

Stress granules and translation are in dynamic equilibrium, but the mechanisms regulating them are not fully defined. Here we show isoform-specific roles for human Gle1: Gle1B functions in mRNA export, whereas, during stress, Gle1A coordinates translation and stress granule formation. This gives insight into how mRNA fate is controlled and has implications for human disease.
Abstract
When eukaryotic cells respond to stress, gene expression pathways change to selectively export and translate subsets of mRNAs. Translationally repressed mRNAs accumulate in cytoplasmic foci known as stress granules (SGs). SGs are in dynamic equilibrium with the translational machinery, but mechanisms controlling this are unclear. Gle1 is required for DEAD-box protein function during mRNA export and translation. We document that human Gle1 (hGle1) is a critical regulator of translation during stress. hGle1 is recruited to SGs, and hGLE1 small interfering RNA–mediated knockdown perturbs SG assembly, resulting in increased numbers of smaller SGs. The rate of SG disassembly is also delayed. Furthermore, SG hGle1-depletion defects correlate with translation perturbations, and the hGle1 role in SGs is independent of mRNA export. Interestingly, we observe isoform-specific roles for hGle1 in which SG function requires hGle1A, whereas mRNA export requires hGle1B. We find that the SG defects in hGle1-depleted cells are rescued by puromycin or DDX3 expression. Together with recent links of hGLE1 mutations in amyotrophic lateral sclerosis patients, these results uncover a paradigm for hGle1A modulating the balance between translation and SGs during stress and disease.
INTRODUCTION
Eukaryotic cells modulate gene expression to mount optimal stress responses and ensure cell survival (Lopez-Maury et al., 2008; de Nadal et al., 2011). Translation is one such mechanism that is rapidly and reversibly regulated during stress to inhibit global protein synthesis and selectively translate mRNAs necessary for adaptation and damage repair (Kedersha and Anderson, 2009; Spriggs et al., 2010). The global decrease in protein synthesis is mediated by phosphorylation of the eukaryotic initiation factor 2 alpha subunit (eIF2α), which results in stalled translation initiation (Gebauer and Hentze, 2004; Sonenberg and Hinnebusch, 2009; Spriggs et al., 2010). Moreover, translationally stalled, mRNA–protein complexes (mRNPs) are redirected to non membrane-bound reversible aggregates in cytoplasmic foci known as stress granules (SGs; Buchan and Parker, 2009; Kedersha and Anderson, 2009; Thomas et al., 2011). Current working models view SGs as sites for sequestering stalled mRNPs from active translation machinery, with specific mechanisms controlling storage, decay, or reinitiation of translation (Kedersha and Anderson, 2002; Kedersha et al., 2005; Buchan and Parker, 2009; Thomas et al., 2011). Whether common factors modulate SG dynamics and active translation is not fully resolved.
Several pieces of evidence support the hypothesis that mRNPs in SGs are in dynamic equilibrium with active translation complexes. Treatment of cells with drugs that immobilize polysomes (such as emetine and cycloheximide) prevents SG assembly. In contrast, puromycin-induced disassembly of polysomes promotes SG assembly (Kedersha et al., 2000). A recent study also showed that hepatitis C virus–infected cells oscillate between active and repressed phases of translation, defined by the absence and presence of SGs, respectively (Ruggieri et al., 2012). Aberrant SG formation is linked with various neurological diseases, including amyotrophic lateral sclerosis (ALS) (Bosco et al., 2010; Dewey et al., 2011; Wolozin, 2012; Vanderweyde et al., 2013). In addition, diverse viruses hijack SG machinery to effectively bias active translation for their own protein synthesis (Bosco et al., 2010; Ariumi et al., 2011; Khaperskyy et al., 2012; Lloyd, 2012, 2013). Further work is required to better understand how interplay between translation and SG dynamics is regulated at the molecular level.
Importantly, the molecular composition of SGs is variable and dependent upon the type of stress (Kedersha et al., 1999; Cuesta et al., 2000; Stoecklin et al., 2004; Balagopal and Parker, 2009). Prototypical SG constituents include poly(A)+ mRNA, 40S ribosomal subunits, translation initiation complexes, RNA-binding proteins, RNA-dependent ATPases known as DEAD-box proteins (Dbps), motor proteins, and cell-signaling molecules (Anderson and Kedersha, 2008; Buchan and Parker, 2009; Kedersha and Anderson, 2009). mRNA–protein rearrangements are likely required for changes in mRNPs necessary for SG association versus active translation, and such mRNP remodeling is potentially facilitated by Dbps. However, the molecular mechanisms of how Dbps are regulated to modulate SG exchange of mRNPs requires further investigation.
Gle1 is a conserved essential protein required for regulation of Dbp function during both mRNA export and translation. Originally identified in the budding yeast Saccharomyces cerevisiae (y; Murphy and Wente, 1996), yGle1 functions in mRNA export in association with inositol hexakisphosphate to stimulate the ATPase activity of Dbp5 for mRNP remodeling that confers export directionality at the nuclear pore complex (NPC; Alcazar-Roman et al., 2006; Weirich et al., 2006; Tran et al., 2007; Montpetit et al., 2011). In the cytoplasm, yGle1 interacts with translation initiation factor eIF3 and modulates the function of a different Dbp, Ded1 (for which the human orthologue is DDX3; Yedavalli et al., 2004; Bolger et al., 2008; Bolger and Wente, 2011). In contrast, during translation termination, yGle1-inositol hexakisphosphate interacts with Sup45 (eRF1) to again regulate Dbp5 (Bolger et al., 2008; Bolger and Wente, 2011). Given Gle1's regulation of multiple Dbps at distinct steps in the gene expression pathway, we speculated that its function might be linked to SG dynamics.
In human (h) cells, the GLE1 gene is alternatively spliced to generate at least two protein isoforms—hGle1A and hGle1B (Kendirgi et al., 2003). The hGle1A and hGle1B isoforms shuttle between the nucleus and cytoplasm and differ by only a 39-amino-acid carboxy(C)-terminal extension in hGle1B. These additional residues contain the binding domain for the NPC protein (nucleoporin, Nup) hCG1 (Kendirgi et al., 2003, 2005), and at steady state, hGle1B localizes predominantly at the NPC (Kendirgi et al., 2003, 2005; Rayala et al., 2004; Folkmann et al., 2013). We showed that hGle1B is necessary and sufficient for mRNA export in human tissue culture cells (Folkmann et al., 2013). In contrast, hGle1A is mainly localized in the cytoplasm (Kendirgi et al., 2003), and discrete cellular functions for hGle1A have not been directly tested.
In this paper, we define a specific role for hGle1A during SG formation and translation regulation during environmental stress responses. hGle1 localizes to SGs, and hGle1 depletion causes SG-assembly and SG-disassembly defects. We also find that SG defects in hGle1-depleted cells strongly correlate with deregulation of translation and are only rescued by expression of hGle1A. Additionally, hGle1 interacts with DDX3, and DDX3 is linked to the hGle1 role in SG dynamics. We propose that hGle1A acts to modulate the distribution of mRNPs between active translation and repressed translation in SGs through regulation of DDX3. We also recently reported unexpected links between hGLE1 mutations and ALS (Kaneb et al., 2014), wherein ALS-linked hGLE1 mutations alter the cellular pools of hGle1A and hGle1B. Thus defining critical hGle1A functions in modulating the balance between active translation and SGs reveals connections between pathophysiology and cellular stress responses.
RESULTS
hGle1A and hGle1B isoforms are differentially required during mRNA export
To follow up on our previous study showing hGle1B rescues mRNA export defects in hGle1-depleted cells (Folkmann et al., 2013), we investigated whether hGle1A is sufficient using our established small interfering RNA (siRNA)-mediated knockdown add-back system in HeLa cells. Immunoblotting confirmed the hGle1 depletion and respective expression of enhanced green fluorescent protein (EGFP)-hGle1A or EGFP-hGle1B (Figure 1A). For an assay for mRNA export, the nuclear versus cytoplasmic (N/C) distribution of bulk poly(A)+ mRNA was detected and quantified by oligo-dT in situ hybridization. Consistent with our published report (Folkmann et al., 2013), poly(A)+ RNA accumulated in the nucleus of hGLE1 siRNA-treated cells (Figure 1, B and C). Expression of EGFP alone, EGFP-tagged siRNA-resistant (R) hGLE1A (EGFP-hGLE1AR), or EGFP-tagged siRNA-resistant hGLE1B (EGFP-hGLE1BR) plasmids in scrambled control (CTRL) siRNA cells showed a widespread distribution of poly(A)+ RNA in the nucleus and cytoplasm with similar mean N/C ratios of ∼0.8 (Figure 1, B and C). As reported (Folkmann et al., 2013), expression of EGFP-hGLE1BR in hGLE1 siRNA-treated cells rescued the nuclear poly(A)+ mRNA accumulation (mean N/C ratios ∼1.0). In contrast, neither EGFP-hGLE1AR (mean N/C ratios ∼1.25) nor EGFP alone (mean N/C ratios ∼1.25) were sufficient to rescue the nuclear poly(A)+ RNA accumulation in hGLE1 siRNA cells (Figure 1, B and C).
FIGURE 1:

hGle1A is not required for mRNA export. (A) Endogenous hGle1 protein levels are reduced upon hGLE1 depletion in HeLa cells. hGLE1 or CTRL siRNA-treated cells were transfected with indicated EGFP-tagged plasmids, and cell lysates were analyzed by immunoblotting using anti-hGle1, GFP, and actin antibodies. (B) Expression of EGFP-hGle1B but not EGFP-hGle1A rescues mRNA export defects in hGle1-depleted HeLa cells. Nuclear poly(A)+ mRNA accumulation was detected by Cy3-labeled oligo-dT in situ hybridization in the CTRL or hGLE1 siRNA-treated samples expressing either EGFP, EGFP-hGLE1AR, or EGFP-hGLE1BR plasmids. Scale bar: 10 μm. (C) Quantification of N/C ratio of poly(A)+ RNA in CTRL and hGLE1 siRNA-treated samples expressing indicated plasmids. Error bars represent mean ± 95% confidence interval from at least three independent experiments. (D) Both hGle1A and hGle1B localize to the cytoplasmic face of NPC in hGle1-depleted cells. hGLE1 or CTRL siRNA-treated HeLa cells were transfected with indicated EGFP-tagged plasmids, and cells were imaged live using superresolution structural microscopy with Pom121-mCherry marking the nuclear envelope. Scale bar: 1 μm.
