

Abstract
Purpose
Formulate phospho-sulindac (P-S, OXT-328) in a Pluronic hydrogel to be used as a topical anti-inflammatory agent and study its efficacy, safety and pharmacokinetics in an arthritis model.
Methods
LEW/crlBR rats with Freund's adjuvant-induced arthritis were treated with P-S formulated in Pluronic hydrogel (PSH). We determined the clinical manifestations of arthritis including the locomotor activity of the rats; evaluated joints for inflammation, bone resorption, cartilage damage, COX-2 expression and NF-κB activation; assayed plasma IL-6 and IL-10 levels; and studied the pharmacokinetics of P-S in rats after topical or oral administration.
Results
PSH applied at the onset of arthritis or when arthritis was fully developed, suppressed it by 56–82%, improved the locomotor activity of the rats 2.1–4.4 fold, suppressed synovial inflammation, bone resorption, cartilage damage, NF-κB activation and COX-2 expression but not plasma IL-6 and IL-10 levels. There were no side effects. PSH produced rapidly high local levels of P-S with < 14% of P-S reaching the circulation, while orally administered PS was rapidly metabolized generating much lower joint levels of P-S.
Conclusions
Topical application of PSH is efficacious and safe in the treatment of Freund's adjuvant-induced arthritis; has a favorable pharmacokinetic profile; and likely acts by suppressing key pro-inflammatory signaling pathways.
Keywords: arthritis, hydrogel, NSAIDs, phospho-sulindac, transdermal
Introduction
Rheumatoid arthritis (RA), an autoimmune disease characterized by chronic inflammation in articular synovial tissue, may result in joint destruction which can be incapacitating (1,2). RA is associated with substantial co-morbidities, mainly cardiovascular, neurological and metabolic. Despite substantial recent progress, RA remains a formidable clinical problem.
Nonsteroidal anti-inflammatory drugs (NSAIDs) are used to treat the inflammation and pain caused by RA. However, their use, especially when prolonged, is associated with gastrointestinal (GI) (3,4), cardiovascular (5–7) and other side effects (8). To overcome these limitations, we developed a novel modification of conventional NSAIDs in which a diethtyl-phosphate moiety is attached to their carboxylic moiety through a spacer molecule. Our data to date indicate that NSAIDs modified in this manner have enhanced efficacy compared to their parent compounds (9–11). Phospho-sulindac (P-S; OXT-328; Fig. 1) is such a compound, which was very efficacious when administered orally to rats with adjuvant-induced arthritis (9). P-S, which is not a prodrug of sulindac (11,12), showed a strong anti-inflammatory effect, inhibiting several pro-inflammatory pathways and reducing prostaglandin E2 levels. We have previously identified the metabolic pathways for P-S in vitro and in vivo (13). After oral delivery P-S undergoes reduction and oxidation reactions yielding P-S sulfide and P-S sulfone. Furthermore, P-S is hydrolyzed releasing sulindac, which generates sulindac sulfide and sulindac sulfone, all of which are glucuronidated. Liver and intestinal microsomes metabolize P-S extensively but cultured cells convert only 10% of it to P-S sulfide and P-S sulfone.
Fig. 1.

The PK profile of PSH and P-S. Muscle and blood levels of P-S and its metabolites after topical (PSH) or oral administration of P-S determined as in methods. The chemical structure of P-S is also shown. Left upper panel inset: muscle levels of P-S and metabolites after oral administration in the same scale as after topical administration, shown also in an expanded form.
Transdermal topical delivery of drugs offers favorable pharmacokinetics especially for drugs with short biological half-life, including reduced drug circulation, bypassing exposure to gastric fluids, and avoidance of first-pass metabolism (14–16). In addition, transdermal topical delivery is often well-accepted by patients because it is simple and, if needed, can be easily terminated. Indeed, transdermal topical delivery of conventional NSAIDs has been reported for ibuprofen and naproxen but not for sulindac (17–20). Besides higher efficacy, topical delivery of compounds may be accompanied by enhanced safety, as, among others, lower doses are often needed to achieve therapeutic effects comparable to those from systemic drug application (21,22). This is an important consideration for diseases like RA that may require prolonged treatment with anti-inflammatory agents (3,4).
Poloxamers, also known by the trade name Pluronics®, are nonionic triblock copolymers used for various drug delivery applications, including transdermal drug delivery (23). They consist of a central hydrophobic chain of poly (propylene oxide) (PPO) flanked by two hydrophilic chains of poly(ethylene oxide) (PEO). Because of their amphiphilic nature, poloxamers are used to increase the water solubility of hydrophobic molecules like P-S. In aqueous solutions poloxamers form micelles, in which PPO is the core surrounded by hydrophilic PEO molecules (24–26). With increasing polymer concentration, the micelles entangle with each other and eventually form a hydrogel, which is well-suited for topical drug delivery (26–30).
Based on the above considerations, we formulated P-S in a Pluronic P123 hydrogel (PSH) and studied its efficacy as a topically applied anti-inflammatory agent in the rat adjuvant arthritis model. Here, we demonstrate that PSH applied to the skin over the inflamed joints was highly efficacious in treating inflammatory arthritis; had excellent safety, likely explained by its pharmacokinetic (PK) properties; and inhibited COX-2 and NF-κB, two key inflammatory mediators.
Materials and Methods
P-S and Reagents
P-S was provided by Medicon Pharmaceuticals, Inc, Setauket, NY. Pluronic P123, poly(ethylene oxide) (PEG; MW=900.000), tetrahydrofuran (THF), acetonitrile (ACN) of high-performance-liquid-chromatography (HPLC) grade were purchased from Sigma-Aldrich, St. Louis, MO. Dialysis membrane (MWCO 3500) was purchased from Fisher-Scientific Pittsburg, PA.
