

PKCι has an essential role in epithelial polarity. Its knockdown increases epithelial barrier through control of the trafficking of claudin-2. PKCι interacts directly with the small GTPase Rab14, and these proteins colocalize in cells. It is proposed that these proteins act together to regulate claudin-2 trafficking.
Abstract
PKCι is essential for the establishment of epithelial polarity and the normal assembly of tight junctions. We find that PKCι knockdown does not compromise the steady-state distribution of most tight junction proteins but results in increased transepithelial resistance (TER) and decreased paracellular permeability. Analysis of the levels of tight junction components demonstrates that claudin-2 protein levels are decreased. However, other tight junction proteins, such as claudin-1, ZO-1, and occludin, are unchanged. Incubation with an aPKC pseudosubstrate recapitulates the phenotype of PKCι knockdown, including increased TER and decreased levels of claudin-2. In addition, overexpression of PKCι results in increased claudin-2 levels. ELISA and coimmunoprecipitation show that the TGN/endosomal small GTPase Rab14 and PKCι interact directly. Immunolabeling shows that PKCι and Rab14 colocalize in both intracellular puncta and at the plasma membrane and that Rab14 expression is required for normal PKCι distribution in cysts in 3D culture. We showed previously that knockdown of Rab14 results in increased TER and decreased claudin-2. Our results suggest that Rab14 and aPKC interact to regulate trafficking of claudin-2 out of the lysosome-directed pathway.
INTRODUCTION
Epithelial cells contain an apical domain facing the “outside world” and a basolateral domain facing the inside environment. Epithelial sheets provide multicellular organisms with protection from intrusion of pathogens while still acting as a selectively permeable barrier to nutrients and ions. This selective barrier is composed of ionic (pore; Watson et al., 2005) and larger molecule (leak) pathways (Anderson and Van Itallie, 2009; Turner, 2009), which are modulated by the specific components of the tight junctions. The formation and maintenance of tight junctions rely largely on the polarized transport and localization of proteins and lipids mediated by polarity complexes.
Atypical protein kinase C (aPKC) is the enzymatic component of the Par polarity complex. The Par complex intimately associates with tight junctions and plays important roles in the formation and maintenance of tight junctions (Suzuki et al., 2001; Hurd et al., 2003; Sasaki et al., 2003; Shen and Turner, 2005; Horikoshi et al., 2009). Functional inhibition of aPKC by overexpressing dominant-negative forms of PKCι or PKCζ, the two isoforms of aPKC, dramatically delayed the reassembly of tight junction proteins at cell–cell contacts after calcium switch in epithelial cells (Suzuki et al., 2001). PKCζ affects the assembly of tight junctions by direct phosphorylation of tight junction proteins. For example, assembly of occludin into tight junctions requires its phosphorylation at several sites by PKCζ (Jain et al., 2011). Similarly, phosphorylation of JAM-A by PKCζ promotes the retention of JAM-A at the junction and the maturation of tight junctions (Iden et al., 2012). PKCι is a close relative of PKCζ, sharing 86% amino acid sequence identity in the kinase domain. Although expression of the dominant-negative form of PKCι resulted in a similar phenotype to that of PKCζ (Suzuki et al., 2001), PKCι- or PKCζ-knockout mice have different phenotypes, implying that PKCι and PKCζ function in distinct signaling pathways (Leitges et al., 2001; Martin et al., 2002).
Tight junction proteins are highly dynamic and undergo continuous trafficking between the cell surface and intracellular compartments (Ivanov et al., 2004; Bruewer et al., 2005; Utech et al., 2005; Marchiando et al., 2010). As major regulators of membrane trafficking, Rab-family small GTPases are directly involved in the regulation of epithelial apical junctions (Marzesco et al., 2002; Yamamura et al., 2008). Rab13, Rab5, Rab34, and their effectors regulate the trafficking of occludin (Morimoto et al., 2005; Coyne et al., 2007), and Rab11 controls the transport of E-cadherin from the trans-Golgi network to basolateral membranes (Riggs et al., 2003; Lock and Stow, 2005). We found that Rab14 protects claudin-2 from targeting to lysosomes (Lu et al., 2014), increasing the transepithelial electrical resistance (TER) and causing defective morphogenesis in three-dimensional (3D) culture. Despite the role for these molecules in tight junction assembly, reported interactions between Rabs and the polarity machinery are limited. Rab2 was reported to require PKCι to recruit β-COP for vesicle budding (Tisdale, 2000). However, a link between Rab small GTPases, aPKC activity, and junction formation has not been demonstrated.
Here we find that PKCι regulates the protein level of claudin-2. Depletion of PKCι results in increased TER and a reduction of claudin-2 in the cells. As with Rab14, we find that PKCι protects claudin-2 from lysosomal degradation. Furthermore, we show that PKCι binds to Rab14 and that PKCι requires Rab14 for its correct distribution in cells in 3D culture. These results suggest that PKCι and Rab14 work coordinately to modify tight junction structure.
RESULTS
Inhibition of PKCι in epithelial cells results in decreased permeability
Expression of a kinase-dead form of PKCι or PKCζ inhibits reassembly of tight junctions after calcium switch (Suzuki et al., 2001). To investigate further the role of PKCι in the regulation of epithelial integrity, we used lentiviral infection with a plasmid encoding a specific short hairpin RNA (shRNA) for PKCι in MDCK II cells. After transduction, the cells were selected to generate stable cell lines. This shRNA provided efficient knockdown of PKCι (Figure 1, A and B). In addition, quantitative reverse transcriptase PCR (RT-PCR) shows that this shRNA has no effect on message levels of PKCζ (Supplemental Figure S1A). Of interest, this analysis also shows that wild-type Madin–Darby canine kidney (MDCK) cells express at least 10 times more PKCι mRNA than PKCζ mRNA.