The steady-state enrichment of hGle1 at the NPC requires hGle1 self-association and interaction with both hNup155 and hCG1 (Kendirgi et al., 2003, 2005; Rayala et al., 2004; Folkmann et al., 2013). We speculated that hGle1A might not rescue mRNA export defects because it lacks the hCG1-binding region and is not recruited to the NPCs (Kendirgi et al., 2003, 2005). For testing this hypothesis, the localization of EGFP-hGle1AR and EGFP-hGle1BR in CTRL and hGLE1 siRNA-treated cells was monitored using three-dimensional (3D) structural illumination microscopy in living cells coexpressing the NPC protein Pom121-mCherry. Interestingly, in CTRL siRNA cells, EGFP-hGle1B localized at the cytoplasmic face of the nuclear envelope, whereas EGFP-hGle1A was not enriched at the nuclear envelope (Figure 1D). However, in hGLE1 siRNA cells, both the EGFP-hGle1A and EGFP-hGle1B isoforms were localized at the cytoplasmic face of the nuclear envelope (Figure 1D). This indicated that endogenous hGle1B competes with EGFP-hGle1A for recruitment to the NPC in CTRL siRNA cells. Furthermore, hGle1A localization at the NPC was not sufficient for efficient mRNA export, suggesting a specific role for the hGle1-hCG1 interaction during the export mechanism. Overall we concluded that hGle1B and hGle1A play distinct cellular roles.
hGle1 is recruited to stress granules
Given yGle1's roles in translation initiation and termination, we speculated that cytoplasmically localized hGle1A was involved in regulating translation. Because SGs and translation are in dynamic equilibrium, and Dbps such as DDX3, DDX1, and eIF4A that play a role in translation are recruited to SGs (Low et al., 2005; Mazroui et al., 2006; Lai et al., 2008; Onishi et al., 2008; Shih et al., 2012), we asked whether hGle1A and hGle1B are recruited to SGs upon heat shock stress. To test this, we subjected HeLa cells coexpressing mCherry-G3BP and either exogenous EGFP, EGFP-hGLE1AR, or EGFP-hGLE1BR to heat shock at 45°C for 60 min and imaged them using live-cell microscopy. EGFP-hGle1A and EGFP-hGle1B, but not EGFP, were colocalized to cytoplasmic foci with the SG marker mCherry-G3BP (Tourriere et al., 2003; Supplemental Figure S1A). Indirect immunofluorescence microscopy with anti-hGle1 and anti-G3BP antibodies was also used to evaluate endogenous hGle1 localization. At 37°C in HeLa cells, endogenous hGle1 localization was pancellular with distinct nuclear rim staining, whereas G3BP was uniformly distributed throughout the cytoplasm (Figure 2A). On heat shock, hGle1 was detected at cytoplasmic foci that colabeled with G3BP (Figure 2A). Line-scan analysis of individual SGs indicated that anti-hGle1 staining overlapped with anti-G3BP (Figure 2A). Moreover, the hGle1 cytoplasmic foci also colocalized with other SG components, including DDX3, HuR, and FMRP (Gallouzi et al., 2000; Mazroui et al., 2002; Lai et al., 2008; Shih et al., 2012; Supplemental Figure S2A). HeLa cells were treated for 60 min at 37°C with thapsigargin, which induces SGs by causing endoplasmic reticulum (ER) stress, and hGle1 localization to SGs was observed (Supplemental Figure S1C). Furthermore, hGle1 was also recruited to SGs in U2OS and RPE-1 cells upon heat shock (Supplemental Figure S1B). Thus hGle1 localization in SGs was a general phenomenon.
FIGURE 2:

hGle1 is recruited specifically to SGs upon heat shock. (A) Endogenous hGle1 is localized to SGs. HeLa cells were either left untreated or exposed to heat shock at 45°C for 60 min. Cells were processed for immunofluorescence using anti-hGle1 and G3BP antibodies. Scale bar: 10 μm. (B) hGle1 is not a component of P bodies. Immunofluorescence of hGle1 and P-body marker Dcp1a in HeLa cells treated with either heat shock or left untreated. The white line in A and B indicates the position of the line scan to assess colocalization in merged images using ImageJ. Arrow points toward the position of white line showing colocalization. Scale bar: 10 μm.
Processing bodies (P bodies) are another type of RNA granule found in cells under normal and stress conditions (Anderson and Kedersha, 2009; Balagopal and Parker, 2009). Although P bodies share many components with SGs (Kedersha et al., 2005; Kedersha and Anderson, 2007), P bodies are linked with mRNA decay (Parker and Sheth, 2007; Decker and Parker, 2012). To investigate whether hGle1 is also a component of P bodies, we subjected HeLa cells to heat shock and performed indirect immunofluorescence with anti-hGle1 and anti-Dcp1a (a P-body marker). P bodies were detected under normal growth conditions in HeLa cells (Figure 2B), and upon heat shock, P bodies were localized near SGs (Figure 2B). However, anti-hGle1 staining did not overlap with anti-Dcp1a, indicating that hGle1 is not a component of P bodies (Figure 2B). Together these data suggested that hGle1 is a novel component of SGs.
hGle1A specifically functions in SG assembly in response to cellular stress
For determining whether hGle1 plays an active role modulating SGs and/or is sequestered in SGs to regulate its own activity during stress, the effect of hGle1 depletion on SGs was assayed. If hGle1 is involved in SG formation, the absence of hGle1 should perturb SG dynamics; however, if hGle1 is simply sequestered, differences in SG dynamics would not be expected (as is found for sequestered signaling molecules; Kim et al., 2005; Arimoto et al., 2008). The presence of SGs was monitored after 60 min of heat shock in CTRL and hGLE1 siRNA-treated HeLa cells by indirect immunofluorescence with anti-G3BP antibodies. The majority of the hGle1-depleted cells exhibited an increased number of small G3BP-positive SGs compared with CTRL cells (Figure 3A). Other hGle1-depleted cells showed either diffuse cytoplasmic distribution of G3BP or localization to a few disorganized foci (Figure 3A). Similar phenotypes were observed when other SG markers were analyzed (DDX3, HuR, and FMRP; Supplemental Figure S2A). As controls for off-target effects, two independent siRNA sets were used that target different regions of the hGLE1 gene. A similar increase perturbation of SGs was observed with both, indicating that the phenotype is specific to hGle1 depletion (Supplemental Figure S2D). SG changes were also observed in hGle1-depleted HeLa cells treated with thapsigargin and in hGle1-depleted U20S and RPE-1 cells upon heat shock (Supplemental Figure S2, B and C). Thus hGle1 depletion altered SG assembly, and the effects were not limited to stress or cell types.
FIGURE 3:

hGle1 is required for SG assembly and SG disassembly. (A) hGle1-depleted cells show SG-assembly defects. HeLa cells transfected with CTRL or hGLE1 siRNAs were subjected to heat shock at 45°C for 60 min and processed for immunofluorescence using anti-hGle1 and G3BP antibodies. hGle1-depleted cells show either increased numbers of SGs or fail to assemble SGs. Scale bar: 10 μm. (B) Expression of EGFP-hGle1A but not EGFP-hGle1B rescues SG-assembly defects in hGle1-depleted cells. CTRL or hGLE1 siRNA-treated HeLa cells were transfected with EGFP, EGFP-hGLE1AR, or EGFP-hGLE1BR plasmids, heat shocked, and processed for immunofluorescence detection of G3BP and hGle1. Scale bar: 10 μm. (C) Quantification of SG numbers in CTRL and hGLE1 siRNA cells expressing indicated plasmids. (D, E) Analysis of SG formation in CTRL and hGLE1 siRNA-treated samples. hGLE1 siRNA or CTRL siRNA-treated HeLa cells were heat shocked at 45°C. Samples were fixed across a time course of 0–60 min and processed for immunofluorescence detection of G3BP and hGle1. p < 0.000001 for each data pair. (F) SG disassembly is delayed in hGle1-depleted cells. Following heat shock at 45°C for 60 min, cells were incubated at 37°C for the indicated times and processed for immunofluorescence using anti-G3BP and hGle1 antibodies. Error bar represents mean ± SE from three independent experiments. **, p < 0.001.
To analyze whether the SG defects observed with hGle1 depletion were due to altered mRNA export, we conducted knockdown experiments for two other NPC-associated essential mRNA export factors: DDX19B (yDbp5 homologue) and NXF1 (Grüter et al., 1998; Kang and Cullen, 1999; Herold et al., 2000; Hodge et al., 2011). siRNA-mediated knockdown of NXF1 or DDX19B resulted in nuclear accumulation of bulk poly(A)+ RNA indicative of an mRNA export defect (Supplemental Figure S3A). However, neither NXF1 siRNA nor DDX19B siRNA-treated cells showed perturbations of SGs like that seen in hGle1-depleted cells (Supplemental Figure S3, A and B). Thus inhibition of mRNA export was not sufficient for perturbing SG formation.
