

Abstract
Aims
Oxidative stress is present in and contributes to calcification of the aortic valve, but the driving factors behind the initiation of valve oxidative stress are not well understood. We tested whether the valve endothelium acts as an initiator and propagator of oxidative stress in aortic valve disease.
Methods and Results
Calcified human aortic valves showed side-specific elevation of superoxide in the endothelium, co-localized with high VCAM1 expression, linking oxidative stress, inflammation, and valve degeneration. Treatment with inflammatory cytokine TNFα increased superoxide and oxidative stress and decreased eNOS and VE-cadherin acutely over 48 hours in aortic valve endothelial cells (VEC) and chronically over 21 days in ex vivo AV leaflets. Co-treatment of VEC with tetrahydrobiopterin (BH4) but not apocynin mitigated TNFα-driven VEC oxidative stress. Co-treatment of ex vivo AV leaflets with TNFα+BH4 or TNFα+peg-SOD rescued endothelial function and mitigated inflammatory responses. Both BH4 and peg-SOD rescued valve leaflets from the pro-osteogenic effects of TNFα treatment, but only peg-SOD was able to mitigate the fibrogenic effects, including increased collagen and αSMA expression.
Conclusions
Aortic valve endothelial cells are a novel source of oxidative stress in aortic valve disease. TNFα-driven VEC oxidative stress causes loss of endothelial protective function, chronic inflammation, and fibrogenic and osteogenic activation, mitigated differentially by BH4 and peg-SOD. These mechanisms identify new targets for tailored antioxidant therapy focused on mitigation of oxidative stress and restoration of endothelial protection.
Introduction
Aortic valve disease (AVD) causes approximately 15,000 deaths per year in the United States, occurring in 2.8% of Americans over the age of 75 [1]. AVD is an active process driven by complex intercellular interactions [2] that is pathobiologically unique from other cardiovascular diseases [3–5]. Valve endothelial cells (VEC), which line the surface of the valve, are phenotypically different from other endothelial cell populations [6–9] and must be investigated for unique pathological mechanisms. VEC are known to play a unique role in regulating the properties of valve tissue in response to the dynamic cardiac environment [10–11], but their role in early disease is not known. Disruption of VEC function is an early and persistent feature of AVD [12–14], but it is not known if disrupted VEC actively contribute to valve calcification. Development of treatments that would protect against AVD by maintaining VEC protective function and blocking potential active contributions to calcification are hampered by a lack of understanding of the underlying mechanisms of VEC pathology.
VEC production of nitric oxide (NO) is protective against AVD, but is reduced in calcified valves [15–18]. In vascular disease, NO is reduced due to a phenomenon known as eNOS uncoupling [19], however, it is not known if eNOS uncoupling represents a significant source of oxidative stress in AVD. Uncoupled eNOS produces superoxide rather than nitric oxide due to a lack of the eNOS co-factor tetrahydrobiopterin (BH4) [20]. Increased superoxide reacts with NO to form peroxynitrite (ONOO-), further reducing the bioavailability of NO [21], or excess superoxide is dismutated to hydrogen peroxide (H2O2) [22]. Superoxide and hydrogen peroxide are increased in calcifying regions of the aortic valve [23–24] and contribute to osteogenic activation of valve interstitial cells [25]. If eNOS uncoupling is a significant phenomenon in the valve endothelium, VEC-derived oxidative stress could be an important and accessible target in the development of treatments to prevent valve calcification.
Inflammation has long been clinically linked to oxidative stress [26] and eNOS uncoupling [27] in vascular endothelial cells, but it is not known whether a similar link exists in valve endothelial cells as a mechanism of valvular disease. Inflammatory activation of the endothelium correlates proportionally with calcification in AVD [28]. Inflammation induces early and sustained expression of endothelial inflammatory adhesion molecules VCAM-1 and ICAM-1 [29]. Inflammatory cytokine TNFα has been identified as a key effector of pro-inflammatory signaling in valve endothelial cells and of pro-calcific signaling in valve interstitial cells [30–32]. TNFα can also initiate a pro-disease endothelial to mesenchymal transition in adult VEC [33–34]. However, it is not known if TNFα causes VEC to actively contribute pro-calcific oxidative stress to the diseased valve environment.