Hydrogel Preparation and Characterization
Pluronic P123 and P-S were dissolved in THF (1:10 w/w) and the solution was dialyzed for 24 h in phosphate buffer saline (PBS, pH 7.4) at room temperature; the buffer was replaced thrice with fresh solution. After drying the dialysis sack with absorbent paper, we placed it under solid PEG (MW 900.000) until gel formation. There was no drug precipitation for at least 24 h after gel formation. Entrapped P-S was quantified with HPLC Waters Alliance 2695 equipped with a Waters 2998 photodiode array detector (328 nm) (Waters, Milford, MA) and a Thermo BDSHypersil C-18 column (150×4,6 mm, particle size 3 μm) (Thermo Firsher Scientific, Waltham, MA) by dissolving a small quantity of the gel into 1 ml of ACN. The mobile phase followed a gradient between buffer A (H2O, ACN, trifluoroacetic acid 94.9:5:0.1 v/v/v) and buffer B (ACN). A small amount of hydrogel was lyophilized and the polymer content was determined. Empty gel was prepared using the cold method (31). Briefly, 28% w/v of polymer was dispersed slowly in PBS at 2–5°C until the polymer was completely hydrated. The solution was then left at room temperature for gel formation.
Animal Studies
Female LEW/crlBR Lewis rats (Charles River, Wilmington, MA), weighing 110–140 g were kept under constant temperature (23±2°C) and humidity (55±15%) with a 12 h light/dark cycle (lights on at 0700 h) and had free access to food and water. All animal studies were approved by our Institutional Animal Care and Use Committee.
Induction of Arthritis
We induced arthritis in the rats following the protocol of Whitley et al. (32). Briefly, each rat received a single tail injection of 100 μl of 10 mg/ml Mycobacterium butyricum (DIFCO Laboratories, Detroit, MI, USA) suspended in incomplete Freund's adjuvant (FA). The animals presented signs of articular inflammation 12 days later; by day 18 the inflammation was pronounced.
PK Study and Treatment with P-S
For oral administration, P-S was dissolved in corn oil and given by gavage. A restraining collar was placed around their necks of animals with topically applied PSH, to avoid unintentional drug oral consumption.
PK Study
Rats were given a single dose of P-S 50 mg/kg either as PSH applied on both hind legs or by oral gavage. At predetermined time points, we sacrificed two animals per time point and collected blood and muscle tissue from the hind legs to determine drug levels. Determination of muscle levels offers significant analytical advantages over skin levels. Our studies (results not shown) revealed that it was not possible to remove quantitatively the P-S from the topically applied PSH using various approaches, including washing the skin with solvents appropriate for lipophilic compounds such as P-S, e.g. dimethylsulfoxide, or epidermis microdissection. In contrast, muscles close to the skin had no such “contamination” with residual P-S and provided reliable results.
Prevention Protocol
Rats with arthritis induced as above were randomly distributed into the following groups (n=5/group): i) control; ii) control hydrogel; iii) P-S 50 mg/kg orally; and iv) PSH 50 mg/kg topically; All treatments were given once daily starting on the day following FA injection (33). We also included a fifth group of rats without arthritis, i.e., not treated with FA. The progression of arthritis was evaluated using a scoring system in which each paw was assigned a score of 0 to 4. Thus, 0 = no swelling or redness in any joint; 4 = very severe swelling and redness in large and small joints; 1 – 3 = intermediate conditions based on specified criteria (32). At the completion of the study, animals were sacrificed 2 h after the last drug dose and blood was collected by cardiac puncture; the plasma, separated immediately by centrifugation, was stored at –80°C until analyzed. The hindpaws were transected with a guillotine, weighed and the joints were preserved in formalin. Muscles from both the hindpaws and forepaws were harvested after skin removal. The stomach and small intestine were also collected, dissected, rinsed thoroughly with PBS and inspected for mucosal damage and other signs of toxicity. Major organs were inspected for gross signs of toxicity.
Treatment Protocol
Rats with arthritis induced as above were randomly distributed into two groups with 7 animals/group. The first group (control) was treated with empty hydrogel (no P-S) and the second with PSH 17 mg/kg, ×3/day. We also included a third group of normal rats as above. Treatment started on day 12 after FA injection, when arthritis was well developed (33). The progression of arthritis was scored as above. Animals were sacrificed on day 20 after FA injection and blood and tissues were harvested and processed as above.
Evaluation of Locomotor Activity
The motor activity of animals under the treatment protocol was evaluated in open field observation using a frame system per animal with a set of 16 infrared photocells (LE8811; Letica, Barcelona, Spain). Occlusions of the photo beams were recorded and sent in the Seda-Com32 computerized system. The interruption counts (beam breaks) during predefined time internals were used as a measure of horizontal locomotor activity. Two types of movement, slow and fast, of individual study rats was monitored for 24 h based on the speed of beam break as defined by the manufacturer. Starting 2 days after the first hydrogel application, we studied in parallel 4 animals, 2 from each study group.
Histological Evaluation and Immunohistochemistry
After 24 h in formalin, hindpaw joints were placed in a decalcifier (Surgipath, Grayslake, IL) for 48 h to achieve complete decalcification. Then, the joints were transected along their longitudinal plane to two approximately equal halves, which were paraffin embedded. A pathologist, unaware of sample identity, evaluated 4 μM-thick joint sections stained with H&E and scored them for inflammation and bone resorption, as described (34). Two additional sections of each sample containing synovium, cartilage and bone marrow were placed side-by-side on a microscope slide (one served as the negative control). Paraffin-embedded sections were deparaffinized, rehydrated and microwave-heated for 15 min in 0.01 mol/L citrate buffer (pH 6.0) for antigen retrieval, and 3% H2O2 was applied to block endogenous peroxidase activity. After 15 min of blocking with horse serum, the primary antibody or control IgG (dilution 1/50) were applied and incubated overnight at 4°C. Slides were washed thrice with PBS, each for 5 min. The biotinylated secondary antibody and the streptavidinbiotin complex (Invitrogen, Grand Island, NY, USA) were applied, each for 30 min at room temperature, with an interval washing. After rinsing with PBS, the slides were immersed for 5 min in the substrate 3,3′-diaminobenzidine (DAB; Sigma, St. Louis, MO, USA), rinsed with distilled water, counterstained with hematoxylin, dehydrated and coverslipped. We used a rabbit phospho-NF-κB p65 (Ser276) antibody, which recognizes activated NF-κB (Cell Signaling Technology, Beverly, MA, USA) and rabbit anti-COX-2 polyclonal antibody (Cayman Chemical, Ann Arbor, MI, USA); specificity of binding was verified by isotypic control. Five different fields per slide from synovium, cartilage and bone marrow were counted and scored for NF-κB and COX-2 expression; positive staining appears brown. Results were expressed as the percentage of positive cells.