Next the hGLE1 siRNA knockdown and add-back approach was used to test for hGle1A versus hGle1B roles at SGs. CTRL siRNA or hGLE1 siRNA cells were transfected with either EGFP, EGFP-hGLE1AR, or EGFP-hGLE1BR plasmids. After 24 h, cells were subjected to heat shock, and G3BP localization was assessed as an indicator of SG formation. Expression of EGFP, EGFP-hGLE1AR, or EGFP-hGLE1BR plasmids had no impact on the number of SGs formed in CTRL siRNA-treated cells (Figure 3, B and C), and the number of SGs formed was significantly higher in hGLE1 siRNA-treated cells expressing EGFP (Figure 3, B and C). Strikingly, the expression of EGFP-hGLE1AR but not EGFP-hGLE1BR led to a significant rescue of the SG defect induced upon hGle1 depletion (Figure 3, B and C). These results further supported the fact that the SG defects in hGLE1 siRNA-treated cells were not due to off-target effects (based on the hGle1A-alone rescue) or mRNA export defects (based on hGle1B being sufficient for rescuing mRNA export). Overall we concluded that the hGle1A isoform specifically functions in SG assembly.
hGle1 is required for early assembly and disassembly of SGs
SGs first assemble as numerous small cytoplasmic foci, and in response to prolonged stress, the foci coalesce to form a smaller number of large SGs (Kedersha et al., 2000). For pinpointing the role for hGle1 in early or later stages of SG assembly, time-course analysis was conducted by heat shocking in 10-min increments, followed by fixation and indirect immunofluorescence with anti-G3BP antibodies to assess SG formation. CTRL siRNA cells assembled many small SGs after 20 min of heat shock. With increasing time of heat shock treatment, the SGs were larger in size and fewer in number (Figure 3, D and E). Quantification of SG size and number at each time point revealed several important distinctions between CTRL and hGLE1 siRNA-treated cells. First, the number of SGs detected in hGLE1 siRNA-treated cells was significantly higher compared with CTRL siRNA-treated cells at all time points during heat shock (Figure 3D). Second, the average SG size was significantly smaller in hGle1-depleted cells compared with CTRL cells at all time points (Figure 3E). Finally, SG number or size did not change much over the entire course of heat shock for the hGle1-depleted cells compared with CTRL cells (in which SGs became bigger and fewer over time; Figure 3, D and E). These results indicated that hGle1 is involved in early SG-assembly steps that regulate SG number and granule size.
We further investigated whether hGle1 is also required during SG disassembly. Following heat shock for 60 min, CTRL siRNA or hGLE1 siRNA cells were allowed to recover for 10, 20, 30, 40, and 60 min at 37°C, and SG disassembly was monitored by indirect immunofluorescence with anti-G3BP antibodies. Cells were scored as having SGs if G3BP foci were detected. Interestingly, hGle1-depleted cells disassembled SGs. However, SG disassembly occurred more slowly than in CTRL cells, with significantly higher percentages of cells still containing SGs at later time points of recovery (Figure 3F). Collectively hGle1 activity was required for proper SG assembly and disassembly.
hGle1 depletion–induced SG defects persist upon microtubule perturbation
Intact and dynamic microtubule (MT) networks are required for proper SG assembly; thus MT disruption by drugs that depolymerize (nocodazole) or stabilize (Taxol) MTs results in numerous smaller SGs (Ivanov et al., 2003; Chernov et al., 2009; Kolobova et al., 2009; Nadezhdina et al., 2010). Given the similar phenotype, we tested whether the SG phenotypes in hGle1-depleted cells were linked to changes in the MT network. If SG defects in hGle1-depleted cells were dependent upon MTs, we predicted that disrupting MT dynamics in hGle1-depleted cells would not have an additive effect. Conversely, if hGle1-dependent SG defects were independent of MTs, a further increase in the number of SGs should result from both hGle1 depletion and disrupted MT networks. CTRL and hGLE1 siRNA cells were treated with vehicle alone, Taxol, or nocodazole, and the number of SGs was determined based on G3BP localization. Indirect immunofluorescence staining with anti–α-tubulin antibodies confirmed that MT networks were disrupted after treatments with Taxol or nocodazole compared with vehicle alone (Figure 4, A–C). Similar to previous studies (Chernov et al., 2009), treatment of CTRL cells with either Taxol or nocodazole led to smaller and numerous SGs. Addition of Taxol and nocodazole with hGLE1 siRNA treatment led to a further increase in the number of SGs (Figure 4, A–C). Interestingly, quantification of SG numbers revealed a significantly higher level in hGLE1 siRNA cells compared with CTRL cells in the presence of either Taxol or nocodazole (Figure 4D). These results supported the conclusion that hGle1-dependent SG assembly defects are independent of an effect on MTs.
FIGURE 4:

hGle1-dependent SG defects are not linked with microtubules. (A–C) HeLa cells transfected with CTRL and hGLE1 siRNAs were treated with (A) vehicle alone, (B) 5 μM nocodazole, or (C) 100 nM Taxol for 120 min at 37°C followed by heat shock at 45°C for 60 min. Cells were processed for immunofluorescence with anti-G3BP, α-tubulin, and hGle1 antibodies. Scale bar: 10 μm. (D) Quantification of SG numbers in hGLE1 and CTRL siRNA cells treated with indicated drugs after heat shock.
The function of hGle1 in SG assembly is linked to translation
SG assembly is regulated by the available pool of free nontranslated mRNPs (Kedersha et al., 2000). Thus, altering translation could lead to perturbations in SG assembly. Given this and that yGle1 plays a role in translation, we next asked whether hGle1 modulates translation and whether this function is linked with SG formation. For measuring translation in CTRL and hGLE1 siRNA-treated HeLa cells under nonstress and stress conditions, metabolic labeling with l-azidohomoalanine (AHA), a methionine analogue, was conducted for 30 min, and newly synthesized AHA-containing proteins were detected with Alexa Fluor-488 alkyne using click chemistry (Dieterich et al., 2006, 2007). Following the labeling reaction, indirect immunofluorescence microscopy was used to assess hGle1 and G3BP. In CTRL siRNA cells, there was strong signal for AHA-labeled proteins under nonstress conditions (Figure 5A). Strikingly, hGLE1 siRNA-treated cells exhibited reduced AHA-488 signal as compared with CTRL siRNA-treated cells. A 35S metabolic labeling assay was used as an alternative measure of protein synthesis, and again a reduction in nascent protein synthesis was observed in hGLE1 siRNA-treated cells compared with CTRL siRNA cells (Figure 5D). Reduced protein synthesis under nonstress conditions was expected, due to the role of hGle1 in mRNA export. Therefore we tested whether depletion of NXF1 also led to reduced protein synthesis under nonstress conditions using AHA labeling. However, we did not observe significant reduction in AHA signal in NXF1 siRNA-treated cells compared with control cells (Supplemental Figure S3C). To further test whether hGle1 has a role in translation, we performed polysome profiles in hGle1-depleted cells. Strikingly, we observed an increased monosome (80S) peak and reduced polysomes compared with CTRL siRNA cells (Figure 5E). This result was consistent with yGle1, suggesting that hGle1 plays a conserved role in translation initiation (Bolger et al., 2008).
FIGURE 5:

hGle1 modulates SG assembly by regulating translation. (A, B) Nascent protein synthesis is deregulated in hGle1-depleted cells. HeLa cells treated with CTRL and hGLE1 siRNAs were subjected to heat shock at 45°C or left untreated. After 15 min, AHA was added to the incubations, and heat shock treatment was continued for an additional 30 min. Samples were processed by Alexa Fluor-488 alkyne staining followed by immunofluorescence with anti-G3BP and hGle1 antibodies. Scale bar: 10 μm. (C) Quantification of AHA-488 staining. Mean fluorescence intensity of AHA-488 staining in individual cells was calculated in CTRL and hGLE1 siRNA cells using ImageJ. (D) CTRL or hGLE1 siRNA-treated HeLa cells were either heat shocked at 45°C or left untreated followed by metabolic labeling with 100 μCi/ml [35S]methionine/cysteine for 30 min at either 37°C or 45°C. Cells were lysed, and 35S incorporation was measured by liquid scintillation counter. Counts per minutes (cpm) are shown for hGLE1 and CTRL siRNA-treated cells. (E) hGle1-depleted cells have polysome profile defects under normal conditions. CTRL or hGLE1 siRNA cells were lysed, and polysome profiles were generated by subjecting cells to a 7–47% sucrose gradient centrifugation. The 40S, 60S, 80s, and polysome peaks are labeled. (F) Expression of hGle1A but not hGle1B rescues translation defect in hGle1-depleted cells. CTRL or hGLE1 siRNA-treated HeLa cells were transfected with mCherry, mCherry-hGLE1AR, or mCherry-hGLE1BR plasmids, heat shocked, and processed for metabolic labeling using AHA. AHA incorporation was detected with Alexa Fluor-488 alkyne using click chemistry. Scale bar: 10 μm. (G) Quantification of AHA-488 staining in CTRL and hGLE1 siRNA cells expressing the indicated plasmids.
We next examined the effect of hGle1 depletion on translation during heat shock. Following heat shock of CTRL siRNA cells, the AHA-488 labeling decreased (Figure 5B) in agreement with previous reports of global translation down-regulation upon heat shock (Panniers, 1994). In comparison, the AHA-488 signal dramatically increased in heat-shocked hGle1-depleted cells (Figure 5, B and C). Moreover, cycloheximide treatment led to a loss of AHA-488 signal in hGLE1 siRNA cells (Supplemental Figure S4). Thus the AHA signal detected was due to nascent protein synthesis. As a control, NXF1 siRNA-treated cells were tested, and there was no increased AHA labeling compared with CTRL siRNA-treated cells upon heat shock (Supplemental Figure S3C). Therefore the deregulation of translation during heat shock in hGle1-depleted cells was not due to global perturbations in mRNA export. To confirm AHA-labeling results by an alternative method, we measured nascent protein synthesis upon heat shock in CTRL and hGLE1 siRNA cells by a 35S metabolic assay. Consistent with AHA-labeling results, we observed increased incorporation of [35S]methionine/cysteine in hGle1-depleted cells (Figure 5D).
Finally, upon evaluation of the SG defects in the hGle1-depleted cells, the deregulation in translation was found to be strongly correlated with the SG defects. Approximately 70% of hGle1-depleted cells with increased AHA-488 labeling either did not assemble SGs or had an increased number of SGs (Figure 5B). Because hGle1 depletion resulted in defects in translation, we next investigated whether the hGle1A and/or hGle1B isoform rescues translation defects in hGle1-depleted cells. CTRL or hGLE1 siRNA-treated cells were transfected with mCherry alone, mCherry-hGle1AR, or mCherry-hGLE1BR plasmids. Twenty-four hours after plasmid transfection, cells were heat shocked and processed for AHA labeling. Interestingly, expression of mCherry-hGle1A but not mCherry-hGle1B or mCherry rescued translation defects in hGle1-depleted cells upon heat shock (Figure 5, F and G). Together these results suggested that hGle1A modulates SG formation by regulating translation upon heat shock.