FIGURE 1:
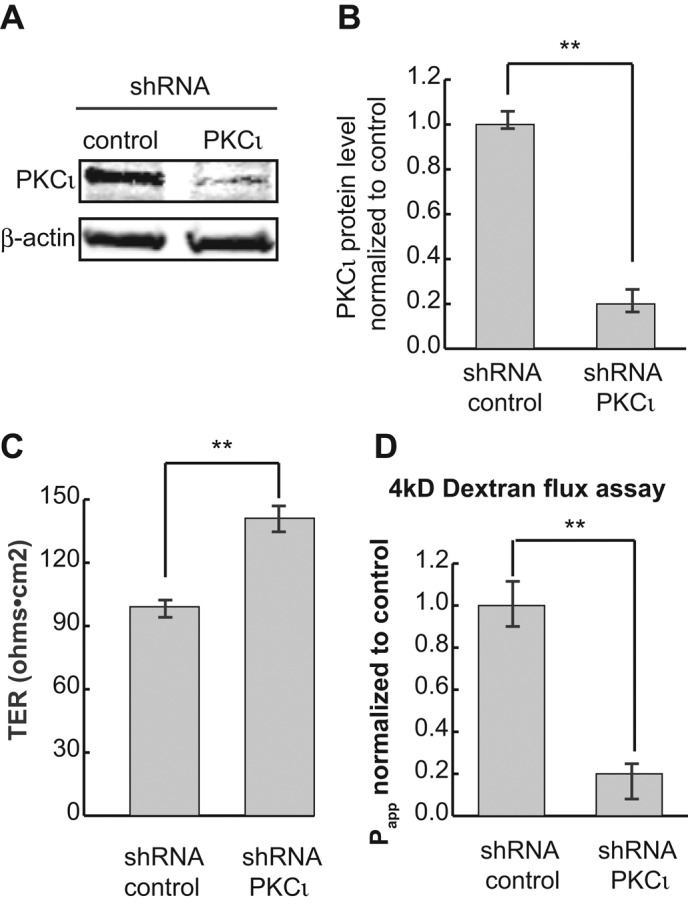
PKCι knockdown decreases the permeability of MDCK II monolayers. (A) PKCι was knocked down by lentiviral shRNA, and cell lysates were analyzed by Western blotting. β-Actin was used as loading control. (B) Quantification of protein levels in A. (C) TER measurement shows PKCι reduction results in a slight but significant increase of TER. (D) The 4- kDa FL-dextran flux assay shows that knockdown of PKCι significantly decreases epithelial permeability. Each experiment was performed in triplicate and repeated at least three times. Error bars represent SEM. **p < 0.001.
To determine whether knockdown of PKCι results in delayed reassembly of junctions, as observed previously with kinase-dead PKCι (Suzuki et al., 2001), we tested the assembly of junctions after calcium switch. As shown in Supplemental Figure S1B, knockdown of PKCι has a similar phenotype to overexpression of the kinase-dead mutant (Suzuki et al., 2001), with delayed targeting of occludin, claudin-1, and ZO-1 to the lateral membrane after return to calcium-containing medium.
To assess the integrity of the epithelium at steady state, we plated PKCι-knockdown cells onto Transwell filters and measured the TER of the monolayers. Surprisingly, knockdown of PKCι resulted in increased TER (Figure 1C), suggesting that PKCι plays a role in the maintenance of the tight junction pore pathway. To further assess this, we tested the permeability of the epithelium using a flux assay with 4-kDa fluorescein dextran. PKCι knockdown resulted in a significant decrease in flux from the apical to the basolateral chamber (Figure 1D), indicating that PKCι also affects the leak pathway.
Changes in permeability may be due to changes in the levels of tight junction proteins. To examine this, we evaluated cell lysates by SDS–PAGE and immunoblotting. The levels of claudin-1, occludin, ZO-1, and E-cadherin are unchanged in knockdown cells compared with control cultures (Figure 2A). However, the level of claudin-2 is significantly decreased, and claudin-4 is increased (Figure 2, A and B). This is consistent with other studies that showed a reciprocal relationship between expression levels of claudin-2 and claudin-4 (Capaldo et al., 2014). To confirm that loss of claudin-2 is due to PKCι knockdown rather than an off-target effect of lentiviral transfection, we transiently transfected MDCK cells with a distinct small interfering RNA (siRNA) against PKCι. As shown in Supplemental Figure S1, C and D, depletion of PKCι by siRNA also decreased claudin-2 protein levels.
FIGURE 2:

PKCι knockdown decreases claudin-2 protein levels. (A) Tight junction proteins claudin-1, -2, and -4, occludin, ZO-1, and adhesion junction protein E-cadherin were analyzed by Western blotting. Claudin-2 is dramatically decreased in PKCι-knockdown cells compared with control, whereas claudin-4 is increased. β-Actin was used as loading control. (B) Representative quantification of claudin-2 and claudin-4 protein levels from at least three independent experiments. Statistics were from three replicates. Error bars represent SEM. **p < 0.001. (C) immunofluorescence microscopy shows normal junctional localization of tight junction proteins. However, much less claudin-2 is present in PKCι-knockdown cells compared with control cells. (D) Western blot showing knockdown of PKCι in MDCK I cells. (E) TER of MDCK I cells after PKCι knockdown. Scale bar, 10 μm (C).
We next immunolabeled cells stably knocked down for PKCι. These cells showed a substantial loss of claudin-2 labeling at the junctions. However, the distribution of occludin and claudin-1 tight junction proteins was the same as in control cells (Figure 2C).
To test whether the changes in TER after PKCι knockdown were primarily due to loss of claudin-2, we performed PKCι knockdown in MDCK type I cells (Figure 2D), which are deficient in claudin-2 (Amasheh et al., 2002). As shown in Figure 2E, knockdown of PKCι in MDCK I cells does not affect the TER.