IL-6 and IL-10 Plasma Levels
Plasma levels were determined using rat IL-6 and IL-10 ELISA kits (Thermo Fisher Scientific, Rockford, IL, USA) and following the manufacturer's instructions.
Results
Hydrogel Characteristics
The amount of P-S entrapped inside the Pluronic hydrogel was 3.3±0.5% (w/w) and its polymer content, determined by lyophilization, was 28±0.7% (w/v). The P-S micellar size prior to and after gel formation was 35±10 nm and the polydispersity index, determined using dynamic light scattering, was 0.30±0.13. Micelles and hydrogel (both loaded with P-S) were reversible. This was established by placing a small quantity of gel in PBS and gently vortexing it until formulation of a drug suspension. Using dynamic light scattering we showed that the size of the micelles prior to gel formation was the same as when they reformed (after they were dissolved in PBS; data not shown).
PK Profile of Topically and Orally Applied P-S
We determined the PK profile of P-S in rats with arthritis both after topical application of PSH and after oral delivery of P-S. Table I and Fig. 1 summarize our findings.
Table I. PK Parameters of P-S After Topical and Oral Application.
| Delivery route | Metabolite | Cmax nmole/g | Tmax h | AUC0–24h (nmole/g)*h | Sum of AUC0–24h (nmole/g)*h | |||||
|---|---|---|---|---|---|---|---|---|---|---|
|
|
|
|
|
|||||||
| Muscle/blood | ||||||||||
| Topical | P-S | 680 | 2 | 1 | 4 | 4109 | 17 | 4697 | 784 | |
| Sulindac | 31 | 18 | 2 | 8 | 399 | 245 | ||||
| S.sulfide | 2 | 10 | 4 | 8 | 18 | 122 | ||||
| S.sulfone | 12 | 23 | 24 | 8 | 170 | 400 | ||||
| Oral | P-S | 11 | 2 | 0.5 | 0.5 | 49 | 18 | 1266 | 1472 | |
| Sulindac | 20 | 33 | 8 | 2 | 296 | 405 | ||||
| S.sulfide | 13 | 13 | 8 | 8 | 158 | 156 | ||||
| S.sulfone | 40 | 52 | 8 | 8 | 764 | 405 | ||||
PS was administered topically and orally in equal doses. Values are the average of two; Individual values were within <9% of each other
After topical application of PSH, the muscle Cmax of P-S was 680 nmole/g tissue, whereas the Cmax values of each of its three main metabolites, sulindac, sulindac sulfide and sulindac sulfone, were ≤31 nmole/g tissue. The Tmax of P-S was 1 h, whereas those of the metabolites ranged between 2 and 24 h, indicating that the hydrolysis of P-S to generate sulindac occurs first, while the reduction reaction generating sulindac sulfide is slower (Tmax04 h), and the oxidation reaction (catalyzed by a different system) producing sulindac sulfone is even slower (Tmax024 h). Of note, the absorption of P-S form PSH is fairly rapid (<1 h). The AUC0–24h of P-S exceeded 4 (μmole/g tissue)*h, while the sum of the corresponding values for the three metabolites was 8 fold lower, indicating that in the area where PSH was applied, PS remains largely intact. Indeed, as Fig. 1 demonstrates, after an initial 66% drop of the muscle P-S concentration, its levels remain fairly high (227–253 nmole/g tissue) for at least 8 h. Most of the topically applied P-S remained locally, as indicated by the far lower levels of P-S and its main metabolites in blood. As shown in Table I, the blood Cmax of P-S was 0.3% of that in muscle. The Tmax of blood P-S and two of its metabolites was significantly prolonged compared to that of muscle, indicating a slow process of drug transfer from the skin to the blood. Indeed, the notion that most of the topically applied P-S remains locally is supported by the fact that, based on AUC0–24h values, only 0.4% of it is in blood; cumulatively, only 14% of P-S and its metabolites is present in the blood over 24 h. The lower levels of P-S in the blood compared to muscle are explained, at least in part, by its rapid hydrolysis by carboxylesterase 1, which is abundant in mouse blood and liver (12).
Oral administration of P-S produced an entirely different PK profile than its topical application. The amount of P-S in the muscle is minuscule; its AUC0–24h is 1.2% of the corresponding value for the topical application of PSH. The Cmax values of P-S and its metabolites in the muscle are either similar to or somewhat smaller than those in blood, indicating that these compounds are likely transported to the muscle from blood. Supporting this notion are findings from our efficacy study in which the levels of P-S and its metabolites were determined in blood, forepaws and hindpaws 2 h after the last dose of P-S before sacrifice. After topical application of PSH on the hindpaws, most of P-S was intact (68.2% of the total), whereas in both the forepaws and the blood P-S was only ∼3.5% of the total.
Taken together, these data indicate that the topical administration of P-S is superior to the oral route in delivering this drug to the joint. This conclusion is further supported by the comparison of the cumulative AUC0–24h of P-S plus its metabolites in muscle; the AUC0–24h after oral administration is only 26.9% of that after topical administration.