In this study, we establish that inflammatory cytokine TNFα drives increased oxidative stress in the valve endothelium, identifying valve endothelial cells as a novel source of elevated oxidative stress in AVD. We show that TNFα-driven endothelial oxidative stress, in part from eNOS uncoupling, causes endothelial dysfunction and exacerbates endothelial inflammatory response and that these effects can be prevented by treatment with TNFα+tetrahydrobiopterin (BH4) or TNFα+peg-SOD. Results from our ex vivo AV leaflet culture show that blocking endothelial oxidative stress also mitigates TNFα-driven myofibroblastic activation, extracellular matrix remodeling, and calcification in the valve tissue. These findings identify a novel pathophysiological mechanism by which TNFα-driven endothelial oxidative stress initiates downstream calcification of the aortic valve. Antioxidant therapies that protect against endothelial-derived increases in oxidative stress can mitigate valve disease progression by preserving the protective capabilities of the endothelium.
Methods
Human aortic valves
Calcified human aortic valves were obtained from adults undergoing planned, non-elective valve replacement surgery at Robert Packer Hospital in Sayre, PA. The Guthrie Institutional Review Board at Robert Packer Hospital and the Institutional Review Board for Human Participants at Cornell University approved all procedures (IRB#0908–24, “Gene expression and phenotypic changes in stenotic aortic valves”). Written informed consent was obtained from all participants. The investigation conformed to the principles outlined in the Declaration of Helsinki. Patient age range was 65–90 years, with a mean age of 76.2 years, 21 samples total. Dihydroethidium (DHE) superoxide detection and immunofluorescence were performed as described in the S1 Supplement.
Aortic valve endothelial cells
Valve endothelial cells (VEC) were harvested from porcine aortic valves (Shirk Meats, Dundee, NY). Use of porcine cells, a widely-accepted human valve analog, provided a large, controlled population of VEC that were screened for phenotype and purity using q-rtPCR and immunofluorescence for CD31, eNOS, VE-cadherin, and absence of αSMA (see S1 Supplement). VEC were found to maintain their endothelial phenotype and purity out to passage six, thus all experiments were conducted between P4 and P6. In all in vitro experiments, VEC were cultured at 100,000 cells/cm2 on 3-D collagen hydrogels recapitulating the native valve environment, as described previously [35].
Endothelial oxidative stress state assessment
Human recombinant TNFα was administered at dosages from 10–100ng/mL and determined to have a significant inflammatory effect (VCAM-1 upregulation, NFκB nuclear translocation) with no statistically significant VEC apoptosis at 30ng/mL, as previously shown [33]. H2O2 dosage (1 μmol/L) was chosen to recapitulate peak increases in secreted H2O2 by VEC stimulated with 30ng/mL TNFα. Acute increases in oxidative stress due to TNFα were measured across 0–4 hours. Peak effect occurred and is presented here at 30 minutes post-treatment. Intracellular oxidative stress was assessed via DHE fluorescence and CM-H2DCF-DA fluorescence, quantified on a microplate reader and with confocal microscopy. Secreted NO was measured using the Griess assay. Secreted H2O2 was measured by the Fluoro H2O2 assay. Mitochondrial reactive oxygen species were measured using MitoSOX Red assay, quantified on a microplate reader and with confocal microscopy. Efficacy of antioxidant administration was optimized for pre-treatment time and dosage. Peak effect was achieved by administering 500μmol/L L-NAME 30 minutes prior to TNFα treatment and all other co-treatments concurrently with TNFα, using 10 μmol/L BH4, 100 μmol/L apocynin (acetovanillone), and 20U/mL Cu-Zn-Superoxide dismutase-polyethylene glycol (peg-SOD).
Ex vivo aortic valve leaflets
Porcine aortic valve leaflets were harvested sterilely from fresh porcine hearts (Shirk Meats, Dundee, NY). Leaflets were immediately rinsed in sterile PBS, transported on ice, and transferred to Dulbecco’s Modified Eagle Medium within two hours. D-MEM was supplemented with 10% fetal bovine serum and 1% penicillin-streptomycin, adjusted to a pH of 7.2. 30ng/mL of TNFα was added directly to the media, supplemented with 10 μmol/L BH4 or 20U/mL peg-SOD, where noted. This culture media was changed every 48 hours for 21 days. TUNEL assay for apoptosis was performed to confirm cell viability and DHE or MitoSOX Red assay were used to detect oxidative stress. Russell-Movat pentachrome staining was used to visualize extracellular matrix components and Alizarin Red S in combination with von Kossa staining was used to assess calcification. Western blot, mRNA isolation, q-rtPCR, and immunofluorescence were performed according to standard practices, as described in the S1 Supplement.
Statistical Analysis
Data is expressed as mean +/- standard error of the mean (SEM). All comparisons between two groups were made using two-tailed, unpaired t-tests assuming unequal variance. Comparisons between multiple groups were made using one-way ANOVA with Tukey’s post hoc tests. Differences between means were considered significant when p < 0.05.
