/S0140-6736(99)01376-8. [DOI] [PubMed] [Google Scholar]
- 22.Bergman RN. Minimal model: perspective from 2005. Horm Res. 2005;64(Suppl 3):8–15. doi: 10.1159/000089312. [DOI] [PubMed] [Google Scholar]
- 23.Palmer ND, Langefeld CD, Campbell JK, et al. Genetic mapping of disposition index and acute insulin response loci on chromosome 11q. The Insulin Resistance Atherosclerosis Study (IRAS) Family Study. Diabetes. 2006 Apr;55(4):911–918. doi: 10.2337/diabetes.55.04.06.db05-0813. [DOI] [PubMed] [Google Scholar]
- 24.Bergman RN, Ader M, Huecking K, Van Citters G. Accurate assessment of beta-cell function: the hyperbolic correction. Diabetes. 2002 Feb;51(Suppl 1):S212–220. doi: 10.2337/diabetes.51.2007.s212. [DOI] [PubMed] [Google Scholar]
- 25.Xiang AH, Peters RK, Kjos SL, et al. Effect of pioglitazone on pancreatic beta-cell function and diabetes risk in Hispanic women with prior gestational diabetes. Diabetes. 2006 Feb;55(2):517–522. doi: 10.2337/diabetes.55.02.06.db05-1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Garcia G, Freeman R, Supiano M, Smith M, Galecki A, Halter J. Glucose metabolism in older adults: a study including subjects more than 80 years of age. J Am Geriatr Soc. 1997;45:813–817. doi: 10.1111/j.1532-5415.1997.tb01507.x. [DOI] [PubMed] [Google Scholar]
- 27.Prigeon RL, Kahn SE, Porte D., Jr Changes in insulin sensitivity, glucose effectiveness, and B-Cell function in regularly exercising subjects. Metabolism. 1995;44:1259–1263. doi: 10.1016/0026-0495(95)90026-8. [DOI] [PubMed] [Google Scholar]
- 28.Spiegel K, Knutson K, Leproult R, Tasali E, Van Cauter E. Sleep loss: a novel risk factor for insulin resistance and Type 2 diabetes. J Appl Physiol. 2005 Nov;99(5):2008–2019. doi: 10.1152/japplphysiol.00660.2005. [DOI] [PubMed] [Google Scholar]
- 29.Spiegel K, Leproult R, L’Hermite-Baleriaux M, Copinschi G, Penev P, Van Cauter E. Leptin levels are dependent on sleep duration: Relationships with sympathovagal balance, carbohydrate regulation, cortisol, and thyrotropin. J Clin Endocrinol Metab. 2004;89:5762–5771. doi: 10.1210/jc.2004-1003. [DOI] [PubMed] [Google Scholar]
- 30.VanHelder T, Symons JD, Radomski MW. Effects of sleep deprivation and exercise on glucose tolerance. Aviat Space Environ Med. 1993 Jun;64(6):487–492. [PubMed] [Google Scholar]
- 31.Youngstedt SD, Kripke DF. Long sleep and mortality: rationale for sleep restriction. Sleep Med Rev. 2004 Jun;8(3):159–174. doi: 10.1016/j.smrv.2003.10.002. [DOI] [PubMed] [Google Scholar]
- 32.Thomas M, Sing H, Belenky G, et al. Neural basis of alertness and cognitive performance impairments during sleepiness. I. Effects of 24 h of sleep deprivation on waking human regional brain activity. J Sleep Res. 2000 Dec;9(4):335–352. doi: 10.1046/j.1365-2869.2000.00225.x. [DOI] [PubMed] [Google Scholar]
- 33.Vgontzas AN, Papanicolaou DA, Bixler EO, et al. Circadian interleukin-6 secretion and quantity and depth of sleep. J Clin Endocrinol Metab. 1999 Aug;84(8):2603–2607. doi: 10.1210/jcem.84.8.5894. [DOI] [PubMed] [Google Scholar]
- 34.Vgontzas AN, Zoumakis E, Bixler EO, et al. Adverse effects of modest sleep restriction on sleepiness, performance, and inflammatory cytokines. Journal of Clinical Endocrinology & Metabolism. 2004;89(5):2119–2126. doi: 10.1210/jc.2003-031562. [DOI] [PubMed] [Google Scholar]