Topically Applied PSH Suppresses FA-Induced Arthritis in Rats
Prevention Protocol
FA-treated rats were treated with P-S applied topically or given orally. P-S successfully controlled the joint inflammation of the hind legs as evidenced by changes in a) the clinical score: 56% reduction by topical administration and 40% reduction by oral administration; and b) the weight of the paws: 65% reduction by topical administration and 60% reduction by oral administration (p < 0.01 for both compared to their respective controls; Fig. 2). We also evaluated in the hindpaws three additional parameters, bone resorption, synovial inflammation and cartilage damage (34). As shown in Fig. 2, the induction of arthritis by FA was accompanied by significant inflammation, bone resorption and cartilage damage. PSH inhibited inflammation by 39% (p < 0.05), bone resorption by 72% (p=0.01) and cartilage damage by 36% (p > 0.05) compared to controls. The effect of orally administered P-S on all there parameters was less pronounced by inhibiting inflammation by 38% (p < 0.05), bone resorption 40% (p > 0.05) and had no effect on cartilage damage.
Fig. 2.
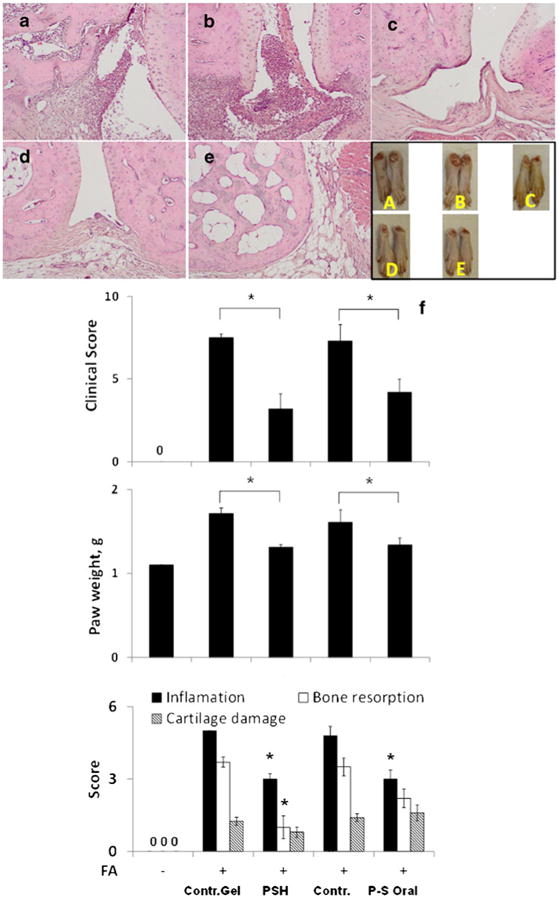
Efficacy of PSH and oral PS in the treatment of adjuvant-induced arthritis. (upper panel) Representative histological sections (H&E) of tissues from the hind legs of (a) control; (b) control hydrogel; (c) P-S hydrogel; (d) oral P-S; and (e) normal groups. (lower panel) (f) The response to PSH and P-S of the involved joints of rats treated under the prevention protocol as in Methods according to clinical score, paw weights and histological assessment. FA, Freund's adjuvant. All values are average ± SEM. *, p < 0.01 compared to the corresponding control group.
Treatment Model
Treatment with PSH 17 mg/kg 3×/day was initiated on day 12 when the signs of arthritis were pronounced. At the end of the study, on day 20, PSH successfully controlled arthritis, reducing the clinical score by 82% and paw weight by 78% compared to their controls (p < 0.01 for both). In addition, PSH inhibited synovial inflammation by 48% and bone resorption by 67% compared to controls (p < 0.01 for both), whereas its effect on cartilage damage by 69% (p < 0.05, Fig. 3).
Fig. 3.
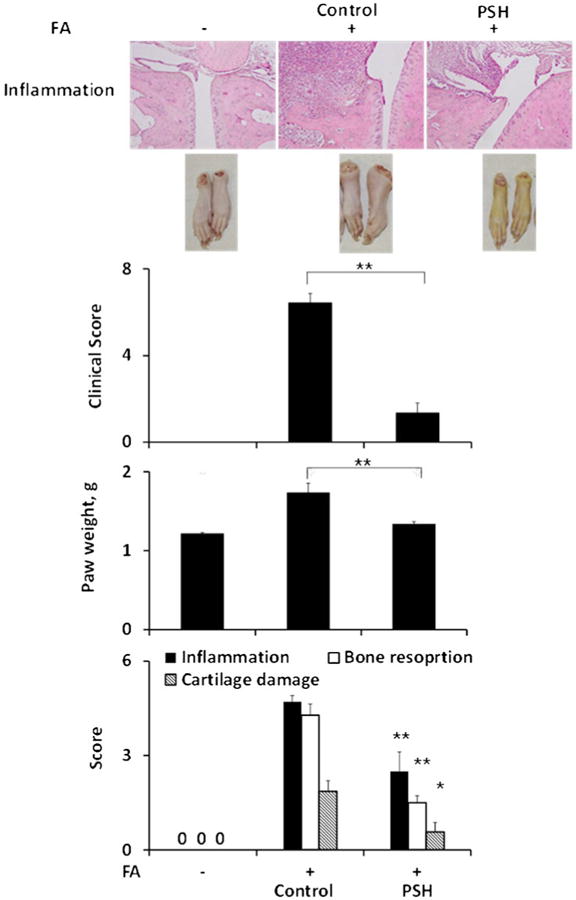
Efficacy of PSH in the treatment of adjuvant-induced arthritis. Representative histological sections (H&E) of joint tissues from the hind legs of rats treated under the treatment protocol as in Methods. The effect of PSH on the clinical score, paw weight and histologically assessed inflammation, bone resorption and cartilage damage are shown. FA, Freund's adjuvant. All values are average ± SEM. **, p < 0.01, *, p < 0.05 compared to the control group.