To assess the role of the kinase activity of aPKC on claudin-2 expression, we incubated wild-type cells with the aPKC pseudosubstrate zeta inhibitory peptide (ZIP; Laudanna et al., 1998) and measured TER and claudin-2 levels. As shown in Figure 3A, inhibition of aPKC kinase activity resulted in slightly decreased levels of claudin-2, accompanied by a substantial increase in TER (Figure 3B). Although the loss of claudin-2 is not as great as observed with PKCι knockdown, these results are consistent with the knockdown data. However, our results differ from previous reports using this drug in MDCK cells (Jain et al., 2011; Mashukova et al., 2011; Ragupathy et al., 2014). This may be due to different concentrations of the inhibitor (10 μM in this study vs. 25–50 μM in the other studies) or different cell type. Of importance, inhibition of protein synthesis did not affect the TER, indicating that the effects of inhibition of PKCι were not due to changes in claudin-2 transcription (Figure 3B). In addition, ZIP treatment did not affect the level of message for claudin-2 (Supplemental Figure S1E). It is important to note that the increase in TER seen with ZIP treatment likely results from effects on more than claudin-2 levels, as the change is far greater than that seen with complete loss of claudin-2.
FIGURE 3:

(A) MDCK II cells were lysed after ZIP treatment and claudin-2 protein levels analyzed. β-Actin was used as loading control (top). Quantification shows claudin-2 protein level slightly but significantly decreased (bottom). (B) TER of confluent cell monolayers was measured after 2 h of ZIP treatment in the presence of cycloheximide. ZIP treatment causes a dramatic increase in TER, but this is unaffected by cycloheximide. (C) Less claudin-2 localizes to the cell–cell junction after ZIP treatment. Random lines were drawn across cell–cell junctions, and the plot profiles were analyzed with ImageJ (middle). Pixel intensity of the peaks was averaged and plotted (right). Pixel intensity of claudin-2 at junctions of treated cells is significantly lower than in controls. In contrast, junctional localization of claudin-1 was not different in untreated and treated cells. Scale bar, 10 μm. (D) MDCK cells were transfected with green fluorescent protein (GFP) or PKCι-GFP. Claudin-2 protein levels were analyzed by immunoblot. β-Actin was used as loading control (top). Claudin-2 protein level increases more than twofold in cells overexpressing PKCι-GFP (bottom). Error bars represent SEM; ** p< 0.001.
We also performed immunofluorescence analysis of cells after ZIP treatment and found that there is less claudin-2 localized to junctions of ZIP-treated cells (Figure 3C). Although not as dramatic as PKCι knockdown, measurement of pixel intensity across the junction confirmed a significant loss of claudin-2 labeling at the junctions after ZIP treatment.
If PKCι is important for regulating claudin-2 levels, overexpression of PKCι might be expected to increase claudin-2 levels. To test this, we overexpressed PKCι by transient transfection and determined claudin-2 levels by immunoblotting. As shown in Figure 3, overexpression of full-length PKCι resulted in substantially increased levels of claudin-2 (Figure 3D).
PKCι knockdown increases lysosomal degradation of claudin-2
To determine whether loss of claudin-2 in PKCι-knockdown cells is due to faster degradation through targeting to lysosomes, we incubated the cells with the lysosomotropic amine NH4Cl. After 24 h of treatment, the claudin-2 protein levels are significantly increased (Figure 4A, left and middle) in both control and PKCι-knockdown cells. Immunofluorescence data show that claudin-2 accumulates in lysosome-associated membrane protein 1 (LAMP1)–labeled lysosomes after NH4Cl treatment (Figure 4B). Claudin-2 is increased in both control and PKCι-knockdown cells under these conditions, suggesting rapid turnover of claudin-2. However, proportionately, there is a greater increase of claudin-2 in PKCι-knockdown cells (twofold) than in control cells (1.35-fold; Figure 4A, right), implying that claudin-2 undergoes faster degradation in PKCι-knockdown cells. Of interest, incubation of the cells with ammonium chloride slightly reverses the increase in TER (Supplemental Figure S1F), indicating that some of the claudin-2 can escape the degradative pathway under these conditions.
FIGURE 4:

NH4Cl treatment restores claudin-2 in PKCι-knockdown cells. (A) Cells were incubated in 20 mM NH4Cl for 24 h and lysed. Claudin-2 was blotted (left) and protein levels were quantified (right). Claudin-2 protein levels increased twofold in PKCι-knockdown cells, in contrast to a 1.35-fold increase in control cells. Data were obtained from samples run in triplicate and three independent experiments. β-Actin was used as loading control. Error bars represent SEM; **p < 0.001. (B) Claudin-2 localizes with LAMP1 after 4-h incubation with 20 mM NH4Cl (arrows). Scale bar, 10 μm.
Rab14 and PKCι interact and colocalize
We showed that Rab14 modulates junction integrity through regulation of claudin-2 trafficking. To determine whether PKCι could act coordinately with Rab14, we tested whether Rab14 and PKCι interact directly, using an enzyme-linked immunosorbent assay (ELISA) and purified proteins. ELISA analysis indicates that Rab14 binds to PKCι in a saturable and high-affinity manner (Figure 5A). Furthermore, competitive ELISA shows that the binding between Rab14 and PKCι is specific (Figure 5B). Of interest, the GTP-bound state of Rab14 did not affect the interaction with PKCι, as the affinity of binding Rab14 in the presence of nonhydrolyzable GTP to PKCι was similar (Supplemental Figure S1G). To determine whether PKCι and Rab14 interact in cells, we immunoprecipitated Rab14-GFP from HEK cells and found that PKCι is specifically immunoprecipitated with Rab14 (Figure 5C).
FIGURE 5:

(A) ELISA shows that PKCι interacts with Rab14 in a high-affinity and saturable manner. Bovine serum albumin was used as control. (B) Competitive ELISA shows that unlabeled Rab14 competes with labeled Rab14 for binding to PKCι. (C) Rab14-GFP coimmunoprecipitates with PKCι.
To determine the subcellular localization of PKCι and Rab14 and where in the cell they might interact, we labeled endogenous Rab14 and PKCι in MDCK cells. Both Rab14 and PKCι reside in cytoplasmic puncta, and some colocalization of the two labels is observed in these puncta. In addition, there is substantial colocalization at cell–cell contacts (Figure 6A).