During cell-stress conditions, various signaling pathways converge to result in the phosphorylation of eIF2α at Ser-51, which inhibits eIF2 recycling and results in a global reduction in translation (Wek et al., 2006; Spriggs et al., 2010). To gain insight into the mechanisms for how hGle1 depletion alters translation, we analyzed the phosphorylation levels of eIF2α. Immunoblotting with anti-eIF2α P [Ser-51] antibody revealed an increase in eIF2α phosphorylation in CTRL siRNA-treated cells after 30 min of heat shock, which persisted to the 60-min time point (Figure 6, A and B). Strikingly, the relative eIF2α phosphorylation level was reduced modestly (∼36%) but significantly in hGle1-depleted cells compared with CTRL siRNA cells (Figure 6, A and B). These results indicated that reduced eIF2α phosphorylation might be linked to translation deregulation in hGle1-depleted cells upon heat shock.
FIGURE 6:
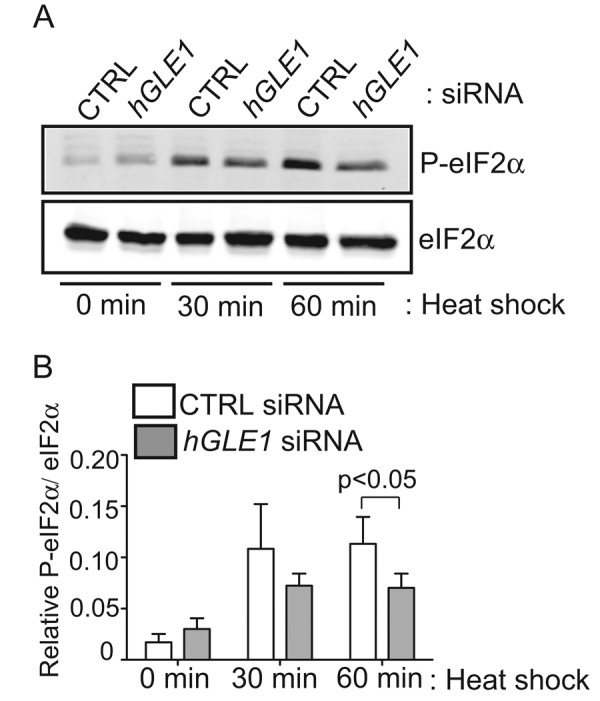
Phosphorylation of eIF2α is reduced in hGle1-depleted cells. (A, B) HeLa cells transfected with either CTRL or hGLE1 siRNA were left untreated or heat shocked at 45°C for 30 min or 60 min. Lysates were analyzed by immunoblotting using anti-eIF2α and anti–phospho-eIF2α (Ser-51) antibodies. Phospho-eIF2α levels were quantified by densitometry and normalized to total eIF2α protein levels. Error bar represents SD from mean from four independent experiments.
hGle1 modulates the dynamic balance between translation and SGs upon stress
Several pharmacological agents are known to change the dynamic equilibrium between SGs and translation. For example, cycloheximide treatment prevents SG formation, whereas puromycin results in larger SGs upon stress (Kedersha et al., 2000). If hGle1 regulates the equilibrium between SGs and active translation, hGle1 depletion might shift the balance of mRNPs toward active translation, limiting the free mRNP pool and inhibiting SG assembly. In this model, SG-assembly defects induced by hGle1 depletion should be rescued by shifting the mRNP balance toward SG formation by addition of puromycin. The number of SGs in CTRL or hGLE1 siRNA-treated cells was determined in the presence of either vehicle alone or puromycin. Treatment of CTRL siRNA cells with either puromycin or vehicle alone did not change the number of SGs significantly (Figure 7, A and B). Notably, treatment of hGle1-depleted cells with either 0.1 mg/ml or 0.5 mg/ml puromycin significantly reduced the number of SGs (Figure 7, A and B). Thus puromycin partially rescued the SG defects in hGle1-depleted cells upon heat shock. As a control, we tested whether puromycin rescues the increase in SGs observed in nocodazole-treated cells and found it did not (Supplemental Figure S5, A and B). Because puromycin failed to rescue MT-dependent SG defects, puromycin and nocodazole were affecting SG formation at distinct steps. We concluded that puromycin rescues the SG phenotype in hGle1-depleted cells by increasing the free mRNP pool.
FIGURE 7:

hGle1 regulates balance between active and stalled translation upon stress. (A, B) Puromycin rescues SG-assembly defects in hGle1-depleted cells. CTRL or hGLE1 siRNA HeLa cells were treated with vehicle alone, 0.1 mg/ml, or 0.5 mg/ml puromycin for 60 min at 45°C. Cells were processed for immunofluorescence using anti-hGle1 and G3BP antibodies. Scale bar: 10 μm. (C, D) EGFP-hGle1A but not EGFP-hGle1B overexpression results in bigger SGs: HeLa cells were transfected with either EGFP, EGFP-hGLE1AR, or EGFP-hGLE1BR, and either (C) left untreated or (D) heat shocked for 60 min at 45°C. After heat shock, cells were processed for immunofluorescence using anti-hGle1 and G3BP antibodies. Data represent mean ± SE from three independent experiments. Scale bar: 10 μm.
If hGle1 levels impact the free mRNP pool by changing the distribution of mRNPs between active translation and SGs, then overexpression of hGle1 should increase the free mRNP pool and result in larger SGs (similar to puromycin treatment alone). HeLa cells transfected with either EGFP, EGFP-hGLE1AR, or EGFP-hGLE1BR plasmids were heat shocked and analyzed for SG formation. G3BP-positive SGs assembled in cells expressing either EGFP, EGFP-hGLE1AR, or EGFP-hGLE1BR plasmids (Figure 7D). Overexpression of hGle1A caused a modest but significant increase in SG size (mean = 1.34 μm2) compared with EGFP (mean = 1.16 μm2) or hGle1B (mean = 1.15 μm2) (Figure 7D). Thus hGle1A but not hGle1B overexpression resulted in larger SG formation. TTP, TIA-1, and G3BP proteins play a role in early assembly of SGs, and their overexpression drives constitutive SG formation under nonstress conditions (Tourriere et al., 2003; Gilks et al., 2004). However, in nonstress conditions, expression of either EGFP, EGFP-hGLE1AR, or EGFP-hGLE1BR plasmids in HeLa cells showed pancellular cytoplasmic staining for G3BP, suggesting that hGle1 cannot nucleate SG formation in the absence of stress (Figure 7C).
DDX3 suppresses loss of hGle1 function
Based on the published connections between yGle1 and Ded1 during translation initiation (Alcazar-Roman et al., 2006; Bolger et al., 2008; Bolger and Wente, 2011; Folkmann et al., 2014), the roles for Ded1 in releasing mRNA from SGs and promoting translation (Hilliker et al., 2011), and the recruitment of the Ded1 human homologue DDX3 to SGs (Lai et al., 2008; Hilliker et al., 2011; Shih et al., 2012), we evaluated whether hGle1 function in SGs is mediated through interaction with DDX3. First, coimmunoprecipitation experiments were conducted with HeLa cell lysates from untreated or heat-shocked cultures. hGle1 and DDX3 were coisolated in the presence or absence of stress (Figure 8A). We also examined whether expression of DDX3 rescues hGle1-dependent SG defects. CTRL or hGLE1 siRNA-treated cells were transfected with EGFP alone, EGFP-DDX19B, or DDX3-EGFP. DDX3-EGFP was expressed at low levels, so the cells did not form constitutive SGs (Supplemental Figure S6). Cells were subjected to heat shock for 60 min at 45°C. Notably, only DDX3-EGFP expression partially rescued SG defects in hGle1-depleted cells (Figure 8, B and C). Next we tested whether DDX3 also rescues translation defects in hGle1-depleted cells upon heat shock. CTRL or hGLE1 siRNA cells were transfected with mCherry alone or mCherry-DDX3 plasmids and heat shocked; this was followed by metabolic labeling with AHA. Importantly, addition of mCherry-DDX3 but not mCherry alone partially rescued the translation defect in hGle1-depleted cells (Figure 8, D and E). Overall hGle1 function in SG assembly and translation might be linked to DDX3 regulation.
FIGURE 8:

DDX3 rescues hGle1-dependent SG and translation defects. (A) DDX3 coimmunoprecipitates with hGle1. HeLa cells were either left untreated or heat shocked for 30 min or 60 min, and cell lysates were immunoprecipitated using anti-hGle1 or IgG control antibodies and immunoblotted with anti-hGle1 or DDX3 antibodies. (B) DDX3 partially rescues SG defects in hGle1-depleted cells. CTRL or hGLE1 siRNA-treated cells were transfected with EGFP, EGFP-DDX19B, or DDX3-EGFP plasmids and heat shocked. Cells were processed for immunofluorescence detection of G3BP and hGle1. Scale bar: 10 μm. (C) SG numbers in CTRL and hGLE1 siRNA cells expressing indicated plasmids were quantified. (D) DDX3 partially rescues translation defects in hGle1-depleted cells. CTRL or hGLE1 siRNA-treated HeLa cells transfected with mCherry or mCherry-DDX3 plasmids were heat shocked at 45°C. After 15 min of heat shock, AHA was added to the incubations, and heat shock treatment was continued for an additional 30 min. Samples were processed for detection of AHA-labeled proteins with Alexa Fluor-488 alkyne using click chemistry. Scale bar: 10 μm. (E) Quantification of AHA-488 staining. Mean fluorescence intensity of AHA-488 staining in individual cells was calculated in CTRL and hGLE1 siRNA cells expressing the indicated plasmids.
DISCUSSION
Localization of mRNA in SGs plays a role in temporal and spatial regulation of gene expression. In this report, an essential role for hGle1 in SG biology is revealed. This function is distinct from hGle1's action during mRNA export at the NPC. hGle1 is recruited to SGs upon stress and also interacts with the translation initiation factor DDX3. Moreover, hGle1 depletion results in both defective SG assembly and altered translation during stress conditions, suggesting that hGle1 depletion shifts the distribution of mRNPs toward translation. This is further supported by our observation that hGle1-dependent SG defects are rescued by addition of the translational inhibitor puromycin. Finally, hGle1A overexpression does not induce SGs in the absence of stress but does increase the size of SGs in response to stress. Similarly, puromycin is not sufficient alone to nucleate SGs but induces larger granule formation in the presence of stress. Thus hGle1A induces the formation of larger SGs by modulating translation. In sum, hGle1 plays a key role in the exchange of mRNPs between SGs and the active translation machinery, thereby serving as a critical factor during the cellular stress response.