- 35.Tasali E, Leproult R, Ehrmann DA, Van Cauter E. Slow-wave sleep and the risk of type 2 diabetes in humans. Proc Natl Acad Sci U S A. 2008 Jan 22;105(3):1044–1049. doi: 10.1073/pnas.0706446105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Flint J, Kothare SV, Zihlif M, et al. Association between inadequate sleep and insulin resistance in obese children. J Pediatr. 2007 Apr;150(4):364–369. doi: 10.1016/j.jpeds.2006.08.063. [DOI] [PubMed] [Google Scholar]
- 37.Chaput JP, Despres JP, Bouchard C, Tremblay A. Association of sleep duration with type 2 diabetes and impaired glucose tolerance. Diabetologia. 2007 Nov;50(11):2298–2304. doi: 10.1007/s00125-007-0786-x. [DOI] [PubMed] [Google Scholar]
- 38.Knutson KL, Ryden AM, Mander BA, Van Cauter E. Role of sleep duration and quality in the risk and severity of type 2 diabetes mellitus. Arch Intern Med. 2006 Sep 18;166(16):1768–1774. doi: 10.1001/archinte.166.16.1768. [DOI] [PubMed] [Google Scholar]
- 39.Jennings JR, Muldoon MF, Hall M, Buysse DJ, Manuck SB. Self-reported sleep quality is associated with the metabolic syndrome. Sleep. 2007 Feb 1;30(2):219–223. doi: 10.1093/sleep/30.2.219. [DOI] [PubMed] [Google Scholar]
- 40.Kawakami N, Takatsuka N, Shimizu H. Sleep disturbance and onset of type 2 diabetes. Diabetes Care. 2004 Jan;27(1):282–283. doi: 10.2337/diacare.27.1.282. [DOI] [PubMed] [Google Scholar]
- 41.Nilsson PM, Roost M, Engstrom G, Hedblad B, Berglund G. Incidence of diabetes in middle-aged men is related to sleep disturbances. Diabetes Care. 2004 Oct;27(10):2464–2469. doi: 10.2337/diacare.27.10.2464. [DOI] [PubMed] [Google Scholar]
- 42.Mallon L, Broman JE, Hetta J. High incidence of diabetes in men with sleep complaints or short sleep duration: a 12-year follow-up study of a middle-aged population. Diabetes Care. 2005 Nov;28(11):2762–2767. doi: 10.2337/diacare.28.11.2762. [DOI] [PubMed] [Google Scholar]
- 43.Bjorkelund C, Bondyr-Carlsson D, Lapidus L, et al. Sleep disturbances in midlife unrelated to 32-year diabetes incidence: the prospective population study of women in Gothenburg. Diabetes Care. 2005 Nov;28(11):2739–2744. doi: 10.2337/diacare.28.11.2739. [DOI] [PubMed] [Google Scholar]
- 44.Meisinger C, Heier M, Loewel H. Sleep disturbance as a predictor of type 2 diabetes mellitus in men and women from the general population. Diabetologia. 2005 Feb;48(2):235–241. doi: 10.1007/s00125-004-1634-x. [DOI] [PubMed] [Google Scholar]
- 45.Yaggi HK, Araujo AB, McKinlay JB. Sleep duration as a risk factor for the development of type 2 diabetes. Diabetes Care. 2006 Mar;29(3):657–661. doi: 10.2337/diacare.29.03.06.dc05-0879. [DOI] [PubMed] [Google Scholar]
- 46.Gangwisch JE, Heymsfield SB, Boden-Albala B, et al. Sleep Duration as a Risk Factor for Diabetes Incidence in a Large US Sample. Sleep. 2007;30(12):1667–1673. doi: 10.1093/sleep/30.12.1667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gale SM, Castracane VD, Mantzoros CS. Energy homeostasis, obesity and eating disorders: recent advances in endocrinology. J Nutr. 2004 Feb;134(2):295–298. doi: 10.1093/jn/134.2.295. [DOI] [PubMed] [Google Scholar]
- 48.van der Lely A, Tschop M, Heiman M, Ghigo E. Biological, physiological, pathophysiological, and pharmacological aspects of ghrelin. Endocr Rev. 2004;25(3):426–457. doi: 10.1210/er.2002-0029. [DOI] [PubMed] [Google Scholar]