Results
Calcified human aortic valves show elevated endothelial superoxide in a side specific manner, co-localized with inflammatory activation
We first examined superoxide levels in calcified (cHAV) human aortic valves, using DHE intensity. Superoxide levels were significantly increased in calcified regions of the valves (Fig 1A) and in the fibrosa endothelium, but not in non-calcified regions or the ventricularis endothelium (Fig 1A and 1B). Elevated endothelial superoxide was associated with the fibrosa side of the stenotic human aortic valve, where calcific lesions are known to form. Across cHAV samples, regions of the endothelium with high levels of superoxide were positive for both VCAM-1 and CD31 (Fig 1C), indicating a link between inflammatory endothelial activation and elevated endothelial superoxide. The interstitial cells directly beneath the human VEC (hVEC) did not have significantly increased superoxide (arrows). hVEC on the ventricularis side were positive for CD31, but did not have significantly increased superoxide or elevated VCAM-1 expression (Fig 1C).
Fig 1. Superoxide is elevated in the endothelium of calcified human aortic valves.

A, Superoxide staining (DHE) of calcified human aortic valve leaflets. Asterisks indicate calcific nodule. Intensity of superoxide staining is colorimetrically scaled, with yellow indicating most intense (inset triangle). Colored boxes indicate region magnified in lower panels; f indicates fibrosa, v indicates ventricularis. Scale bar = 1 mm. B, Quantification of DHE intensity (superoxide) across all valve samples, presented as DHE intensity in different regions of calcified valves (cHAV), n = 21. * indicates p < 0.05 between indicated groups. C, Co-localization of elevated superoxide with VCAM-1 and CD31 expression in calcified human aortic valve leaflet endothelium. Representative images from n = 21 valves. Scale bar = 20μm.
Side-specific expression of endothelial superoxide dismutase in calcified human aortic valves
The fibrosa endothelium of diseased valves showed little to no SOD1 expression, while the ventricularis endothelium retained strong expression of SOD1 (Fig 2A). Quantitative analysis of SOD1 expression in the CD31+ endothelial cells of cHAV confirmed that the fibrosa endothelium showed significantly less SOD1 than the ventricularis endothelium (Fig 2B).
Fig 2. Calcified human aortic valves have lower expression of SOD on the fibrosa endothelium than on the ventricularis.
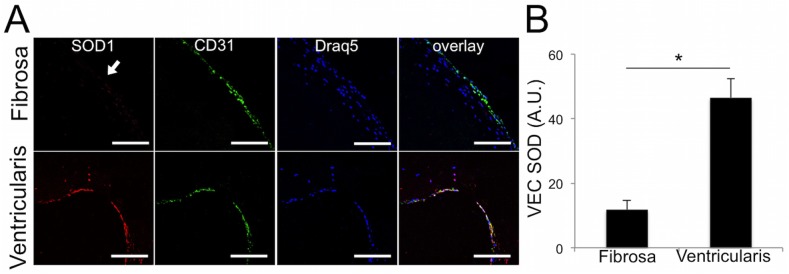
A. Immunofluorescence for endothelial protein CD31 and SOD1 revealed little to no expression of SOD1 (arrow) on the fibrosa endothelium of calcified aortic valve leaflets. The ventricularis showed strong SOD1 expression in the endothelium. B, Quantification of the pixel intensity of SOD1 in the CD31+ endothelial cells in cHAV, n = 21. The fibrosa endothelium had significantly less SOD1 than the ventricularis in cHAV. * indicates p < 0.05 between groups, Student’s t-test.
TNFα causes increased oxidative stress and decreased NO in VEC
TNFα caused a significant increase in VEC intracellular oxidative stress (Fig 3A), superoxide (S1 Fig), and secreted H2O2 (Fig 3B), 30 minutes after treatment. After 48 hours, TNFα or H2O2 treatment decreased VEC secretion of nitric oxide (Fig 3C) and decreased expression of eNOS and VE-cadherin proteins (Fig 3D and S2 Fig).
Fig 3. TNFα drives increased oxidative stress in aortic valve endothelial cells via eNOS uncoupling.