Previously we defined functions for Gle1 as a regulator of Dbps during mRNA export and translation (Murphy and Wente, 1996; Tran et al., 2007; Bolger et al., 2008; Bolger and Wente, 2011; Noble et al., 2011). Given the effects here on translation in the hGle1-depleted cells and the interactions between hGle1 and DDX3, a role for Gle1 in translation is likely conserved between S. cerevisiae and human cells. Multiple lines of evidence suggest that the impact of hGle1 on SGs is not due to its role in mRNA export. Depletion of the mRNA export factor DDX19B or NXF1 does not result in SG defects. Moreover, hGle1A rescues SG defects yet fails to rescue the mRNA export defects. It is clear that the hGleA and hGle1B isoforms play distinct separable functions in the cell: hGle1A in cytoplasmic SG function and hGle1B in mRNA export at the NPC.
This work supports a model wherein hGle1 exists in at least two distinct pools in the cell, allowing for a repertoire of independent functions and an ability to simultaneously regulate multiple steps in the gene expression pathway. It is possible that isoform-specific interactions with their unique protein partners might determine localization and/or functions. For instance, hGle1B but not hGle1A interacts with hCG1. Interestingly, only EGFP-hGle1B localizes to the NPC when transiently expressed in HeLa cells. EGFP-Gle1A does not localize to the NPC in the presence of endogenous hGle1. However, we find here that EGFP-hGle1A can in fact localize to NPCs when endogenous hGle1 is depleted; yet mRNA export is not efficient with hGle1A alone. We conclude that hGle1B binding to hCG1 is required for a step in mRNA export that is distinct from strict localization at NPCs. Indeed, our very recent studies support this conclusion, as interactions between the C-terminal domain of Nup42 (human homologue hCG1) and yGle1 are required for mRNP remodeling in yeast (Adams et al., 2014).
How mRNPs move in and out of SGs is a long-standing question in the field (Kedersha et al., 2005; Anderson and Kedersha, 2008; Zhang et al., 2011). To transition between these different states, mRNP rearrangements potentially occur, and thus RNA-binding proteins and mRNP remodelers are expected to play a role in directing mRNPs for storage in SG or translation. In yeast, the Dbp Ded1 regulates release of mRNA from SGs and promotes translation (Hilliker et al., 2011). We propose that hGle1 might function in distribution of mRNPs between SGs and translation through its regulation of DDX3. We find hGle1 interacts with DDX3, and the hGle1-dependent SG and translation defects are rescued by expression of DDX3. Interestingly, Ded1 and DDX3 assemble SGs in an ATP-independent manner (Hilliker et al., 2011; Shih et al., 2012). However, Ded1's ATPase activity is required for disassembly of SGs and re-entry into translation (Hilliker et al., 2011). Here we show that hGle1 depletion results in SG-assembly defects and slows disassembly of SGs. Moreover, hGle1 is involved in regulating the exchange between SGs and translation in a manner similar to that reported for Ded1. Thus lack of proper hGle1-mediated control of DDX3 could lead to improper SG dynamics.
Surprisingly, eIF2α phosphorylation at Ser-51 is modestly reduced in hGle1-depleted cells upon stress. This is consistent with the continued translation observed upon heat shock in hGle1-depleted cells. As expression of nonphosphorylatable serine to alanine mutant at residue 51 of eIF2α allows continued protein synthesis upon heat shock in CHO cells (Murtha-Riel et al., 1993), eIF2α phosphorylation is likely one of the primary mechanisms to inhibit global protein synthesis upon stress (Gebauer and Hentze, 2004; Wek et al., 2006; Sonenberg and Hinnebusch, 2009). It is intriguing that hGle1 depletion affects eIF2α phosphorylation and that cells maintain translation under stress. The eIF2 kinase, PKR is often activated by its association with double-stranded RNAs, and upon heat shock, Alu RNA can form stable complexes with PKR that result in its activation (Chu et al., 1998; Williams, 1999). Thus, as hGle1 regulates Dbps for mRNP remodeling, this could be a link to a potential activation of PKR kinase. Alternatively, there might be other mechanisms that play a role in regulating eIF2α phosphorylation. Additionally, translation perturbations observed in hGle1-depleted cells could be due to other regulatory pathways that may or may not be linked with eIF2α. Further studies are required to determine precisely how hGle1 levels influence eIF2α and modulate translation.
Deregulation of SG assembly, disassembly, and their clearance is associated with various neurological diseases. An emerging theme is that hyperassembly of SGs acts as an intermediate for disease progression. For instance, ALS-linked TDP-43 and FUS mutants form larger and more stable SGs under normal stress conditions (Dewey et al., 2011; Parker et al., 2012; Wolozin, 2012; Buchan et al., 2013; Vance et al., 2013). The SG phenotype during hGle1A overexpression is remarkably similar to that for the ALS-linked TDP-43 and FUS mutants. Our very recent work has linked mutations in hGLE1 with ALS (Kaneb et al., 2014). These ALS-linked hGLE1 mutants lack the hCG1 binding site and fail to localize at the NPC in a manner similar to the hGle1A isoform. Thus the ALS-linked hGLE1 mutations alter the cellular pools of hGle1A and hGle1B isoforms, and this may contribute to disease phenotypes. Our work here presents further evidence that depleting specific hGle1 isoforms differentially affects mRNA export and translation. Additionally, mutations in hGLE1 linked with inherited human diseases LCCS1 and LAAHD (Nousiainen et al., 2008) are due to disregulated mRNA export (Folkmann et al., 2013, 2014).
Taking these observations together, we speculate that the ALS hGLE1 mutations could impact both mRNA export (by decreasing the pool of hGle1B available) and translation (by altering the pool of hGle1A and SG dynamics). Moreover, other disease states could arise from alterations in hGle1A function specifically that impact translational regulation. Investigating the hGle1 mechanisms that drive the assembly and disassembly of SGs under normal conditions will guide the understanding of how pathological aggregates form.
MATERIALS AND METHODS
Cell culture, treatments, and plasmids
HeLa, U2OS, and RPE-1 cells were grown in complete DMEM or DMEM-F12 (Life Technologies, Grand Island, NY) supplemented with 10% fetal bovine serum (FBS; Atlanta Biologicals, Norcross, GA) at 37°C in 5% CO2. Cells were incubated at 45°C in a non-CO2 air incubator for 60 min to induce heat stress unless indicated otherwise. For ER-induced stress, cells were exposed to 10 μM thapsigargin (Sigma-Aldrich, St. Louis, MO) for 60 min at 37°C. Puromycin (Sigma-Aldrich) was used at either 0.1 mg/ml or 0.5 mg/ml for 60 min. Cycloheximide (Sigma-Aldrich) was used at 10 μg/ml. For MT perturbations, cells were incubated with 5 μM nocodazole (Sigma-Aldrich) or 500 nM Taxol (Sigma-Aldrich) for 120 min at 37°C; this was followed by heat shock at 45°C for 60 min. Plasmids expressing EGFP-C1, EGFP-hGle1BR (pSW3908; Folkmann et al., 2013), EGFP-hGLE1A (pSW1409; Kendirgi et al., 2003), and Pom121-mCherry (Mor et al., 2010) were described previously. siRNA-resistant EGFP-hGLE1AR, mCherry-hGLE1AR, and mCherry-hGLE1BR were generated by introduction of three silent mutations at the hGLE1 siRNA-targeting regions of EGFP-hGLE1A (pSW3909), mCherry-hGLE1A, and mCherry-hGLE1B plasmids by site-directed mutagenesis, respectively. DDX3-EGFP plasmid was PCR amplified from a cDNA (MGC:20129) purchased from GeneCopoeia (Rockville, MD) and cloned into pEGFP-N3 vector using XhoI and BamHI sites (pSW4105). mCherry-DDX3 was generated by PCR amplification of DDX3 cDNA and cloned into pmCherry-C1 vector.
siRNAs and plasmids transfections
Negative control siRNA and hGLE1 siRNAs were purchased from Qiagen (Valencia, CA). NXF1 and DDX19B SMARTpool siRNAs were purchased from Dharmacon (Lafayette, CO). Cells were reverse-transfected with indicated 20 nM siRNAs using HiPerFect (Qiagen) following the manufacturer's instructions. Plasmid transfections were performed using Fugene 6 (Promega, Madison, WI) following the manufacturer's instructions. For rescue of SG and translation phenotype, siRNA-resistant (“R”) EGFP, EGFP-hGLE1BR, and EGFP-hGLE1AR expression constructs were transfected after 48 h of siRNA transfections.
Coimmunoprecipitation
HeLa cells were plated in 100-mm dishes (Fisher Scientific, Pittsburgh, PA) and either left untreated or heat shocked for the indicated times at 45°C. After treatment, cells were washed with 1× phosphate-buffered saline (PBS) and lysed in buffer containing 0.5% NP-40, 50 mM Tris-Cl (pH 7.5), 100 mM NaCl, 1 mM EDTA, 50 mM NaF, 1 mM phenylmethylsulfonyl fluoride, and protease inhibitor cocktail (Roche, Nutley, NJ). Cell lysates were spun at 13,000 rpm for 10 min at 4°C, and the supernatants were incubated with control immunoglobulin G (IgG) or affinity-purified guinea pig polyclonal antibodies raised against recombinant N-terminus of hGle1 amino acids 1–360 (ASW48; Covance) for 5 h at 4°C. Protein A–Sepharose (GE Healthcare, Pittsburgh, PA) beads were added, and lysates were incubated for an additional 1 h at 4°C. Beads were washed with lysis buffer five times, and immune complexes were eluted with 50 μl 2× SDS-laemmli buffer. The eluted complexes were resolved on 7.5% SDS–PAGE and subjected to immunoblotting using anti-hGle1, 1:1000 (ASW48), and anti-DDX3, 1:1000 (Bethyl Laboratories, Montgomery, TX), antibodies.