In these rats, we also evaluated the effect of P-S on spontaneous locomotor activity in an open field environment, performing 24-h measurements of their slow and fast movements (Fig. 4). Compared to controls, PSH improved both types of locomotor activity, increasing the slow movements by 1.6–2.6 fold and the fast movements by 2.4–9.4 fold; all changes were statistically significant (p < 0.01). The improved mobility of the arthritic rats was consistent throughout the treatment period. Nevertheless, we calculated the cumulative effect of PSH on locomotor activity and observed that overall the slow movements were increased 2.1 fold and the fast ones 4.4 fold over untreated controls.
Fig. 4.

The effect of PSH on the locomotor activity of rats with arthritis. Both the slow (top) and fast (middle) movements of rats under the treatment protocol as in Methods, recorded over 24 h and expressed per 5 min time units. The cumulative values of slow and fast movements for each group are shown in the bottom panel. All values are average ± SEM. All differences between control and treated rats in the top two panels are statistically significant (p < 0.01–0.02). *, p < 0.01 compared to the control group.
Drug Safety
In both studies, the gastrointestinal tract was inspected carefully at sacrifice under a magnifying lens and the mucosa appeared normal, with no evident damage in all groups. In addition, all major organs were inspected and no evidence of toxicity was present.
PSH Inhibits COX-2 and NF-κB But Has No Effect on IL-6 and IL-10
The COX-2 and NF-κB signaling cascades are critical mediators of inflammation, playing a prominent role in RA. Indeed, NF-κB, considered the master switch of inflammation, regulates the expression of almost 400 different genes, which include COX-2 and cytokines such as IL-6 and IL-10 (35). Therefore, we evaluated whether PSH reduced RA by modulating these molecular targets. The expression of COX-2 and NF-κB was evaluated by immunohistochemistry in joint tissues and the levels of cytokines were determined in blood.
The induction of arthritis was accompanied by a marked activation of NF-κB and a strong expression of COX-2 in inflammatory cells, as determined by immunohistochemistry (Fig. 5). Compared to arthritis controls, PSH reduced NF-κB activation by 30% (p < 0.05) and COX-2 expression by 45% (p < 0.01). In contrast, the blood levels of IL-6 and IL-10, both of which were increased in arthritic rats compared to those not treated with FA, remained essentially unchanged in response to PSH.
Fig. 5.
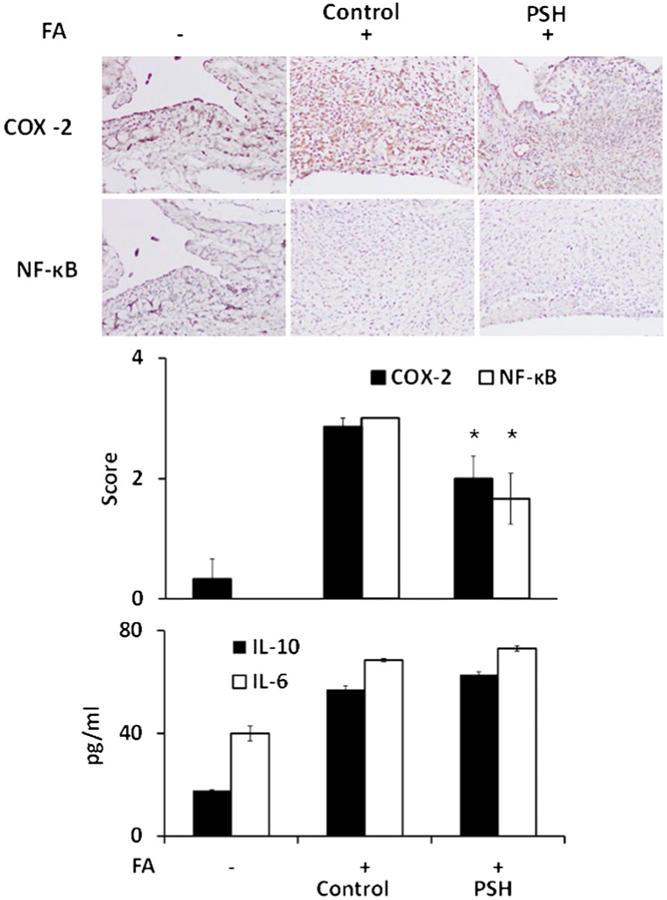
The effect of PSH treatment on COX-2, NF-κB and cytokines in rats. Upper panels: Representative images of sections from the involved joints of rats stained immuno-histochemically for the expression of COX-2 and NF-κB activation (phospho-NF-κB p65 (Ser276) antibody). PSH was applied under the treatment protocol as in Methods. Lower panels: Histograms of the scores of COX-2 expression and NF-κB activation and plasma cytokine levels of the three study groups. FA, Freund's adjuvant. All values are average ± SEM. *, p < 0.01 compared to the control group with arthritis (FA+).
Discussion
The long-term treatment of RA with anti-inflammatory agents requires a method of drug delivery that minimizes systemic drug exposure to avoid side effects while maintaining or improving their therapeutic efficacy. The transdermal delivery of P-S has the potential to satisfy these requirements. Our results demonstrate that PSH topically applied to rats with experimental arthritis markedly suppresses arthritis and its clinical manifestations including diminished locomotor activity, while ensuring excellent drug safety. Underlying these effects is a profound inhibition of synovial inflammation through inhibition of NF-κB and COX-2, key members of two signaling cascades heavily involved in the pathogenesis of arthritis.
The topical application of therapeutic agents, long-employed for the treatment of localized diseases, has undergone a dramatic change in recent years with the introduction of drug nanocarriers that not only overcome problems with drug solubility but also enhance drug delivery through the skin barrier (30,36). In this study, the pluronic hydrogel demonstrated desirable drug formulation properties. For example, it maintains its physical properties, regardless of its state (micelle vs. hydrogel). The value of the hydrogel formulation of P-S was further demonstrated by our pharmacokinetic study, which established its superior ability to deliver P-S locally, better than oral administration. As our data show, topically applied PSH delivers rapidly a substantial portion of P-S (in less than 1 h) to the area of the joint, where it remains for a prolonged period of time largely intact, undergoing minimal metabolism to sulindac and sulindac's two main metabolites, sulindac sulfide and sulindac sulfone. This is a point of particular importance, since P-S is significantly more potent pharmacologically than its parent compound, sulindac (11). Of perhaps equal interest is the finding that only a miniscule amount of P-S reaches the circulation, where it is hydrolyzed by carboxylesterases (12).