- 49.Schoeller DA, Cella LK, Sinha MK, Caro JF. Entrainment of the diurnal rhythm of plasma leptin to meal timing. J Clin Invest. 1997;100:1882–1887. doi: 10.1172/JCI119717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Simon C, Gronfier C, Schlienger JL, Brandenberger G. Circadian and ultradian variations of leptin in normal man under continuous enteral nutrition: Relationship to sleep and body temperature. J Clin Endocrinol Metab. 1998;83:1893–1899. doi: 10.1210/jcem.83.6.4864. [DOI] [PubMed] [Google Scholar]
- 51.Dzaja A, Dalal MA, Himmerich H, Uhr M, Pollmacher T, Schuld A. Sleep enhances nocturnal plasma ghrelin levels in healthy subjects. Am J Physiol Endocrinol Metab. 2004 Jun;286(6):E963–967. doi: 10.1152/ajpendo.00527.2003. [DOI] [PubMed] [Google Scholar]
- 52.Danguir J, Nicolaidis S. Dependence of sleep on nutrients’ availability. Physiology & Behavior. 1979;22(4):735–740. doi: 10.1016/0031-9384(79)90240-3. [DOI] [PubMed] [Google Scholar]
- 53.Rechtschaffen A, Bergmann BM. Sleep deprivation in the rat by the disk-over-water method. Behav Brain Res. 1995;69(1–2):55–63. doi: 10.1016/0166-4328(95)00020-t. [DOI] [PubMed] [Google Scholar]
- 54.Taheri S, Zeitzer JM, Mignot E. The role of hypocretins (orexins) in sleep regulation and narcolepsy. Annu Rev Neurosci. 2002;25:283–313. doi: 10.1146/annurev.neuro.25.112701.142826. [DOI] [PubMed] [Google Scholar]
- 55.Sakurai T. Roles of orexin/hypocretin in regulation of sleep/wakefulness and energy homeostasis. Sleep Med Rev. 2005 Aug;9(4):231–241. doi: 10.1016/j.smrv.2004.07.007. [DOI] [PubMed] [Google Scholar]
- 56.Saper CB, Chou TC, Elmquist JK. The need to feed: homeostatic and hedonic control of eating. Neuron. 2002 Oct 10;36(2):199–211. doi: 10.1016/s0896-6273(02)00969-8. [DOI] [PubMed] [Google Scholar]
- 57.Samson WK, Taylor MM, Ferguson AV. Non-sleep effects of hypocretin/orexin. Sleep Med Rev. 2005 Aug;9(4):243–252. doi: 10.1016/j.smrv.2004.07.006. [DOI] [PubMed] [Google Scholar]
- 58.Harris GC, Wimmer M, Aston-Jones G. A role for lateral hypothalamic orexin neurons in reward seeking. Nature. 2005 Sep 22;437(7058):556–559. doi: 10.1038/nature04071. [DOI] [PubMed] [Google Scholar]
- 59.Harris GC, Aston-Jones G. Arousal and reward: a dichotomy in orexin function. Trends Neurosci. 2006 Oct;29(10):571–577. doi: 10.1016/j.tins.2006.08.002. [DOI] [PubMed] [Google Scholar]
- 60.Modirrousta M, Mainville L, Jones BE. Orexin and MCH neurons express c-Fos differently after sleep deprivation vs. recovery and bear different adrenergic receptors. Eur J Neurosci. 2005 May;21(10):2807–2816. doi: 10.1111/j.1460-9568.2005.04104.x. [DOI] [PubMed] [Google Scholar]
- 61.Everson CA, Crowley WR. Reductions in circulating anabolic hormones induced by sustained sleep deprivation in rats. Am J Physiol Endocrinol Metab. 2004 Jun;286(6):E1060–1070. doi: 10.1152/ajpendo.00553.2003. [DOI] [PubMed] [Google Scholar]
- 62.Koban M, Swinson KL. Chronic REM-sleep deprivation of rats elevates metabolic rate and increases UCP1 gene expression in brown adipose tissue. Am J Physiol Endocrinol Metab. 2005 Jul;289(1):E68–74. doi: 10.1152/ajpendo.00543.2004. [DOI] [PubMed] [Google Scholar]
- 63.Koban M, Le WW, Hoffman GE. Changes in hypothalamic corticotropin-releasing hormone, neuropeptide Y, and proopiomelanocortin gene expression during chronic rapid eye movement sleep deprivation of rats. Endocrinology. 2006 Jan;147(1):421–431. doi: 10.1210/en.2005-0695. [DOI] [PubMed] [Google Scholar]