A, TNFα increases oxidative stress in VEC at 30 minutes. B, TNFα increases hydrogen peroxide (H2O2) secretion from VEC at 30 minutes. C, TNFα or H2O2 decrease nitric oxide secretion from VEC at 48 hours (n = 4). D, TNFα or H2O2 decrease eNOS and VE-cadherin expression in VEC at 48 hours. Representative western blot images (inset) and blot quantification. E, L-NAME, BH4, or peg-SOD but not apocynin block increases in superoxide (DHE) in VEC caused by TNFα, at 30 minutes. F, L-NAME, apocynin, and peg-SOD mitigate increases in general oxidative stress (DCF) caused by TNFα at 30 minutes, but only BH4 completely blocks superoxide increase, maintaining control levels. G, L-NAME, BH4, or peg-SOD but not apocynin block increases in H2O2 secreted by VEC at 30 minutes caused by TNFα at 30 minutes. H, TNFα drives increased mtROS, mitigated only by co-treatment with SOD. I, BH4, or peg-SOD but not L-NAME or apocynin block decreases in nitric oxide secretion in VEC caused by TNFα at 48 hours. * indicates p < 0.05 versus control. # indicates p < 0.05 versus TNFα. ^ indicates p < 0.05 versus apocynin. N = 4.
To elucidate the source of increases in endothelial oxidative stress and the resulting endothelial dysfunction, we administered NOS inhibitor L-NAME, eNOS co-factor BH4, NADPH oxidases inhibitor apocynin, or peg-SOD in conjunction with TNFα treatment. At 30 minutes, L-NAME, BH4, and peg-SOD blocked TNFα-stimulated increases in VEC superoxide, but apocynin did not (Fig 3E). L-NAME and apocynin reduced overall oxidative stress compared to TNFα alone, and peg-SOD lowered oxidative stress further still. Only eNOS co-factor BH4 restored VEC oxidative stress back to control levels (Fig 3F). Similar trends were observed in H2O2 secretion: co-administration of L-NAME, BH4, or peg-SOD blocked increases in secreted H2O2 by VEC, but apocynin did not (Fig 3G). TNFα similarly drove increases in VEC mitochondrial reactive oxygen species (mtROS), which were only abrogated by peg-SOD (Fig 3H). Over 48 hours, only BH4 or peg-SOD maintained control levels of nitric oxide secretion (Fig 3I). Catalase co-treatment was able to reduce general oxidative stress state in VEC, but was found to cause a significant increase in apoptosis, loss of VE-cadherin, nuclear translocation of NFκB, and VCAM-1 expression (S3 Fig). Thus, catalase treatment had off-target effects on VEC that were significant enough to prevent further investigation of its viability as a pro-endothelial valve treatment against inflammation and calcification.
Mitigating the effects of TNFα-oxidative stress rescues endothelial function and abates VEC inflammatory response
We then examined the protein-level effects of BH4 and peg-SOD on VEC when applied in conjunction with TNFα treatment. TNFα decreased VE-cadherin and eNOS and increased VCAM-1 protein expression in VEC (Fig 4A, arrows). Co-treatment of VEC with BH4 or peg-SOD protected against loss of VE-cadherin and eNOS and reduced VCAM-1 response to TNFα. Western blot analysis confirmed this result (Fig 4B and 4C and S4 Fig).
Fig 4. Blocking eNOS uncoupling mitigates TNFα-induced endothelial dysfunction and inflammatory response.

A, TNFα decreases VE-cadherin and eNOS expression in VEC over 48 hours (arrows), rescued by BH4 or peg-SOD. TNFα increases VCAM-1 expression in VEC over 48 hours, mitigated by BH4 or peg-SOD. B, Western blot analysis of VE-cadherin (VE-cad), eNOS, and VCAM-1 protein in same conditions as A, with GAPDH housekeeping protein. C, Quantification of western blot results, normalized to GAPDH and control, n = 3. * indicates p < 0.05 versus control, # indicates p < 0.05 versus TNFα. Scale bar = 20μm. Images representative of three independent experiments.
TNFα increases VEC oxidative stress in ex vivo aortic valve leaflets, rescued by antioxidants
We then used ex vivo culture of healthy porcine AV leaflets to examine tissue-level effects of TNFα-driven endothelial oxidative stress and the ability of BH4 or peg-SOD to mitigate TNFα effects. No significant apoptosis was observed in the leaflets throughout the course of the experiment (S5 Fig). Similar to our findings in vitro, over 21 days TNFα increased endothelial superoxide, most distinctly in the fibrosa endothelium (Fig 5A). BH4 treatment mitigated fibrosa but not ventricularis-side increases in superoxide. Peg-SOD lowered endothelial superoxide even further than BH4, significantly decreasing levels on both the fibrosa and ventricularis sides (Fig 5B). To elucidate a potential mechanism behind differential BH4 and peg-SOD effects, we examined the contribution of mitochondrial reactive oxygen species (mtROS) within our ex vivo leaflets. We found that TNFα indeed increased levels of mtROS, specifically within the ventricularis endothelium (Fig 5C). Quantification of mtROS fluorescence revealed a 42±9.3% increase in mtROS only in ventricularis VEC and not in fibrosa VEC or VIC, which was mitigated by co-treatment with SOD, but not BH4 (Fig 5D).