The PK results that we obtained are highly relevant to the safety of P-S. Of note, P-S has no GI toxicity whereas sulindac and sulindac sulfide account for much of sulindac's significant GI toxicity (11). Thus, it is important that < 14% of topically applied P-S reaches the circulation and that sulindac and its metabolites are present in blood at very low levels. These findings explain in part the lack of any gastrointestinal toxicity of P-S in these rats. A corollary to this is that the possibility of cardiac toxicity, which is a major concern for all NSAIDs, including sulindac (37), should be either non-existent or greatly minimized, as circulating drug levels are so low and thus the cardiovascular system should be spared due to P-S's very limited access to it.
The PK study also clearly indicates that in all parameters evaluated, topical P-S generated a more favorable PK profile than oral P-S. Importantly, the AUC0–24h after oral administration of P-S was about a quarter of that achieved by topical application at the targeted tissue.
Topically applied PSH was efficacious in the control of arthritis when administered under both the so-called prevention and treatment protocols, implying that P-S is equally effective when given during the clinical evolution of arthritis or when the disease is fully developed. Although a direct comparison between the two models may not be entirely valid, it appears that dividing the daily dose of P-S into three (17 mg/kg each) may be more efficacious than its single daily administration (50 mg/kg). The former reduced the arthritis score by 82% and paw weight by 78% versus 65% and 56%, respectively, by the later. Both modes of dosing were accompanied by a remarkable ∼70% reduction in bone resorption; that the response was similar in both may reflect the slower pace of this process compared to inflammation per se.
In terms of clinical implications, it was remarkable that PSH dramatically increased the mobility of the animals, including both slow and fast movements. This effect appeared to be somewhat dissociated from the anti-inflammatory effect of P-S, perhaps reflecting, at least to some extent, the considerable analgesic effect of P-S (preliminary data support this notion). Fast movements were improved far more than slow movements, especially during the beginning of treatment when joint inflammation was prominent. Although, we cannot readily explain this difference, we suspect that pain might have been more intense at the beginning, subsiding as the inflammation was resolving.
It is entirely plausible that compounds similar to P-S could be used for the control of arthritis though their topical application. This is especially likely for phospho-ibuprofen and phospho-aspirin, two compounds structurally analogous to P-S that have also shown strong efficacy in rat adjuvantinduced arthritis. Of note, their physico-chemical properties including lipophilicity, and their molecular weight are similar to those of P-S suggesting that they can be formulated in hydrogels.
Mechanistically, the induction of arthritis was accompanied by activation of NF-κB and induction of COX-2 expression. Although the cross-talk between these two prominent signaling players is not clear and may be context-depended, their importance to arthritis is firmly established (38,39). P-S inhibited both of them and the combined effect has likely contributed to its anti-inflammatory efficacy. It is conceivable that additional pathways involved in the complex process of inflammation may have been affected by P-S, which is known from other systems to have multi-targeted mechanistic effects (9). The blood levels of both IL-6 and IL-10 were increased as a result of arthritis but P-S failed to affect either one. Our data cannot exclude that topical changes in their joint levels may have contributed to the control of arthritis by P-S.
Conclusions
Topical application of P-S formulated in Pluronic hydrogel is efficacious in the treatment of FA-induced arthritis in rats evidenced by reduction in macroscopic and microscopic joint inflammation and bone resorption and markedly improved animal mobility. The administration of PSH was accompanied by no apparent side effects; in particular the gastrointestinal mucosa of the rats, the site of much of the toxicity of sulindac, the parent compound of P-S, was normal.
The efficacy and safety of PSH can be explained to a large extent by its PK profile. The topical application of PSH resulted in rapid absorption of P-S, which remained intact locally for prolonged periods of time. Less than 14% of the topically applied P-S reached the circulation, and a significant portion of it was in the form of metabolites devoid of gastrointestinal toxicity, which likely explains its apparent lack of gastrointestinal toxicity, a main limitation of conventional sulindac. The PK profile of topically applied PSH, differs from that of orally administered P-S, and suggests that it should be accompanied by minimal if any cardiac toxicity as well. Mechanistically, the anti-inflammatory effect of PSH is explained by its inhibition of NF-κB activation and COX-2 suppression in the affected joints.
We speculate that topical application of P-S using a hydrogel carrier will be clinically useful in the symptomatic treatment of arthritis and perhaps other forms of localized inflammation when the oral administration of anti-inflammatory agents may be less desirable. Thus, PSH deserves further evaluation as a promising formulation of P-S to be used as a topical anti-inflammatory agent.
Acknowledgments
This work was supported by the Department of Defense grant W81XWH1110799 and National Institute of Health grant R01CA139454.
This work received financial support from NIH grant R01CA139454 and DOD grant W81XWH 11-1-0799. We also thank William and Jane Knapp for their support.
Abbreviations
- FA
Freund's adjuvant
- GI
gastrointestinal
- NSAIDs
nonsteroidal anti-inflammatory drugs
- PK
pharmacokinetic
- PSH
phospho-sulindac hydrogel
- P-S
phospho-sulindac
- RA
rheumatoid arthritis
Footnotes
Disclosures: The authors have nothing to disclose except for BR, who has an equity position in Medicon Pharmaceuticals, Inc., and NO who is affiliated with the same.