- 64.Bodosi B, Gardi J, Hajdu I, Szentirmai E, Obal F, Jr, Krueger JM. Rhythms of ghrelin, leptin, and sleep in rats: effects of the normal diurnal cycle, restricted feeding, and sleep deprivation. Am J Physiol Regul Integr Comp Physiol. 2004 Nov;287(5):R1071–1079. doi: 10.1152/ajpregu.00294.2004. [DOI] [PubMed] [Google Scholar]
- 65.Laposky AD, Bass J, Kohsaka A, Turek FW. Sleep and circadian rhythms: Key components in the regulation of energy metabolism. FEBS Lett. 2007 Aug 14; doi: 10.1016/j.febslet.2007.06.079. [DOI] [PubMed] [Google Scholar]
- 66.Imaki M, Hatanaka Y, Ogawa Y, Yoshida Y, Tanada S. An epidemiological study on relationship between the hours of sleep and life style factors in Japanese factory workers. J Physiol Anthropol Appl Human Sci. 2002 Mar;21(2):115–120. doi: 10.2114/jpa.21.115. [DOI] [PubMed] [Google Scholar]
- 67.Ohida T, Kamal AM, Uchiyama M, et al. The influence of lifestyle and health status factors on sleep loss among the Japanese general population. Sleep. 2001 May 1;24(3):333–338. doi: 10.1093/sleep/24.3.333. [DOI] [PubMed] [Google Scholar]
- 68.Mullington JM, Chan JL, Van Dongen HP, et al. Sleep loss reduces diurnal rhythm amplitude of leptin in healthy men. J Neuroendocrinol. 2003;15(9):851–854. doi: 10.1046/j.1365-2826.2003.01069.x. [DOI] [PubMed] [Google Scholar]
- 69.Spiegel K, Tasali E, Penev P, Van Cauter E. Sleep curtailment in healthy young men is associated with decreased leptin levels, elevated ghrelin levels and increased hunger and appetite. Ann Intern Med. 2004;141(11):846–850. doi: 10.7326/0003-4819-141-11-200412070-00008. [DOI] [PubMed] [Google Scholar]
- 70.Taheri S, Lin L, Austin D, Young T, Mignot E. Short sleep duration is associated with reduced leptin, elevated ghrelin, and increased body mass index. PLoS Medicine. 2004;1(3):e62. doi: 10.1371/journal.pmed.0010062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Littman AJ, Vitiello MV, Foster-Schubert K, et al. Sleep, ghrelin, leptin and changes in body weight during a 1-year moderate-intensity physical activity intervention. Int J Obes (Lond) 2006 Aug 15;31(3):466–475. doi: 10.1038/sj.ijo.0803438. [DOI] [PubMed] [Google Scholar]
- 72.Chen K, Li F, Li J, et al. Induction of leptin resistance through direct interaction of C-reactive protein with leptin. Nat Med. 2006 Apr;12(4):425–432. doi: 10.1038/nm1372. [DOI] [PubMed] [Google Scholar]
- 73.Meier-Ewert HK, Ridker PM, Rifai N, et al. Effect of sleep loss on C-reactive protein, an inflammatory marker of cardiovascular risk. J Am Coll Cardiol. 2004 Feb 18;43(4):678–683. doi: 10.1016/j.jacc.2003.07.050. [DOI] [PubMed] [Google Scholar]
- 74.Dinges D, Chugh D. Physiological correlates of sleep deprivation. In: Kinney J, Tucker H, editors. Physiology, Stress, and Malnutrition: Functional Correlates, Nutritional Intervention. Philadelphia, PA: Lippincott Williams & Wilkins; 1997. p. 668. [Google Scholar]
- 75.Vioque J, Torres A, Quiles J. Time spent watching television, sleep duration and obesity in adults living in Valencia, Spain. Int J Obes Relat Metab Disord. 2000;24(12):1683–1688. doi: 10.1038/sj.ijo.0801434. [DOI] [PubMed] [Google Scholar]
- 76.Shigeta H, Shigeta M, Nakazawa A, Nakamura N, Yoshikawa T. Lifestyle, obesity, and insulin resistance. Diabetes Care. 2001 Mar;24(3):608. doi: 10.2337/diacare.24.3.608. [DOI] [PubMed] [Google Scholar]