FIGURE 6:

PKCι colocalizes with Rab14 in puncta and at cell–cell contacts in MDCK cells. (A) Endogenous Rab14 is present in cytoplasmic puncta and at cell junctions. There is some colocalization with PKCι at both locations. Pearson's colocalization is 0.47 ± 0.07; N = 17 independent images. Scale bar, 10 μm. (B) Knockdown of Rab14 results in decreased PKCι. (C) Knockdown of PKCι results in decreased Rab14. Representative blots from three independent experiments in B and C, and β-actin was used as loading control. Statistics were acquired from three replicate sample loadings. Error bars represent SEM; **p < 0.001.
Because of the interaction between Rab14 and PKCι observed through both biochemical and morphological analysis, we next tested the effects of knockdown of either PKCι or Rab14 on the expression of Rab14 (PKCι KD) or PKCι (Rab14 KD). As shown in Figure 6, knockdown of Rab14 results in less PKCι protein (Figure 6B), and knockdown of PKCι results in less Rab14 protein (Figure 6C). These results suggest that Rab14 and PKCι form a complex and stabilize each other in the cell.
Rab14 knockdown results in redistribution of aPKC in epithelial cysts
Epithelial cysts normally exhibit apical and junctional labeling of aPKC (Martin-Belmonte et al., 2007), and knockdown of Rab14 results in failure to form single lumen cysts (Lu et al., 2014). To test the distribution of PKCι in cysts generated from Rab14-knockdown cells, we fixed Rab14-knockdown cysts and labeled them for PKCι and the apical marker podocalyxin. Although some apical localization of PKCι is maintained after Rab14 knockdown, there is some mislocalization of PKCι to the basolateral cytoplasm (Figure 7), suggesting that the interaction of PKCι with Rab14 is essential for the normal localization of PKCι and may provide an additional reason why Rab14-knockdown cells do not form single-lumen cysts.
FIGURE 7:

Rab14 knockdown results in mislocalized aPKC. Matrigel-grown cysts were labeled for gp135 (podocalyxin, an apical membrane marker) and aPKC. In Rab14-knockdown cells, aPKC is mislocalized from its normal apical location to the basal domain (arrows). Scale bar, 10 μm.
DISCUSSION
Establishment and maintenance of epithelial polarity require the coordinated regulation of polarity complexes and membrane trafficking. Our results suggest that these pathways intersect at the endosomal compartment through the interaction of the membrane trafficking regulator Rab14 and the polarity protein PKCι. Whereas compromise of PKCι kinase activity (Suzuki et al., 2001) or PKCι knockdown and pharmacological inhibition by ZIP (this study) slowed assembly of tight junctions, reflected as delayed recruitment of occludin, claudin-1, and ZO-1 to the plasma membrane after calcium switch, at steady state, these tight junction proteins appear to be unaffected. However, we observe depletion of claudin-2, suggesting that the targeting of this molecule is regulated by phosphorylation by PKCι.
Depletion of PKCι results in increased TER and decreased paracellular permeability. These changes indicate that both the pore and leak pathways are regulated by PKCι. The increase in TER can be attributed to loss of claudin-2, as it is in the class of “leaky” claudins that mediate increased movement of ions across epithelia (Amasheh et al., 2002; Rosenthal et al., 2010). The leak pathway is regulated by ZO-1 (Van Itallie et al., 2009) and occludin (Balda et al., 1996; Yu et al., 2005), and we do not detect a change in distribution of these tight junction proteins at steady state. However, studies demonstrated an interaction between occludin and claudin-2 (Raleigh et al., 2011), and loss of claudin-2 may limit trafficking of occludin, resulting in less permeability of the leak pathway.
Decreased claudin-2 with PKCι knockdown is due to changes in trafficking of this protein, and our study provides evidence that PKCι normally functions to protect it from lysosomal targeting. Of interest, we also see a change in the transcription of claudin-2 with PKCι knockdown (unpublished data). The mechanism for this is unclear, but it was also seen with knockdown of Rab14 (unpublished data). This effect on the transcription of integrins is observed with Rab25 knockdown (Krishnan et al., 2013), suggesting that trafficking of these molecules provides signals that regulate gene transcription.
Phosphorylation and dephosphorylation of tight junction proteins is a common mechanism of regulation of targeting to the plasma membrane (D'souza et al., 2005, 2007; Ikari et al., 2006, 2008; Aono and Hirai, 2008; Van Itallie et al., 2012). PKCζ phosphorylates occludin and JAM-A, affecting their distribution in the cell (Jain et al., 2011; Iden et al., 2012). Claudin-2 is phosphorylated on S208, and this phosphorylation promotes its membrane retention, whereas dephosphorylated claudin-2 is targeted to lysosomes for degradation (Van Itallie et al., 2012). PKC pseudosubstrate inhibition of kinase activity recapitulated the phenotype of PKCι knockdown, indicating that the kinase activity is required for the regulation of claudin-2 trafficking. The amino acids surrounding S208 in claudin-2 meet the requirement for a consensus PKCι site (Kang et al., 2012), indicating that claudin-2 could be a direct substrate of PKCι.
Rab GTPases are major organizers of intracellular membrane trafficking in eukaryotic cells (Stenmark, 2009). Rab13 and Rab14 play important roles in tight junction formation and maintenance (Marzesco et al., 2002; Morimoto et al., 2005; Yamamura et al., 2008; Lu et al., 2014), and knockdown of Rab14 results in loss of claudin-2 from the cells (Lu et al., 2014). However, there are limited reports of an interaction between Rabs and atypical PKCs. Rab2 interacts with PKCι and stimulates its association with membrane. Furthermore, both Rab2 and PKCι are essential for β-COP recruitment to the transport vesicles (Tisdale, 2000). We show that Rab14 interacts directly with PKCι and that they colocalize in cells. Of interest, these proteins may stabilize each other, as knockdown of one results in decreased levels of the other. Furthermore, it is likely that this interaction is functional, as the normal apical distribution of PKCι is disrupted in cysts that have Rab14 knockdown. These findings, together with similar effects upon claudin-2 levels with knockdown of either PKCι or Rab14, suggest that these proteins work in concert to regulate tight junction structure.