References
- 1.Alamanos Y, Drosos AA. Epidemiology of adult rheumatoid arthritis. Autoimmun Rev. 2005;4:130–6. doi: 10.1016/j.autrev.2004.09.002. [DOI] [PubMed] [Google Scholar]
- 2.Eurenius E, Stenstrom CH. Physical activity, physical fitness, and general health perception among individuals with rheumatoid arthritis. Arthritis Rheum. 2005;53:48–55. doi: 10.1002/art.20924. [DOI] [PubMed] [Google Scholar]
- 3.Rahme E, Bernatsky S. NSAIDs and risk of lower gastrointestinal bleeding. Lancet. 2010;376:146–8. doi: 10.1016/S0140-6736(10)60839-2. [DOI] [PubMed] [Google Scholar]
- 4.Sostres C, Gargallo CJ, Arroyo MT, Lanas A. Adverse effects of non-steroidal anti-inflammatory drugs (NSAIDs, aspirin and coxibs) on upper gastrointestinal tract. Best Pract Res Clin Gastroenterol. 2010;24:121–32. doi: 10.1016/j.bpg.2009.11.005. [DOI] [PubMed] [Google Scholar]
- 5.Farkouh ME, Greenberg BP. An evidence-based review of the cardiovascular risks of nonsteroidal anti-inflammatory drugs. Am J Cardiol. 2009;103:1227–37. doi: 10.1016/j.amjcard.2009.01.014. [DOI] [PubMed] [Google Scholar]
- 6.McGettigan P, Henry D. Cardiovascular risk and inhibition of cyclooxygenase: a systematic review of the observational studies of selective and nonselective inhibitors of cyclooxygenase 2. JAMA J Am Med Assoc. 2006;296:1633–44. doi: 10.1001/jama.296.13.jrv60011. [DOI] [PubMed] [Google Scholar]
- 7.Ray WA, Stein CM, Daugherty JR, Hall K, Arbogast PG, Griffin MR. COX-2 selective non-steroidal anti-inflammatory drugs and risk of serious coronary heart disease. Lancet. 2002;360:1071–3. doi: 10.1016/S0140-6736(02)11131-7. [DOI] [PubMed] [Google Scholar]
- 8.Rainsford KD. Profile and mechanisms of gastrointestinal and other side effects of nonsteroidal anti-inflammatory drugs (NSAIDs) Am J Med. 1999;107:27S–35. doi: 10.1016/s0002-9343(99)00365-4. discussion 35S–36S. [DOI] [PubMed] [Google Scholar]
- 9.Huang L, Mackenzie G, Ouyang N, Sun Y, Xie G, Johnson F, et al. The novel phospho-non-steroidal anti-inflammatory drugs, OXT-328, MDC-22 and MDC-917, inhibit adjuvant-induced arthritis in rats. Br J Pharmacol. 2011;162:1521–33. doi: 10.1111/j.1476-5381.2010.01162.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Huang L, Zhu C, Sun Y, Xie G, Mackenzie GG, Qiao G, et al. Phospho-sulindac (OXT-922) inhibits the growth of human colon cancer cell lines: a redox/polyamine-dependent effect. Carcinogenesis. 2010;31:1982–90. doi: 10.1093/carcin/bgq149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mackenzie GG, Sun Y, Huang L, Xie G, Ouyang N, Gupta RC, et al. Phospho-sulindac (OXT-328), a novel sulindac derivative, is safe and effective in colon cancer prevention in mice. Gastroenterology. 2010;139:1320–32. doi: 10.1053/j.gastro.2010.06.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wong CC, Cheng KW, Xie G, Zhou D, Zhu CH, Constantinides PP, et al. Carboxylesterases 1 and 2 hydrolyze phosphononsteroidal anti-inflammatory drugs: relevance to their pharmacological activity. J Pharmacol Exp Ther. 2012;340:422–32. doi: 10.1124/jpet.111.188508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Xie G, Nie T, Mackenzie GG, Sun Y, Huang L, Ouyang N, et al. The metabolism and pharmacokinetics of phospho-sulindac (OXT-328) and the effect of difluoromethylornithine. Br J Pharmacol. 2012;165:2152–66. doi: 10.1111/j.1476-5381.2011.01705.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kalia YN, Naik A, Garrison J, Guy RH. Iontophoretic drug delivery. Adv Drug Deliv Rev. 2004;56:619–58. doi: 10.1016/j.addr.2003.10.026. [DOI] [PubMed] [Google Scholar]
- 15.Naik A, Kalia YN, Guy RH. Transdermal drug delivery: overcoming the skin's barrier function. Pharm Sci Technol Today. 2000;3:318–26. doi: 10.1016/s1461-5347(00)00295-9. [DOI] [PubMed] [Google Scholar]
- 16.Prausnitz MR, Mitragotri S, Langer R. Current status and future potential of transdermal drug delivery. Nat Rev Drug Discov. 2004;3:115–24. doi: 10.1038/nrd1304. [DOI] [PubMed] [Google Scholar]
- 17.Rhee YS, Chang SY, Park CW, Chi SC, Park ES. Optimization of ibuprofen gel formulations using experimental design technique for enhanced transdermal penetration. Int J Pharm. 2008;364:14–20. doi: 10.1016/j.ijpharm.2008.07.029. [DOI] [PubMed] [Google Scholar]
- 18.Farrington K, Regan F. Molecularly imprinted sol gel for ibuprofen: an analytical study of the factors influencing selectivity. Talanta. 2009;78:653–9. doi: 10.1016/j.talanta.2008.12.013. [DOI] [PubMed] [Google Scholar]
- 19.Ustundag Okur N, Apaydin S, Karabay Yavasoglu NU, Yavasoglu A, Karasulu HY. Evaluation of skin permeation and anti-inflammatory and analgesic effects of new naproxen micro-emulsion formulations. Int J Pharm. 2011;416:136–44. doi: 10.1016/j.ijpharm.2011.06.026. [DOI] [PubMed] [Google Scholar]