- 77.Vorona R, Winn M, Babineau T, Eng B, Feldman H, Ware J. Overweight and Obese Patients in a Primary Care Population Report Less Sleep Than Patients With a Normal Body Mass Index. Arch Intern Med. 2005;165:25–30. doi: 10.1001/archinte.165.1.25. [DOI] [PubMed] [Google Scholar]
- 78.Singh M, Drake CL, Roehrs T, Hudgel DW, Roth T. The association between obesity and short sleep duration: A population-based study. Journal of Clinical Sleep Medicine. 2005;1(4):357–363. [PubMed] [Google Scholar]
- 79.Patel SR, Ayas NT, Malhotra MR, et al. A prospective study of sleep duration and mortality risk in women. Sleep. 2004;27(3):440–444. doi: 10.1093/sleep/27.3.440. [DOI] [PubMed] [Google Scholar]
- 80.Patel SR, Malhotra A, White DP, Gottlieb DJ, Hu FB. Association between Reduced Sleep and Weight Gain in Women. Am J Epidemiol. 2006 Nov 15;164(10):947–954. doi: 10.1093/aje/kwj280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Cournot M, Ruidavets JB, Marquie JC, Esquirol Y, Baracat B, Ferrieres J. Environmental factors associated with body mass index in a population of Southern France. Eur J Cardiovasc Prev Rehabil. 2004 Aug;11(4):291–297. doi: 10.1097/01.hjr.0000129738.22970.62. [DOI] [PubMed] [Google Scholar]
- 82.Kohatsu ND, Tsai R, Young T, et al. Sleep duration and body mass index in a rural population. Arch Intern Med. 2006 Sep 18;166(16):1701–1705. doi: 10.1001/archinte.166.16.1701. [DOI] [PubMed] [Google Scholar]
- 83.Bjorvatn B, Sagen IM, Oyane N, et al. The association between sleep duration, body mass index and metabolic measures in the Hordaland Health Study. J Sleep Res. 2007 Mar;16(1):66–76. doi: 10.1111/j.1365-2869.2007.00569.x. [DOI] [PubMed] [Google Scholar]
- 84.Ko GT, Chan JC, Chan AW, et al. Association between sleeping hours, working hours and obesity in Hong Kong Chinese: the ‘better health for better Hong Kong’ health promotion campaign. Int J Obes (Lond) 2007 Feb;31(2):254–260. doi: 10.1038/sj.ijo.0803389. [DOI] [PubMed] [Google Scholar]
- 85.Rontoyanni VG, Baic S, Cooper AR. Association between nocturnal sleep duration, body fatness, and dietary intake in Greek women. Nutrition. 2007 Nov-Dec;23(11–12):773–777. doi: 10.1016/j.nut.2007.07.005. [DOI] [PubMed] [Google Scholar]
- 86.Chaput JP, Lord C, Aubertin-Leheudre M, Dionne IJ, Khalil A, Tremblay A. Is overweight/obesity associated with short sleep duration in older women? Aging Clin Exp Res. 2007 Aug;19(4):290–294. doi: 10.1007/BF03324704. [DOI] [PubMed] [Google Scholar]
- 87.Gangwisch JE, Malaspina D, Boden-Albala B, Heymsfield SB. Inadequate Sleep as a Risk Factor for Obesity: Analyses of the NHANES I. Sleep. 2005;28(10):1289–1296. doi: 10.1093/sleep/28.10.1289. [DOI] [PubMed] [Google Scholar]
- 88.Hasler G, Buysse D, Klaghofer R, et al. The Association Between Short Sleep duration and Obesity in Young Adults: a 13-Year Prospective Study. Sleep. 2004;27(4):661–666. doi: 10.1093/sleep/27.4.661. [DOI] [PubMed] [Google Scholar]
- 89.Chaput JP, Brunet M, Tremblay A. Relationship between short sleeping hours and childhood overweight/obesity: results from the ‘Quebec en Forme’ Project. Int J Obes (Lond) 2006 Mar 14;30(7):1080–1085. doi: 10.1038/sj.ijo.0803291. [DOI] [PubMed] [Google Scholar]
- 90.Locard E, Mamelle N, Billette A, Miginiac M, Munoz F, Rey S. Risk factors of obesity in a five year old population. Parental versus environmental factors. International Journal of Obesity & Related Metabolic Disorders: Journal of the International Association for the Study of Obesity. 1992;16(10):721–729. [PubMed] [Google Scholar]