MATERIALS AND METHODS
Reagents and antibodies
Common chemicals and reagents were purchased from Sigma-Aldrich (St. Louis, MO). ZIP was from AnaSpec (Fremont, CA). The following antibodies were used: rabbit or mouse anti–claudin-1, rabbit or mouse anti–claudin-2, mouse anti–ZO-1, rabbit or mouse anti-occludin from Life Technologies (Grand Island, NY). Rabbit anti-Rab14, mouse anti–β-actin, mouse anti–E-cadherin and rabbit anti–E-cadherin were purchased from Sigma-Aldrich. Rabbit anti-aPKC was from Santa Cruz Biotechnology (Dallas, TX), and mouse anti-PKCι was from BD Biosciences (San Jose, CA). Alexa Fluor secondary antibodies were from Life Technologies.
Plasmids
pLKO shRNA construct against PKCι is AAATCTGGCATGTTCTTCAGG. pLKO empty vector was used as control. Cells were infected with this vector on four independent occasions.
The siRNA GCAGGUGGUACCUCCCUUUAAACCA and scramble negative control were synthesized by Integrated DNA Technologies (San Diego, CA).
Cell culture
Cell culture reagents were purchased from Mediatech (Manassas, VA). MDCK II cells were used in all experiments, except that HEK cells were used for coimmunoprecipitations. Cells were cultured in high-glucose DMEM with 10% fetal bovine serum (Atlanta Biologicals, Lawrenceville, GA), 1% nonessential amino (Mediatech), and 1% penicillin/streptomycin/l-glutamine (Sigma-Aldrich) under 5% CO2 at 37°C.
For transient transfection, polyethylenimine (PEI; Fisher Scientific, Waltham, MA) was used as DNA carrier. Briefly, PEI and DNA were mixed in Opti-MEM (Mediatech) at a ratio of 4:1. After incubation at room temperature for 10 min, the mixture was added to cells. Medium was replaced with fresh normal growth medium after 16 h.
For lentiviral transduction, cells were infected with lentivirus in the presence of 6 μg/ml Polybrene overnight, followed by selection in growth medium containing 2 μg/ml puromycin. Controls were transduced with empty vector.
For drug treatments, cells were treated with 10 μm ZIP (AnaSpec, Fremont, CA) in the presence of 10 μg/ml cycloheximide for 2 h at 37°C.
Transepithelial electrical resistance measurement
For TER measurements, cells were plated at 1.3 × 105 cells/cm2 on filter inserts (Corning, Corning, NY) and cultured for 4–5 d. TER was measured with a Millicell epithelial volt-ohm meter (EMD Millipore, Billerica, MA). TER measurements are expressed as Ω⋅cm2. TER measurements were performed in triplicate, and values are expressed as mean ± SEM.
Western blotting
Cells were lysed in RIPA buffer (25 mM Tris, pH 7.4, 150 mM NaCl, 1 mM EDTA, 1% Triton X-100). Samples were separated by SDS–PAGE and transferred to cellulose membrane. Transferred membranes were probed with primary antibodies diluted in LI-COR (Lincoln, NE) blocking buffer (with 0.05% Tween-20). Secondary antibodies were rabbit or mouse IR-Dye 680 or 800 (LI-COR). Membranes were imaged using a LI-COR Odyssey scanner. Boxes were manually placed around each band of interest, which returned near-infrared fluorescence values of raw intensity, with intralane background subtracted using LI-COR Odyssey 3.0 analytical software. The fluorescence value for each protein of interest was normalized to the in-lane value of β-actin, and this normalized ratio was averaged from duplicate or triplicate lanes. Data were analyzed using the t test. Measures were considered significant when p < 0.05. Error bars are SEM.
ELISAs
Polystyrene high-binding microwell plates (Sigma-Aldrich) were coated with 100 μl/well (0.5 μg/ml) of active PKCι (EMD Millipore) in 0.1 M carbonate coating buffer (pH 9.6) and incubated overnight at 4°C. The wells were blocked with 1% bovine albumin fraction V (Sigma-Aldrich) in wash buffer (20 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid [HEPES], pH 7.4, 80 mM KCl, 2 mM MgCl2, 0.2% bovine albumin fraction V, 0.05% Tween) for 1 h at 37°C. Rab14-6His (100 μl) in a series of concentrations (0, 1, 5, 10, 25, 50, 75, 100, 150, 250 nM) in binding buffer (20 mM HEPES, pH 7.4, 80 mM KCl, 2 mM MgCl2, 0.2% bovine albumin fraction V) was added to the wells and incubated at room temperature for 1 h. After washing, mouse anti-hexahistidine (6His) was added for 1 h at room temperature. Plates were washed and incubated with anti-mouse immunoglobulin G (IgG) conjugated to alkaline phosphatase. The enzyme reaction was developed with 100 μl/well of p-nitrophenyl phosphate (pNPP) as chromogenic substrates. pNPP buffer was prepared by dissolving one tablet of phosphatase substrate (Sigma-Aldrich) in 5 ml of substrate buffer (0.1 M glycine, 1 mM MgCl2, 1 mM ZnCl2, pH 10.4). Plates were analyzed by measuring optical density at 405 nm on a microtiter plate reader (Tecan, San Jose, CA). Samples were tested in triplicate, and each run included negative controls (bovine albumin fraction V; Sigma-Aldrich). Values were analyzed using Prism software (GraphPad, La Jolla, CA).