- 20.Huang ZJ, Hsu E, Li HC, Rosner AL, Rupert RL, Song XJ. Topical application of compound Ibuprofen suppresses pain by inhibiting sensory neuron hyperexcitability and neuroinflammation in a rat model of intervertebral foramen inflammation. J Pain Off J Am Pain Soc. 2011;12:141–52. doi: 10.1016/j.jpain.2010.06.008. [DOI] [PubMed] [Google Scholar]
- 21.Taboada M, Santen R, Lima J, Hossain J, Singh R, Klein KO, et al. Pharmacokinetics and pharmacodynamics of oral and transdermal 17beta estradiol in girls with Turner syndrome. J Clin Endocrinol Metab. 2011;96:3502–10. doi: 10.1210/jc.2011-1449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yerramsetty KM, Rachakonda VK, Neely BJ, Madihally SV, Gasem KA. Effect of different enhancers on the transdermal permeation of insulin analog. Int J Pharm. 2010;398:83–92. doi: 10.1016/j.ijpharm.2010.07.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pillai O, Panchagnula R. Transdermal delivery of insulin from poloxamer gel: ex vivo and in vivo skin permeation studies in rat using iontophoresis and chemical enhancers. J Control Release Off J Control Release Soc. 2003;89:127–40. doi: 10.1016/s0168-3659(03)00094-4. [DOI] [PubMed] [Google Scholar]
- 24.Wan DH, Zheng O, Zhou Y, Wu LY. Solubilization of Ibuprofen in Pluronic Block Copolymer F127 Micelles. Acta Phys-Chim Sin. 2010;26:3243–8. [Google Scholar]
- 25.Foster B, Cosgrove T, Hammouda B. Pluronic triblock copolymer systems and their interactions with ibuprofen. Langmuir. 2009;25:6760–6. doi: 10.1021/la900298m. [DOI] [PubMed] [Google Scholar]
- 26.Wanka G, Hoffmann H, Ulbricht W. Phase-diagrams and aggregation behavior of poly(oxyethylene)-poly(oxypropylene)-poly(oxyethylene) triblock copolymers in aqueous-solutions. Macromolecules. 1994;27:4145–59. [Google Scholar]
- 27.Alexandridis P. Poly(ethylene oxide) poly(propylene oxide) block copolymer surfactants. Curr Opin Colloid Interface Sci. 1997;2:478–89. [Google Scholar]
- 28.Cabana A, AitKadi A, Juhasz J. Study of the gelation process of polyethylene oxide(a) polypropylene oxide(b) polyethylene oxide(a) copolymer (Poloxamer 407) aqueous solutions. J Colloid Interface Sci. 1997;190:307–12. doi: 10.1006/jcis.1997.4880. [DOI] [PubMed] [Google Scholar]
- 29.Anderson BC, Pandit NK, Mallapragada SK. Understanding drug release from poly(ethylene oxide)-b-poly(propylene oxide)-b-poly(-ethylene oxide) gels. J Control Release Off J Control Release Soc. 2001;70:157–67. doi: 10.1016/s0168-3659(00)00341-2. [DOI] [PubMed] [Google Scholar]
- 30.Cheng KW, Mattheolabakis G, Wong CC, Ouyang N, Huang L, Constantinides PP, et al. Topical phospho-sulindac (OXT-328) is effective in the treatment of non-melanoma skin cancer. Int J Oncol. 2012;41:1199–1203. doi: 10.3892/ijo.2012.1577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schmolka IR. Artificial skin. I. Preparation and properties of pluronic F-127 gels for treatment of burns. J Biomed Mater Res. 1972;6:571–82. doi: 10.1002/jbm.820060609. [DOI] [PubMed] [Google Scholar]
- 32.Whiteley PE, Dalrymple SA. Models of inflammation: adjuvant-induced arthritis in the rat. Curr Protoc Pharmacol. 2001;Chapter 5(Unit5 5) doi: 10.1002/0471141755.ph0505s13. [DOI] [PubMed] [Google Scholar]
- 33.Bendele A. Animal models of rheumatoid arthritis. J Musculoskelet Neuronal Interact. 2001;1:377–85. [PubMed] [Google Scholar]
- 34.Bendele A, McComb J, Gould T, McAbee T, Sennello G, Chlipala E, et al. Animal models of arthritis: relevance to human disease. Toxicol Pathol. 1999;27:134–42. doi: 10.1177/019262339902700125. [DOI] [PubMed] [Google Scholar]
- 35.Ahn KS, Aggarwal BB. Transcription factor NF-kappaB: a sensor for smoke and stress signals. Ann N Y Acad Sci. 2005;1056:218–33. doi: 10.1196/annals.1352.026. [DOI] [PubMed] [Google Scholar]
- 36.Mattheolabakis G, Lagoumintzis G, Panagi Z, Papadimitriou E, Partidos CD, Avgoustakis K. Transcutaneous delivery of a nano-encapsulated antigen: induction of immune responses. Int J Pharm. 2010;385:187–93. doi: 10.1016/j.ijpharm.2009.10.033. [DOI] [PubMed] [Google Scholar]
- 37.Lione A, Scialli AR. The developmental toxicity of indomethacin and sulindac. Reprod Toxicol. 1995;9:7–20. doi: 10.1016/0890-6238(94)00051-w. [DOI] [PubMed] [Google Scholar]
- 38.Roman-Blas JA, Jimenez SA. NF-kappa B as a potential therapeutic target in osteoarthritis and rheumatoid arthritis. Osteoarthr Cartil. 2006;14:839–48. doi: 10.1016/j.joca.2006.04.008. [DOI] [PubMed] [Google Scholar]
- 39.Anderson GD, Hauser SD, McGarity KL, Bremer ME, Isakson PC, Gregory SA. Selective inhibition of cyclooxygenase (COX)-2 reverses inflammation and expression of COX-2 and interleukin 6 in rat adjuvant arthritis. J Clin Invest. 1996;97:2672–9. doi: 10.1172/JCI118717. [DOI] [PMC free article] [PubMed] [Google Scholar]