- 91.von Kries R, Toschke AM, Wurmser H, Sauerwald T, Koletzko B. Reduced risk for overweight and obesity in 5- and 6-y-old children by duration of sleep--a cross-sectional study. Int J Obes Relat Metab Disord. 2002;26(5):710–716. doi: 10.1038/sj.ijo.0801980. [DOI] [PubMed] [Google Scholar]
- 92.Sekine M, Yamagami T, Handa K, et al. A dose-response relationship between short sleeping hours and childhood obesity: results of the Toyama Birth Cohort Study. Child Care, Health and Development. 2002;28(2):163–170. doi: 10.1046/j.1365-2214.2002.00260.x. [DOI] [PubMed] [Google Scholar]
- 93.Seicean A, Redline S, Seicean S, et al. Association between short sleeping hours and overweight in adolescents: results from a US Suburban High School survey. Sleep Breath. 2007 Dec;11(4):285–293. doi: 10.1007/s11325-007-0108-z. [DOI] [PubMed] [Google Scholar]
- 94.Dieu HT, Dibley MJ, Sibbritt D, Hanh TT. Prevalence of overweight and obesity in preschool children and associated socio-demographic factors in Ho Chi Minh City, Vietnam. Int J Pediatr Obes. 2007;2(1):40–50. doi: 10.1080/17477160601103922. [DOI] [PubMed] [Google Scholar]
- 95.Agras WS, Hammer LD, McNicholas F, Kraemer HC. Risk factors for childhood overweight: a prospective study from birth to 9.5 years. J Pediatr. 2004 Jul;145(1):20–25. doi: 10.1016/j.jpeds.2004.03.023. [DOI] [PubMed] [Google Scholar]
- 96.Reilly J, Armstrong J, Dorosty A, et al. Early life risk factors for obesity in childhood: cohort study. Br Med J. 2005;330(7504):1357. doi: 10.1136/bmj.38470.670903.E0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Lumeng JC, Somashekar D, Appugliese D, Kaciroti N, Corwyn RF, Bradley RH. Shorter sleep duration is associated with increased risk for being overweight at ages 9 to 12 years. Pediatrics. 2007 Nov;120(5):1020–1029. doi: 10.1542/peds.2006-3295. [DOI] [PubMed] [Google Scholar]
- 98.Knutson KL. Sex differences in the association between sleep and body mass index in adolescents. J Pediatr. 2005 Dec;147(6):830–834. doi: 10.1016/j.jpeds.2005.07.019. [DOI] [PubMed] [Google Scholar]
- 99.Eisenmann JC, Ekkekakis P, Holmes M. Sleep duration and overweight among Australian children and adolescents. Acta Paediatr. 2006 Aug;95(8):956–963. doi: 10.1080/08035250600731965. [DOI] [PubMed] [Google Scholar]
- 100.Gupta NK, Mueller WH, Chan W, Meininger JC. Is obesity associated with poor sleep quality in adolescents? American Journal of Human Biology. 2002;14(6):762–768. doi: 10.1002/ajhb.10093. [DOI] [PubMed] [Google Scholar]
- 101.Gaina A, Sekine M, Chen X, Hamanishi S, Kagamimori S. Validity of child sleep diary questionnaire among junior high school children. J Epidemiol. 2004 Jan;14(1):1–4. doi: 10.2188/jea.14.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Westerterp KR. Pattern and intensity of physical activity. Nature. 2001 Mar 29;410(6828):539. doi: 10.1038/35069142. [DOI] [PubMed] [Google Scholar]
- 103.Rising R, Harper IT, Fontvielle AM, Ferraro RT, Spraul M, Ravussin E. Determinants of total daily energy expenditure: variability in physical activity. Am J Clin Nutr. 1994 Apr;59(4):800–804. doi: 10.1093/ajcn/59.4.800. [DOI] [PubMed] [Google Scholar]