Immunoblotting and immunofluorescence
For immunoblotting, cells were plated in 60-mm dishes (Fisher Scientific) and reverse transfected with 20 nM indicated siRNAs. After 72 h post-siRNA transfections, cells were lysed in buffer containing 1% NP-40, 150 mM NaCl, 50 mM Tris-Cl (pH 7.5), and protease and phosphatase inhibitor cocktail. For add-back experiments, indicated plasmids were transfected after 24 h of siRNA treatments. Proteins were resolved on 7.5% SDS–PAGE and immunoblotted with anti-hGle1, 1:1000 (Folkmann et al., 2013), anti-GFP, 1:1000 (Life Technologies), anti-actin, 1:5000 (Sigma-Aldrich), anti-eIF2α, 1:1000 (Cell Signaling Technology, Danvers, MA), and anti–phospho-eIF2α (Ser-51), 1:1000 (Cell Signaling Technology). Far-red dye–conjugated secondary antibodies were used for detection, and blots were scanned using a Li-Cor Odyssey scanner. For immunofluorescence, cells were plated on 1.5-mm round coverslips in a 24-well plate (Fisher Scientific). Following treatment, cells were fixed with 4% paraformaldehyde for 15 min and permeabilized with 0.2% Triton X-100 for 5 min. Coverslips were blocked with 10% FBS/PBS for 1 h at room temperature. The following primary antibodies were used: anti-hGle1, 1:300 (ASW48), anti-G3BP, 1:300 (BD Transduction, San Jose, CA), anti-DDX3, 1:300 (Bethyl Laboratories), anti-HuR, 1:100 (Santa Cruz Biotechnology, Dallas, TX), anti-FMRP, 1:300 (Millipore, Billerica, MA), and anti–α-tubulin, 1:500 (Abcam, Cambridge, MA), for ∼3 h at room temperature. Alexa Fluor–conjugated secondary antibodies (Life Technologies) were used, and slides were mounted using Prolong Gold AntiFade (Life Technologies). Cells were imaged using a 63×/1.4 NA oil-immersion objective on a confocal microscope (Leica TCS SP5).
In situ hybridization
HeLa cells were fixed with 4% paraformaldehyde for 15 min. Cells were washed with 1× PBS followed by permeablization with 0.2% Triton X-100/1× PBS for 5 min. Cells were incubated with 1 ng/μl of Cy3-conjugated oligo-dT in hybridization buffer containing 125 μg/ml tRNA, 1 mg/ml single-stranded DNA, 1% bovine serum albumin (BSA), 10% dextran sulfate, 50% formamide, and 5× saline sodium citrate (SSC), for 1.5 h at 37°C. After staining, coverslips were washed with 2× SSC two times followed by washes with 1× SSC. Coverslips were finally rinsed with 1× PBS, and slides were mounted using Prolong Gold AntiFade. When combined with immunofluorescence, in situ hybridization was performed following secondary antibody incubations. Images were acquired using 63×/1.4 NA oil-immersion objective on a Leica TCS SP5 confocal microscope (Leica Microsystems, Buffalo Grove, IL). Images were processed using ImageJ (National Institutes of Health, Bethesda, MD). For each EGFP-positive cell, mean Cy3 intensity was determined for the cytoplasmic and nuclear compartments, and N/C ratios were calculated.
Live-cell microscopy
HeLa cells were cotransfected with plasmids encoding mCherry-G3BP and either EGFP, EGFP-hGle1BR, or EGFP-hGle1AR using Fugene 6. Before imaging, culture medium was replaced with phenol-free DMEM supplemented with 25 mM HEPES and 10% FBS. Cells were imaged live on a heated stage at 45°C using a 63×/1.4 NA oil-immersion objective lens on a Leica SP5 microscope. For 3D structural illumination microscopy, CTRL or hGLE1 siRNA cells were cotransfected with Pom121-mCherry and either EGFP, EGFP-hGLE1BR, or EGFP-hGLE1AR plasmids. Images were acquired in 3D structural-illumination microscopy mode on a heated stage at 37°C using a Delta Vision OMX microscope (Applied Precision, Pittsburgh, PA).
Metabolic labeling for protein synthesis
CTRL and hGLE1 siRNA-transfected cells were washed with DMEM without cysteine and methionine and incubated in the same medium for 45 min at 37°C. Cells were either left untreated or subjected to heat shock at 45°C for 15 min. The methionine analogue AHA (50 μM; Life Technologies) was added to the incubations, and cells were either left untreated or heat shocked at 45°C for an additional 30 min. Following treatment, cells were fixed with 4% paraformaldehyde and permeabilized with 0.2% Triton X-100. Cells were washed with 2% BSA/PBS three times, and AHA-labeled proteins were detected with 1 μM alkyne-tagged Alexa Fluor-488 (Life Technologies) using Click-iT cell detection reagent (Life Technologies) for 45 min at room temperature. Cells were further processed for immunofluorescence as described above. For 35S metabolic labeling, CTRL and hGLE1 siRNA-treated cells were washed with DMEM without cysteine and methionine and incubated in the same medium for 45 min at 37°C. Radioactive [35S]methionine/cysteine (100 μCi/ml) (Perkin Elmer, Waltham, MA) was added to the medium, and cells were incubated for an additional 30 min at 37°C. Cells were lysed in RIPA buffer and protease and phosphatase inhibitor cocktail. Protein concentrations were determined using a DC protein assay kit (Bio-Rad, Hercules, CA). For liquid scintillation counts, equal amounts of labeled cell lysates were precipitated with 1 ml of 10% trichloroacetic acid (TCA) and BSA as a carrier protein for 30 min on ice. TCA precipitates were filtered onto glass microfiber filter disks grade GF/C in a filtration apparatus under vacuum. Disks were washed with 10% TCA followed by 100% ice-cold ethanol. Filters were air dried, and then counts were determined using a liquid scintillation counter (Beckman Coulter, Brea, CA)
Measurement of SG size and number
Post–image processing was done using ImageJ software. ImageJ plug-in 3D objects counter was used to calculate size and numbers of SGs of individual cells. The 3D objects counter measurement parameters were set to “surface,” and the minimum size filter was set to 3. Surface measurements were used to calculate sizes of SGs. SG sizes and numbers were represented using box plot generated in Prism6 (GraphPad, La Jolla, CA). The percentages of cells with SGs were calculated as 100 × [(number of cells with G3BP-positive foci)/(total number of cells)]. All data are the result of at least three independent experiments. Statistical significance was determined using Student's t test (Microsoft Excel, Redmond, WA) and Fisher's exact test (GraphPad) where appropriate.
Polysome profiles
Cells were grown in 10-cm dishes and lysed in 10 mM Tris-Cl (pH 7.5), 100 mM NaCl, 30 mM MgCl2, 200 μg heparin/ml, 100 μg/ml cycloheximide, and 1% Triton X-100. The lysates were cleared by centrifugation at 5000 rpm for 5 min. Cleared lysates were layered on 7–47% sucrose gradients cast in 50 mM Tris (pH 7.5), 50 mM NH4Cl, and 12 mM MgCl2, 100 μg/ml cycloheximide in ultracentrifuge tubes (Beckman). Gradients were centrifuged in a Beckman SW-41 rotor for 3 h at 28,800 rpm. An absorbance profile was collected from the gradients at 254 nm.
Supplementary Material
Acknowledgments
We thank Renee Dawson for technical assistance and critical reading of the manuscript; the Wente laboratory for discussions; Yaron Shav-Tal for the Pom121-mCherry plasmid; Andrew Link for assistance with polysome profiling; and James R. Goldenring for the mCherry-G3BP plasmid and for sharing reagents. This work was supported in part by the National Cancer Institute's Cancer Center Support grant P30CA068485 using the Cell Imaging Shared Resource and by grants from the National Institutes of Health (5R37GM051219 to S.R.W.; F31NS070431 to A.W.F.). A. was supported in part by the Vanderbilt International Scholar Program.
Abbreviations used:
- 3D
three-dimensional
- AHA
l-azidohomoalanine
- ALS
amyotrophic lateral sclerosis
- BSA
bovine serum albumin
- CTRL
scrambled control siRNA
- Dbp
DEAD-box protein
- EGFP
enhanced green fluorescent protein
- eIF2α
eukaryotic initiation factor 2 alpha
- FBS
fetal bovine serum
- IgG
immunoglobulin G
- mRNP
mRNA–protein complex
- MT
microtubule
- N/C
nuclear/cytoplasmic
- NPC
nuclear pore complex
- Nup
nucleoporin
- P bodies
processing bodies
- PBS
phosphate-buffered saline
- SG
stress granule
- siRNA
small interfering RNA
- TCA
trichloroacetic acid.
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E14-11-1523) on February 18, 2015.