To test for effect of the nucleotide binding state of Rab14 on the interaction with PKCι, microwell plates were coated and blocked as described. Rab14-6His at increasing amounts (0.001, 0.002, 0.005, 0.01, 0.02, 0.05, 0.1, 0.2, 0.5 μg) with or without 1 mM GTPγs in the binding buffer were used to bind PKCι.
Competitive ELISA
Microwell plates were coated and blocked as described. A series of concentrations of Rab14-6His (0, 0.1, 1, 10, 50, 100, 1000 nM) were mixed with 1 nM biotin-Rab14-6His and added to the wells for 1 h at room temperature. After washing, binding of biotin-Rab14-6His was assessed by the addition of 100 μl/well of alkaline phosphatase–conjugated anti-biotin IgG (Sigma-Aldrich) at a 1:30,000 dilution for 1 h. Substrates were developed and analyzed as described.
Cyst culture
Single cells were plated on the top of 100% Matrigel (BD Biosciences)–coated, glass-bottom, eight-well chambers (155409; Lab-Tek, Rochester, NY) and cultured in DMEM with 2% Matrigel. Cysts were fixed and labeled 4 or 5 d after plating.
Dextran flux assay
Confluent cell monolayers on Transwells were rinsed three times with Hank's balanced saline solution (HBSS) and equilibrated in HBSS for 30 min at 37°C. Apical Hank's was replaced with 0.2 ml fresh HBSS containing 1.0 mg/ml 4-kDa fluorescein dextran (Sigma-Aldrich) and incubated at 37°C for 3 h. A 100-μl HBSS sample was collected from the basolateral chamber, and fluorescein signal was read on a Varioskan Flash plate reader (Thermo Fisher Scientific, Waltham, MA). Fluxes were calculated using the equation for apparent permeability coefficient Papp = (dQ/dt)/AC0 (Van Itallie et al., 2008), where Q is the amount of 4-kDa FL-Dextran present in the basolateral chamber, A is the filter area, and C0 is the initial 4-kDa FL-dextran concentration in the apical chamber.
Calcium switch
Confluent cells plated on Transwells were incubated in calcium-free medium overnight. Regular medium was replaced, and cells were fixed 1 h after replacement. Cells without medium replacement were used as nonswitch control.
Immunofluorescence microscopy and image analysis
Cells plated on coverslips or Transwells were fixed in methanol at −20°C for 10 min or in 4% formaldehyde freshly depolymerized from paraformaldehyde for 15 min at room temperature. After permeabilization, cells were incubated in primary antibody at 4°C overnight. Cells were washed and incubated with appropriate secondary antibody, and samples were mounted onto glass slides with ProLong Gold (Life Technologies). Imaging was performed on an Olympus FluoView1200 confocal microscope with a 60× (numerical aperture 1.4) oil immersion objective. Images were processed and merged using Photoshop (Adobe, San Jose, CA) or ImageJ (Schneider et al., 2012).
Coimmunoprecipitation
HEK cells were transfected with Rab14-GFP and Flag-PKCι. Cells were harvested and lysed in Pierce Lysis Buffer (25 mM Tris, 150 mM NaCl, 1 mM EDTA, 1% NP-40, 5% glycerol, pH 7.4; Thermo Scientific, Rockford, IL). Lysates were cleared by incubating with control agarose resin for 1 h at 4°C and then incubated with 5 μg of rabbit anti-Rab14 antibody or rabbit IgG as control overnight at 4°C, followed by incubation with Pierce Protein A/G Agarose (Thermo Scientific). Input and immunoprecipitation fractions were analyzed using anti-PKCι antibody.
Supplementary Material
Acknowledgments
We thank members of the Wilson lab for helpful discussions. This work was supported by National Institutes of Health Grant DK084047 (J.M.W.).
Abbreviations used:
- PKC
protein kinase C
- TER
transepithelial electrical resistance
- ZIP
zeta inhibitory peptide.
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E14-12-1613) on February 18, 2015.
REFERENCES
- Amasheh S, Meiri N, Gitter AH, Schoneberg T, Mankertz J, Schulzke JD, Fromm M. Claudin-2 expression induces cation-selective channels in tight junctions of epithelial cells. J Cell Sci. 2002;115:4969–4976. doi: 10.1242/jcs.00165. [DOI] [PubMed] [Google Scholar]
- Anderson JM, Van Itallie CM. Physiology and function of the tight junction. Cold Spring Harb Perspect Biol. 2009;1:a002584. doi: 10.1101/cshperspect.a002584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aono S, Hirai Y. Phosphorylation of claudin-4 is required for tight junction formation in a human keratinocyte cell line. Exp Cell Res. 2008;314:3326–3339. doi: 10.1016/j.yexcr.2008.08.012. [DOI] [PubMed] [Google Scholar]
- Balda MS, Whitney JA, Flores C, Gonzalez S, Cereijido M, Matter K. Functional dissociation of paracellular permeability and transepithelial electrical resistance and disruption of the apical-basolateral intramembrane diffusion barrier by expression of a mutant tight junction membrane protein. J Cell Biol. 1996;134:1031–1049. doi: 10.1083/jcb.134.4.1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruewer M, Utech M, Ivanov AI, Hopkins AM, Parkos CA, Nusrat A. Interferon-gamma induces internalization of epithelial tight junction proteins via a macropinocytosis-like process. FASEB J. 2005;19:923–933. doi: 10.1096/fj.04-3260com. [DOI] [PubMed] [Google Scholar]