- 104.Levine JA, Lanningham-Foster LM, McCrady SK, et al. Interindividual variation in posture allocation: possible role in human obesity. Science. 2005 Jan 28;307(5709):584–586. doi: 10.1126/science.1106561. [DOI] [PubMed] [Google Scholar]
- 105.Hill JO, Wyatt HR. Role of physical activity in preventing and treating obesity. J Appl Physiol. 2005 Aug;99(2):765–770. doi: 10.1152/japplphysiol.00137.2005. [DOI] [PubMed] [Google Scholar]
- 106.Weaver TE, Laizner AM, Evans LK, et al. An instrument to measure functional status outcomes for disorders of excessive sleepiness. Sleep. 1997 Oct;20(10):835–843. [PubMed] [Google Scholar]
- 107.Briones B, Adams N, Strauss M, et al. Relationship Between Sleepiness and General Health Status. Sleep. 1996;19(7):583–588. doi: 10.1093/sleep/19.7.583. [DOI] [PubMed] [Google Scholar]
- 108.Everson CA, Bergmann BM, Rechtschaffen A. Sleep Deprivation in the Rat: III. Total Sleep Deprivation. Sleep. 1989;12(1):13–21. doi: 10.1093/sleep/12.1.13. [DOI] [PubMed] [Google Scholar]
- 109.Rechtschaffen A, Bergmann BM. Sleep deprivation in the rat: an update of the 1989 paper. Sleep. 2002 Feb 1;25(1):18–24. doi: 10.1093/sleep/25.1.18. [DOI] [PubMed] [Google Scholar]
- 110.Scarpace PJ, Matheny M, Pollock BH, Tumer N. Leptin increases uncoupling protein expression and energy expenditure. Am J Physiol. 1997 Jul;273(1 Pt 1):E226–230. doi: 10.1152/ajpendo.1997.273.1.E226. [DOI] [PubMed] [Google Scholar]
- 111.Tang-Christensen M, Vrang N, Ortmann S, Bidlingmaier M, Horvath TL, Tschop M. Central administration of ghrelin and agouti-related protein (83–132) increases food intake and decreases spontaneous locomotor activity in rats. Endocrinology. 2004 Oct;145(10):4645–4652. doi: 10.1210/en.2004-0529. [DOI] [PubMed] [Google Scholar]
- 112.Halaas JL, Boozer C, Blair-West J, Fidahusein N, Denton DA, Friedman JM. Physiological response to long-term peripheral and central leptin infusion in lean and obese mice. Proc Natl Acad Sci U S A. 1997 Aug 5;94(16):8878–8883. doi: 10.1073/pnas.94.16.8878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Hukshorn CJ, Saris WH, Westerterp-Plantenga MS, Farid AR, Smith FJ, Campfield LA. Weekly subcutaneous pegylated recombinant native human leptin (PEG-OB) administration in obese men. J Clin Endocrinol Metab. 2000 Nov;85(11):4003–4009. doi: 10.1210/jcem.85.11.6955. [DOI] [PubMed] [Google Scholar]
- 114.Westerterp-Plantenga MS, Saris WH, Hukshorn CJ, Campfield LA. Effects of weekly administration of pegylated recombinant human OB protein on appetite profile and energy metabolism in obese men. Am J Clin Nutr. 2001 Oct;74(4):426–434. doi: 10.1093/ajcn/74.4.426. [DOI] [PubMed] [Google Scholar]
- 115.Hukshorn CJ, Westerterp-Plantenga MS, Saris WH. Pegylated human recombinant leptin (PEG-OB) causes additional weight loss in severely energy-restricted, overweight men. Am J Clin Nutr. 2003 Apr;77(4):771–776. doi: 10.1093/ajcn/77.4.771. [DOI] [PubMed] [Google Scholar]
- 116.Mackintosh RM, Hirsch J. The effects of leptin administration in non-obese human subjects. Obes Res. 2001 Aug;9(8):462–469. doi: 10.1038/oby.2001.60. [DOI] [PubMed] [Google Scholar]
- 117.Rosenbaum M, Murphy EM, Heymsfield SB, Matthews DE, Leibel RL. Low dose leptin administration reverses effects of sustained weight-reduction on energy expenditure and circulating concentrations of thyroid hormones. J Clin Endocrinol Metab. 2002 May;87(5):2391–2394. doi: 10.1210/jcem.87.5.8628. [DOI] [PubMed] [Google Scholar]