REFERENCES
- Adams RL, Terry LJ, Wente SR. Nucleoporin FG domains facilitate mRNP remodeling at the cytoplasmic face of the nuclear pore complex. Genetics. 2014;197:1213–1224. doi: 10.1534/genetics.114.164012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alcazar-Roman AR, Tran EJ, Guo S, Wente SR. Inositol hexakisphosphate and Gle1 activate the DEAD-box protein Dbp5 for nuclear mRNA export. Nat Cell Biol. 2006;8:711–716. doi: 10.1038/ncb1427. [DOI] [PubMed] [Google Scholar]
- Anderson P, Kedersha N. Stress granules: the Tao of RNA triage. Trends Biochem Sci. 2008;33:141–150. doi: 10.1016/j.tibs.2007.12.003. [DOI] [PubMed] [Google Scholar]
- Anderson P, Kedersha N. RNA granules: post-transcriptional and epigenetic modulators of gene expression. Nat Rev Mol Cell Biol. 2009;10:430–436. doi: 10.1038/nrm2694. [DOI] [PubMed] [Google Scholar]
- Arimoto K, Fukuda H, Imajoh-Ohmi S, Saito H, Takekawa M. Formation of stress granules inhibits apoptosis by suppressing stress-responsive MAPK pathways. Nat Cell Biol. 2008;10:1324–1332. doi: 10.1038/ncb1791. [DOI] [PubMed] [Google Scholar]
- Ariumi Y, Kuroki M, Kushima Y, Osugi K, Hijikata M, Maki M, Ikeda M, Kato N. Hepatitis C virus hijacks P-body and stress granule components around lipid droplets. J Virol. 2011;85:6882–6892. doi: 10.1128/JVI.02418-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balagopal V, Parker R. Polysomes, P bodies and stress granules: states and fates of eukaryotic mRNAs. Curr Opin Cell Biol. 2009;21:403–408. doi: 10.1016/j.ceb.2009.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolger TA, Folkmann AW, Tran EJ, Wente SR. The mRNA export factor Gle1 and inositol hexakisphosphate regulate distinct stages of translation. Cell. 2008;134:624–633. doi: 10.1016/j.cell.2008.06.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolger TA, Wente SR. Gle1 is a multifunctional DEAD-box protein regulator that modulates Ded1 in translation initiation. J Biol Chem. 2011;286:39750–39759. doi: 10.1074/jbc.M111.299321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosco DA, Lemay N, Ko HK, Zhou H, Burke C, Kwiatkowski TJ, Jr, Sapp P, McKenna-Yasek D, Brown RH, Jr, Hayward LJ. Mutant FUS proteins that cause amyotrophic lateral sclerosis incorporate into stress granules. Hum Mol Genet. 2010;19:4160–4175. doi: 10.1093/hmg/ddq335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchan JR, Kolaitis RM, Taylor JP, Parker R. Eukaryotic stress granules are cleared by autophagy and Cdc48/VCP function. Cell. 2013;153:1461–1474. doi: 10.1016/j.cell.2013.05.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchan JR, Parker R. Eukaryotic stress granules: the ins and outs of translation. Mol Cell. 2009;36:932–941. doi: 10.1016/j.molcel.2009.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chernov KG, Barbet A, Hamon L, Ovchinnikov LP, Curmi PA, Pastre D. Role of microtubules in stress granule assembly: microtubule dynamical instability favors the formation of micrometric stress granules in cells. J Biol Chem. 2009;284:36569–36580. doi: 10.1074/jbc.M109.042879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu WM, Ballard R, Carpick BW, Williams BR, Schmid CW. Potential Alu function: regulation of the activity of double-stranded RNA-activated kinase PKR. Mol Cell Biol. 1998;18:58–68. doi: 10.1128/mcb.18.1.58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuesta R, Laroia G, Schneider RJ. Chaperone hsp27 inhibits translation during heat shock by binding eIF4G and facilitating dissociation of cap-initiation complexes. Genes Dev. 2000;14:1460–1470. [PMC free article] [PubMed] [Google Scholar]
- Decker CJ, Parker R. P-bodies and stress granules: possible roles in the control of translation and mRNA degradation. Cold Spring Harb Perspect Biol. 2012;4:a012286. doi: 10.1101/cshperspect.a012286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Nadal E, Ammerer G, Posas F. Controlling gene expression in response to stress. Nat Rev Genet. 2011;12:833–845. doi: 10.1038/nrg3055. [DOI] [PubMed] [Google Scholar]
- Dewey CM, Cenik B, Sephton CF, Dries DR, Mayer P, III, Good SK, Johnson BA, Herz J, Yu G. TDP-43 is directed to stress granules by sorbitol, a novel physiological osmotic and oxidative stressor. Mol Cell Biol. 2011;31:1098–1108. doi: 10.1128/MCB.01279-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dieterich DC, Lee JJ, Link AJ, Graumann J, Tirrell DA, Schuman EM. Labeling, detection and identification of newly synthesized proteomes with bioorthogonal non-canonical amino-acid tagging. Nat Protoc. 2007;2:532–540. doi: 10.1038/nprot.2007.52. [DOI] [PubMed] [Google Scholar]
- Dieterich DC, Link AJ, Graumann J, Tirrell DA, Schuman EM. Selective identification of newly synthesized proteins in mammalian cells using bioorthogonal noncanonical amino acid tagging (BONCAT) Proc Natl Acad Sci USA. 2006;103:9482–9487. doi: 10.1073/pnas.0601637103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Folkmann AW, Collier SE, Zhan X, Aditi, Ohi MD, Wente SR. Gle1 functions during mRNA export in an oligomeric complex that is altered in human disease. Cell. 2013;155:582–593. doi: 10.1016/j.cell.2013.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Folkmann AW, Dawson TR, Wente SR. Insights into mRNA export-linked molecular mechanisms of human disease through a Gle1 structure-function analysis. Adv Biol Regul. 2014;54:74–91. doi: 10.1016/j.jbior.2013.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallouzi I-E, Brennan CM, Stenberg MG, Swanson MS, Eversole A, Maizels N, Steitz JA. HuR binding to cytoplasmic mRNA is perturbed by heat shock. Proc Natl Acad Sci USA. 2000;97:3073–3078. doi: 10.1073/pnas.97.7.3073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gebauer F, Hentze MW. Molecular mechanisms of translational control. Nat Rev Mol Cell Biol. 2004;5:827–835. doi: 10.1038/nrm1488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilks N, Kedersha N, Ayodele M, Shen L, Stoecklin G, Dember LM, Anderson P. Stress granule assembly is mediated by prion-like aggregation of TIA-1. Mol Biol Cell. 2004;15:5383–5398. doi: 10.1091/mbc.E04-08-0715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grüter P, Tabernero C, von Kobbe C, Schmitt C, Saavedra C, Bachi A, Wilm M, Felber BK, Izaurralde E. TAP, the human homolog of Mex67p, mediates CTE-dependent RNA export from the nucleus. Mol Cell. 1998;1:649–659. doi: 10.1016/s1097-2765(00)80065-9. [DOI] [PubMed] [Google Scholar]
- Herold A, Suyama M, Rodrigues JP, Braun IC, Kutay U, Carmo-Fonseca M, Bork P, Izaurralde E. TAP (NXF1) belongs to a multigene family of putative RNA export factors with a conserved modular architecture. Mol Cell Biol. 2000;20:8996–9008. doi: 10.1128/mcb.20.23.8996-9008.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilliker A, Gao Z, Jankowsky E, Parker R. The DEAD-box protein Ded1 modulates translation by the formation and resolution of an eIF4F-mRNA complex. Mol Cell. 2011;43:962–972. doi: 10.1016/j.molcel.2011.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodge CA, Tran EJ, Noble KN, Alcazar-Roman AR, Ben-Yishay R, Scarcelli JJ, Folkmann AW, Shav-Tal Y, Wente SR, Cole CN. The Dbp5 cycle at the nuclear pore complex during mRNA export I: dbp5 mutants with defects in RNA binding and ATP hydrolysis define key steps for Nup159 and Gle1. Genes Dev. 2011;25:1052–1064. doi: 10.1101/gad.2041611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanov PA, Chudinova EM, Nadezhdina ES. Disruption of microtubules inhibits cytoplasmic ribonucleoprotein stress granule formation. Exp Cell Res. 2003;290:227–233. doi: 10.1016/s0014-4827(03)00290-8. [DOI] [PubMed] [Google Scholar]
- Kaneb HM, Folkmann AW, Belzil VV, Jao LE, Leblond CS, Girard SL, Daoud H, Noreau A, Rochefort D, Hince P, et al. Deleterious mutations in the essential mRNA metabolism factor, hGle1, in amyotrophic lateral sclerosis. Hum Mol Genet. 2014;24:1363–1373. doi: 10.1093/hmg/ddu545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang Y, Cullen BR. The human Tap protein is a nuclear mRNA export factor that contains novel RNA-binding and nucleocytoplasmic transport sequences. Genes Dev. 1999;13:1126–1139. doi: 10.1101/gad.13.9.1126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kedersha N, Anderson P. Stress granules: sites of mRNA triage that regulate mRNA stability and translatability. Biochem Soc Trans. 2002;30:963–969. doi: 10.1042/bst0300963. [DOI] [PubMed] [Google Scholar]
- Kedersha N, Anderson P. Mammalian stress granules and processing bodies. Methods Enzymol. 2007;431:61–81. doi: 10.1016/S0076-6879(07)31005-7. [DOI] [PubMed] [Google Scholar]
- Kedersha N, Anderson P. Regulation of translation by stress granules and processing bodies. Prog Mol Biol Transl Sci. 2009;90:155–185. doi: 10.1016/S1877-1173(09)90004-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kedersha N, Cho MR, Li W, Yacono PW, Chen S, Gilks N, Golan DE, Anderson P. Dynamic shuttling of TIA-1 accompanies the recruitment of mRNA to mammalian stress granules. J Cell Biol. 2000;151:1257–1268. doi: 10.1083/jcb.151.6.1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kedersha N, Stoecklin G, Ayodele M, Yacono P, Lykke-Andersen J, Fritzler MJ, Scheuner D, Kaufman RJ, Golan DE, Anderson P. Stress granules and processing bodies are dynamically linked sites of mRNP remodeling. J Cell Biol. 2005;169:871–884. doi: 10.1083/jcb.200502088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kedersha NL, Gupta M, Li W, Miller I, Anderson P. RNA-binding proteins TIA-1 and TIAR link the phosphorylation of eIF-2 alpha to the assembly of mammalian stress granules. J Cell Biol. 1999;147:1431–1442. doi: 10.1083/jcb.147.7.1431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kendirgi F, Barry DM, Griffis ER, Powers MA, Wente SR. An essential role for hGle1 nucleocytoplasmic shuttling in mRNA export. J Cell Biol. 2003;160:1029–1040. doi: 10.1083/jcb.200211081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kendirgi F, Rexer DJ, Alcazar-Roman AR, Onishko HM, Wente SR. Interaction between the shuttling mRNA export factor Gle1 and the nucleoporin hCG1: a conserved mechanism in the export of Hsp70 mRNA. Mol Biol Cell. 2005;16:4304–4315. doi: 10.1091/mbc.E04-11-0998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khaperskyy DA, Hatchette TF, McCormick C. Influenza A virus inhibits cytoplasmic stress granule formation. FASEB J. 2012;26:1629–1639. doi: 10.1096/fj.11-196915. [DOI] [PubMed] [Google Scholar]