- Capaldo CT, Farkas AE, Hilgarth RS, Krug SM, Wolf MF, Benedik JK, Fromm M, Koval M, Parkos C, Nusrat A. Proinflammatory cytokine-induced tight junction remodeling through dynamic self-assembly of claudins. Mol Biol Cell. 2014;25:2710–2719. doi: 10.1091/mbc.E14-02-0773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coyne CB, Shen L, Turner JR, Bergelson JM. Coxsackievirus entry across epithelial tight junctions requires occludin and the small GTPases Rab34 and Rab5. Cell Host Microbe. 2007;2:181–192. doi: 10.1016/j.chom.2007.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'souza T, Agarwal R, Morin PJ. Phosphorylation of claudin-3 at threonine 192 by cAMP-dependent protein kinase regulates tight junction barrier function in ovarian cancer cells. J Biol Chem. 2005;280:26233–26240. doi: 10.1074/jbc.M502003200. [DOI] [PubMed] [Google Scholar]
- D'souza T, Indig FE, Morin PJ. Phosphorylation of claudin-4 by PKCepsilon regulates tight junction barrier function in ovarian cancer cells. Exp Cell Res. 2007;313:3364–3375. doi: 10.1016/j.yexcr.2007.06.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horikoshi Y, Suzuki A, Yamanaka T, Sasaki K, Mizuno K, Sawada H, Yonemura S, Ohno S. Interaction between PAR-3 and the aPKC-PAR-6 complex is indispensable for apical domain development of epithelial cells. J Cell Sci. 2009;122:1595–1606. doi: 10.1242/jcs.043174. [DOI] [PubMed] [Google Scholar]
- Hurd TW, Gao L, Roh MH, Macara IG, Margolis B. Direct interaction of two polarity complexes implicated in epithelial tight junction assembly. Nat Cell Biol. 2003;5:137–142. doi: 10.1038/ncb923. [DOI] [PubMed] [Google Scholar]
- Iden S, Misselwitz S, Peddibhotla SS, Tuncay H, Rehder D, Gerke V, Robenek H, Suzuki A, Ebnet K. aPKC phosphorylates JAM-A at Ser285 to promote cell contact maturation and tight junction formation. J Cell Biol. 2012;196:623–639. doi: 10.1083/jcb.201104143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikari A, Matsumoto S, Harada H, Takagi K, Hayashi H, Suzuki Y, Degawa M, Miwa M. Phosphorylation of paracellin-1 at Ser217 by protein kinase A is essential for localization in tight junctions. J Cell Sci. 2006;119:1781–1789. doi: 10.1242/jcs.02901. [DOI] [PubMed] [Google Scholar]
- Ikari A, Okude C, Sawada H, Yamazaki Y, Sugatani J, Miwa M. TRPM6 expression and cell proliferation are up-regulated by phosphorylation of ERK1/2 in renal epithelial cells. Biochem Biophys Res Commun. 2008;369:1129–1133. doi: 10.1016/j.bbrc.2008.03.002. [DOI] [PubMed] [Google Scholar]
- Ivanov AI, Nusrat A, Parkos CA. Endocytosis of epithelial apical junctional proteins by a clathrin-mediated pathway into a unique storage compartment. Mol Biol Cell. 2004;15:176–188. doi: 10.1091/mbc.E03-05-0319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain S, Suzuki T, Seth A, Samak G, Rao R. Protein kinase Czeta phosphorylates occludin and promotes assembly of epithelial tight junctions. Biochem J. 2011;437:289–299. doi: 10.1042/BJ20110587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang JH, Toita R, Kim CW, Katayama Y. Protein kinase C (PKC) isozyme-specific substrates and their design. Biotechnol Adv. 2012;30:1662–1672. doi: 10.1016/j.biotechadv.2012.07.004. [DOI] [PubMed] [Google Scholar]
- Krishnan M, Lapierre LA, Knowles BC, Goldenring JR. Rab25 regulates integrin expression in polarized colonic epithelial cells. Mol Biol Cell. 2013;24:818–831. doi: 10.1091/mbc.E12-10-0745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laudanna C, Mochly-Rosen D, Liron T, Constantin G, Butcher EC. Evidence of zeta protein kinase C involvement in polymorphonuclear neutrophil integrin-dependent adhesion and chemotaxis. J Biol Chem. 1998;273:30306–30315. doi: 10.1074/jbc.273.46.30306. [DOI] [PubMed] [Google Scholar]
- Leitges M, Sanz L, Martin P, Duran A, Braun U, Garcia JF, Camacho F, Diaz-Meco MT, Rennert PD, Moscat J. Targeted disruption of the zetaPKC gene results in the impairment of the NF-kappaB pathway. Mol Cell. 2001;8:771–780. doi: 10.1016/s1097-2765(01)00361-6. [DOI] [PubMed] [Google Scholar]
- Lock JG, Stow JL. Rab11 in recycling endosomes regulates the sorting and basolateral transport of E-cadherin. Mol Biol Cell. 2005;16:1744–1755. doi: 10.1091/mbc.E04-10-0867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu R, Johnson DL, Stewart L, Waite K, Elliott D, Wilson JM. Rab14 regulation of claudin-2 trafficking modulates epithelial permeability and lumen morphogenesis. Mol Biol Cell. 2014;25:1744–1754. doi: 10.1091/mbc.E13-12-0724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchiando AM, Shen L, Graham WV, Weber CR, Schwarz BT, Austin JR, 2nd, Raleigh DR, Guan Y, Watson AJ, Montrose MH, Turner JR. Caveolin-1-dependent occludin endocytosis is required for TNF-induced tight junction regulation in vivo. J Cell Biol. 2010;189:111–126. doi: 10.1083/jcb.200902153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin P, Duran A, Minguet S, Gaspar ML, Diaz-Meco MT, Rennert P, Leitges M, Moscat J. Role of zeta PKC in B-cell signaling and function. EMBO J. 2002;21:4049–4057. doi: 10.1093/emboj/cdf407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin-Belmonte F, Gassama A, Datta A, Yu W, Rescher U, Gerke V, Mostov K. PTEN-mediated apical segregation of phosphoinositides controls epithelial morphogenesis through Cdc42. Cell. 2007;128:383–397. doi: 10.1016/j.cell.2006.11.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marzesco AM, Dunia I, Pandjaitan R, Recouvreur M, Dauzonne D, Benedetti EL, Louvard D, Zahraoui A. The small GTPase Rab13 regulates assembly of functional tight junctions in epithelial cells. Mol Biol Cell. 2002;13:1819–1831. doi: 10.1091/mbc.02-02-0029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mashukova A, Wald FA, Salas PJ. Tumor necrosis factor alpha and inflammation disrupt the polarity complex in intestinal epithelial cells by a posttranslational mechanism. Mol Cell Biol. 2011;31:756–765. doi: 10.1128/MCB.00811-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morimoto S, Nishimura N, Terai T, Manabe S, Yamamoto Y, Shinahara W, Miyake H, Tashiro S, Shimada M, Sasaki T. Rab13 mediates the continuous endocytic recycling of occludin to the cell surface. J Biol Chem. 2005;280:2220–2228. doi: 10.1074/jbc.M406906200. [DOI] [PubMed] [Google Scholar]
- Ragupathy S, Esmaeili F, Paschoud S, Sublet E, Citi S, Borchard G. Toll-like receptor 2 regulates the barrier function of human bronchial epithelial monolayers through atypical protein kinase C zeta, and an increase in expression of claudin-1. Tissue Barriers. 2014;2:e29166. doi: 10.4161/tisb.29166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raleigh DR, Boe DM, Yu D, Weber CR, Marchiando AM, Bradford EM, Wang Y, Wu L, Schneeberger EE, Shen L, Turner JR. Occludin S408 phosphorylation regulates tight junction protein interactions and barrier function. J Cell Biol. 2011;193:565–582. doi: 10.1083/jcb.201010065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riggs B, Rothwell W, Mische S, Hickson GR, Matheson J, Hays TS, Gould GW, Sullivan W. Actin cytoskeleton remodeling during early Drosophila furrow formation requires recycling endosomal components Nuclear-fallout and Rab11. J Cell Biol. 2003;163:143–154. doi: 10.1083/jcb.200305115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenthal R, Milatz S, Krug SM, Oelrich B, Schulzke JD, Amasheh S, Gunzel D, Fromm M. Claudin-2, a component of the tight junction, forms a paracellular water channel. J Cell Sci. 2010;123:1913–1921. doi: 10.1242/jcs.060665. [DOI] [PubMed] [Google Scholar]
- Sasaki H, Matsui C, Furuse K, Mimori-Kiyosue Y, Furuse M, Tsukita S. Dynamic behavior of paired claudin strands within apposing plasma membranes. Proc Natl Acad Sci USA. 2003;100:3971–3976. doi: 10.1073/pnas.0630649100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nat Methods. 2012;9:671–675. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen L, Turner JR. Actin depolymerization disrupts tight junctions via caveolae-mediated endocytosis. Mol Biol Cell. 2005;16:3919–3936. doi: 10.1091/mbc.E04-12-1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stenmark H. Rab GTPases as coordinators of vesicle traffic. Nat Rev Mol Cell Biol. 2009;10:513–525. doi: 10.1038/nrm2728. [DOI] [PubMed] [Google Scholar]
- Suzuki A, Yamanaka T, Hirose T, Manabe N, Mizuno K, Shimizu M, Akimoto K, Izumi Y, Ohnishi T, Ohno S. Atypical protein kinase C is involved in the evolutionarily conserved par protein complex and plays a critical role in establishing epithelia-specific junctional structures. J Cell Biol. 2001;152:1183–1196. doi: 10.1083/jcb.152.6.1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tisdale EJ. Rab2 requires PKC iota/lambda to recruit beta-COP for vesicle formation. Traffic. 2000;1:702–712. doi: 10.1034/j.1600-0854.2000.010903.x. [DOI] [PubMed] [Google Scholar]
- Turner JR. Intestinal mucosal barrier function in health and disease. Nat Rev Immunol. 2009;9:799–809. doi: 10.1038/nri2653. [DOI] [PubMed] [Google Scholar]
- Utech M, Ivanov AI, Samarin SN, Bruewer M, Turner JR, Mrsny RJ, Parkos CA, Nusrat A. Mechanism of IFN-gamma-induced endocytosis of tight junction proteins: myosin II-dependent vacuolarization of the apical plasma membrane. Mol Biol Cell. 2005;16:5040–5052. doi: 10.1091/mbc.E05-03-0193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Itallie CM, Fanning AS, Bridges A, Anderson JM. ZO-1 stabilizes the tight junction solute barrier through coupling to the perijunctional cytoskeleton. Mol Biol Cell. 2009;20:3930–3940. doi: 10.1091/mbc.E09-04-0320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Itallie CM, Holmes J, Bridges A, Gookin JL, Coccaro MR, Proctor W, Colegio OR, Anderson JM. The density of small tight junction pores varies among cell types and is increased by expression of claudin-2. J Cell Sci. 2008;121:298–305. doi: 10.1242/jcs.021485. [DOI] [PubMed] [Google Scholar]
- Van Itallie CM, Tietgens AJ, LoGrande K, Aponte A, Gucek M, Anderson JM. Phosphorylation of claudin-2 on serine 208 promotes membrane retention and reduces trafficking to lysosomes. J Cell Sci. 2012;125:4902–4912. doi: 10.1242/jcs.111237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson CJ, Hoare CJ, Garrod DR, Carlson GL, Warhurst G. Interferon-gamma selectively increases epithelial permeability to large molecules by activating different populations of paracellular pores. J Cell Sci. 2005;118:5221–5230. doi: 10.1242/jcs.02630. [DOI] [PubMed] [Google Scholar]
- Yamamura R, Nishimura N, Nakatsuji H, Arase S, Sasaki T. The interaction of JRAB/MICAL-L2 with Rab8 and Rab13 coordinates the assembly of tight junctions and adherens junctions. Mol Biol Cell. 2008;19:971–983. doi: 10.1091/mbc.E07-06-0551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu AS, McCarthy KM, Francis SA, McCormack JM, Lai J, Rogers RA, Lynch RD, Schneeberger EE. Knockdown of occludin expression leads to diverse phenotypic alterations in epithelial cells. Am J Physiol Cell Physiol. 2005;288:C1231–C1241. doi: 10.1152/ajpcell.00581.2004. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.