- Kim WJ, Back SH, Kim V, Ryu I, Jang SK. Sequestration of TRAF2 into stress granules interrupts tumor necrosis factor signaling under stress conditions. Mol Cell Biol. 2005;25:2450–2462. doi: 10.1128/MCB.25.6.2450-2462.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolobova E, Efimov A, Kaverina I, Rishi AK, Schrader JW, Ham AJ, Larocca MC, Goldenring JR. Microtubule-dependent association of AKAP350A and CCAR1 with RNA stress granules. Exp Cell Res. 2009;315:542–555. doi: 10.1016/j.yexcr.2008.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai MC, Lee YH, Tarn WY. The DEAD-box RNA helicase DDX3 associates with export messenger ribonucleoproteins as well as tip-associated protein and participates in translational control. Mol Biol Cell. 2008;19:3847–3858. doi: 10.1091/mbc.E07-12-1264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lloyd RE. How do viruses interact with stress-associated RNA granules. PLoS Pathog. 2012;8:e1002741. doi: 10.1371/journal.ppat.1002741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lloyd RE. Regulation of stress granules and P-bodies during RNA virus infection. Wiley Interdiscip Rev RNA. 2013;4:317–331. doi: 10.1002/wrna.1162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Maury L, Marguerat S, Bahler J. Tuning gene expression to changing environments: from rapid responses to evolutionary adaptation. Nat Rev Genet. 2008;9:583–593. doi: 10.1038/nrg2398. [DOI] [PubMed] [Google Scholar]
- Low WK, Dang Y, Schneider-Poetsch T, Shi Z, Choi NS, Merrick WC, Romo D, Liu JO. Inhibition of eukaryotic translation initiation by the marine natural product pateamine A. Mol Cell. 2005;20:709–722. doi: 10.1016/j.molcel.2005.10.008. [DOI] [PubMed] [Google Scholar]
- Mazroui R, Huot M-E, Tremblay S, Filion C, Labelle Y, Khandjian EW. Trapping of messenger RNA by fragile X mental retardation protein into cytoplasmic granules induces translation repression. Hum Mol Genet. 2002;11:3007–3017. doi: 10.1093/hmg/11.24.3007. [DOI] [PubMed] [Google Scholar]
- Mazroui R, Sukarieh R, Bordeleau ME, Kaufman RJ, Northcote P, Tanaka J, Gallouzi I, Pelletier J. Inhibition of ribosome recruitment induces stress granule formation independently of eukaryotic initiation factor 2α phosphorylation. Mol Biol Cell. 2006;17:4212–4219. doi: 10.1091/mbc.E06-04-0318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montpetit B, Thomsen ND, Helmke KJ, Seeliger MA, Berger JM, Weis K. A conserved mechanism of DEAD-box ATPase activation by nucleoporins and InsP6 in mRNA export. Nature. 2011;472:238–242. doi: 10.1038/nature09862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mor A, Suliman S, Ben-Yishay R, Yunger S, Brody Y, Shav-Tal Y. Dynamics of single mRNP nucleocytoplasmic transport and export through the nuclear pore in living cells. Nat Cell Biol. 2010;12:543–552. doi: 10.1038/ncb2056. [DOI] [PubMed] [Google Scholar]
- Murphy R, Wente SR. An RNA-export mediator with an essential nuclear export signal. Nature. 1996;383:357–360. doi: 10.1038/383357a0. [DOI] [PubMed] [Google Scholar]
- Murtha-Riel P, Davies MV, Scherer BJ, Choi SY, Hershey JW, Kaufman RJ. Expression of a phosphorylation-resistant eukaryotic initiation factor 2 alpha-subunit mitigates heat shock inhibition of protein synthesis. J Biol Chem. 1993;268:12946–12951. [PubMed] [Google Scholar]
- Nadezhdina ES, Lomakin AJ, Shpilman AA, Chudinova EM, Ivanov PA. Microtubules govern stress granule mobility and dynamics. Biochim Biophys Acta. 2010;1803:361–371. doi: 10.1016/j.bbamcr.2009.12.004. [DOI] [PubMed] [Google Scholar]
- Noble KN, Tran EJ, Alcazar-Roman AR, Hodge CA, Cole CN, Wente SR. The Dbp5 cycle at the nuclear pore complex during mRNA export II: nucleotide cycling and mRNP remodeling by Dbp5 are controlled by Nup159 and Gle1. Genes Dev. 2011;25:1065–1077. doi: 10.1101/gad.2040611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nousiainen HO, Kestila M, Pakkasjarvi N, Honkala H, Kuure S, Tallila J, Vuopala K, Ignatius J, Herva R, Peltonen L. Mutations in mRNA export mediator GLE1 result in a fetal motoneuron disease. Nat Genet. 2008;40:155–157. doi: 10.1038/ng.2007.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onishi H, Kino Y, Morita T, Futai E, Sasagawa N, Ishiura S. MBNL1 associates with YB-1 in cytoplasmic stress granules. J Neurosci Res. 2008;86:1994–2002. doi: 10.1002/jnr.21655. [DOI] [PubMed] [Google Scholar]
- Panniers R. Translational control during heat shock. Biochimie. 1994;76:737–747. doi: 10.1016/0300-9084(94)90078-7. [DOI] [PubMed] [Google Scholar]
- Parker R, Sheth U. P bodies and the control of mRNA translation and degradation. Mol Cell. 2007;25:635–646. doi: 10.1016/j.molcel.2007.02.011. [DOI] [PubMed] [Google Scholar]
- Parker SJ, Meyerowitz J, James JL, Liddell JR, Crouch PJ, Kanninen KM, White AR. Endogenous TDP-43 localized to stress granules can subsequently form protein aggregates. Neurochem Int. 2012;60:415–424. doi: 10.1016/j.neuint.2012.01.019. [DOI] [PubMed] [Google Scholar]
- Rayala HJ, Kendirgi F, Barry DM, Majerus PW, Wente SR. The mRNA export factor human Gle1 interacts with the nuclear pore complex protein Nup155. Mol Cell Proteomics. 2004;3:145–155. doi: 10.1074/mcp.M300106-MCP200. [DOI] [PubMed] [Google Scholar]
- Ruggieri A, Dazert E, Metz P, Hoffmann S, Bergeest JP, Mazur J, Bankhead P, Hiet MS, Kallis S, Alvisi G, et al. Dynamic oscillation of translation and stress granule formation mark the cellular response to virus infection. Cell Host Microbe. 2012;12:71–85. doi: 10.1016/j.chom.2012.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shih JW, Wang WT, Tsai TY, Kuo CY, Li HK, Wu Lee YH. Critical roles of RNA helicase DDX3 and its interactions with eIF4E/PABP1 in stress granule assembly and stress response. Biochem J. 2012;441:119–129. doi: 10.1042/BJ20110739. [DOI] [PubMed] [Google Scholar]
- Sonenberg N, Hinnebusch AG. Regulation of translation initiation in eukaryotes: mechanisms and biological targets. Cell. 2009;136:731–745. doi: 10.1016/j.cell.2009.01.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spriggs KA, Bushell M, Willis AE. Translational regulation of gene expression during conditions of cell stress. Mol Cell. 2010;40:228–237. doi: 10.1016/j.molcel.2010.09.028. [DOI] [PubMed] [Google Scholar]
- Stoecklin G, Stubbs T, Kedersha N, Wax S, Rigby WF, Blackwell TK, Anderson P. MK2-induced tristetraprolin:14–3-3 complexes prevent stress granule association and ARE-mRNA decay. EMBO J. 2004;23:1313–1324. doi: 10.1038/sj.emboj.7600163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas MG, Loschi M, Desbats MA, Boccaccio GL. RNA granules: the good, the bad and the ugly. Cell Signal. 2011;23:324–334. doi: 10.1016/j.cellsig.2010.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tourriere H, Chebli K, Zekri L, Courselaud B, Blanchard JM, Bertrand E, Tazi J. The RasGAP-associated endoribonuclease G3BP assembles stress granules. J Cell Biol. 2003;160:823–831. doi: 10.1083/jcb.200212128. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Tran EJ, Zhou Y, Corbett AH, Wente SR. The DEAD-box protein Dbp5 controls mRNA export by triggering specific RNA:protein remodeling events. Mol Cell. 2007;28:850–859. doi: 10.1016/j.molcel.2007.09.019. [DOI] [PubMed] [Google Scholar]
- Vance C, Scotter EL, Nishimura AL, Troakes C, Mitchell JC, Kathe C, Urwin H, Manser C, Miller CC, Horthobagyi T, et al. ALS mutant FUS disrupts nuclear localization and sequesters wild-type FUS within cytoplasmic stress granules. Hum Mol Genet. 2013;22:2676–2688. doi: 10.1093/hmg/ddt117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanderweyde T, Youmans K, Liu-Yesucevitz L, Wolozin B. Role of stress granules and RNA-binding proteins in neurodegeneration: a mini-review. Gerontology. 2013;59:524–533. doi: 10.1159/000354170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weirich CS, Erzberger JP, Flick JS, Berger JM, Thorner J, Weis K. Activation of the DExD/H-box protein Dbp5 by the nuclear-pore protein Gle1 and its coactivator InsP6 is required for mRNA export. Nat Cell Biol. 2006;8:668–676. doi: 10.1038/ncb1424. [DOI] [PubMed] [Google Scholar]
- Wek RC, Jiang HY, Anthony TG. Coping with stress: eIF2 kinases and translational control. Biochem Soc Trans. 2006;34:7–11. doi: 10.1042/BST20060007. [DOI] [PubMed] [Google Scholar]
- Williams BR. PKR; a sentinel kinase for cellular stress. Oncogene. 1999;18:6112–6120. doi: 10.1038/sj.onc.1203127. [DOI] [PubMed] [Google Scholar]
- Wolozin B. Regulated protein aggregation: stress granules and neurodegeneration. Mol Neurodegener. 2012;7:56. doi: 10.1186/1750-1326-7-56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yedavalli VSRK, Neuveut C, Chi Y-H, Kleiman L, Jeang K-T. Requirement of DDX3 DEAD box RNA helicase for HIV-1 Rev-RRE export function. Cell. 2004;119:381–392. doi: 10.1016/j.cell.2004.09.029. [DOI] [PubMed] [Google Scholar]
- Zhang J, Okabe K, Tani T, Funatsu T. Dynamic association-dissociation and harboring of endogenous mRNAs in stress granules. J Cell Sci. 2011;124:4087–4095. doi: 10.1242/jcs.090951. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.