Fig 5. TNFα induces side-specific endothelial oxidative stress in ex vivo aortic valve leaflets.

A, TNFα causes increased superoxide in the fibrosa endothelium of ex vivo porcine aortic valve leaflets cultured for 21 days, revealed by DHE staining. Superoxide is mitigated on the fibrosa by co-treatment with BH4 and on both the fibrosa and ventricularis by peg-SOD. Pixel intensity of superoxide levels is scaled colorimetrically (inset triangle) to show regions of highest superoxide. Colored boxes indicate regions magnified on right, showing endothelium on fibrosa and ventricularis sides of valve. Scale bar is 1mm in left images, 20μm in magnified images. B, Quantification of endothelial superoxide in ex vivo aortic valves stained with DHE reveals side-specific rescue-effects of BH4 and more pronounced mitigation of superoxide on both sides of the valve by peg-SOD. C, TNFα increases mtROS in the ventricularis VEC of ex vivo AV leaflets. D, Quantification of mtROS fluorescenc. Ventricularis-specific increases in mtROS are mitigated by peg-SOD but not BH4. * indicates p < 0.05 versus control condition. # indicates p < 0.05 versus fibrosa endothelium in same condition. & indicates p < 0.05 versus same side endothelium in TNFα condition. ^ indicates p < 0.05 versus same side endothelium in TNFα+BH4. N = 6.
Antioxidants improve endothelial function and repress valve interstitial cell myofibroblastic activation in aortic valve leaflets
AV leaflets cultured in TNFα for 21 days showed significant endothelial dysfunction, with decreases in CD31, VE-cadherin, and eNOS on both the fibrosa and the ventricularis. These effects were rescued by co-treatment with BH4 or peg-SOD. TNFα also caused upregulation of VCAM-1 on both sides of the valve, which was mitigated almost entirely on both the fibrosa and ventricularis by co-treatment with BH4 or peg-SOD. TNFα significantly increased αSMA in both the endothelium and the underlying interstitium on both sides of the valve, decreased on the fibrosa by peg-SOD and on the ventricularis by BH4 or peg-SOD (Fig 6A). Loss of VE-cadherin and eNOS and increases in αSMA due to TNFα and the protective effect of antioxidants were confirmed via western blot analysis (Fig 6B and S6 Fig).
Fig 6. TNFα induces endothelial dysfunction, increased inflammatory activation, and myofibroblastic activation ex vivo, rescued by antioxidants.

A, In ex vivo porcine aortic valve leaflets, TNFα decreased endothelial proteins CD31, VE-cadherin, and eNOS on both sides of the valve, rescued by BH4 or peg-SOD. TNFα increased VCAM1 on both sides of the valve, mitigated by BH4 or peg-SOD. TNFα increased αSMA on both sides of the valve, which was mitigated on the fibrosa by peg-SOD but not by BH4 (arrow) and on the ventricularis by either BH4 or SOD. B, Western blot quantification confirmed loss of VE-cadherin and eNOS and increase in αSMA in TNFα condition that is rescued by BH4 or peg-SOD. * indicates p < 0.05 versus CTL, # indicates p < 0.05 versus TNFα. Scale bar = 20μm. N = 6.
TNFα causes extracellular matrix disorganization and calcification in aortic valve leaflets, rescued by antioxidants
Ex vivo aortic valve leaflets treated with TNFα for 21 days showed significant fibrogenesis and osteogenesis compared to controls. Control AV leaflets showed distinct trilaminar structure of the extracellular matrix (ECM), with well-defined organization of elastin fibers within the ventricularis (arrow, Fig 7A). Valve leaflets treated with TNFα showed ECM disorganization, marked by significantly decreased elastin and glycosaminoglycans (GAG), and significantly increased collagen. q-rtPCR analysis of the valve tissue showed increased synthesis of COL1A1 and COL3A1 mRNA with TNFα (Fig 7B). BH4 protected against loss of elastin and GAG, but allowed similar increases in collagen as in TNFα alone. Peg-SOD protected against loss of elastin and GAG and provided significant protection against increases in collagen, maintaining collagen expression at control levels. Peg-SOD blocked increases in COL1A1 and COL3A1 mRNA. COL2A1 was decreased in all treatment conditions.
Fig 7. TNFα induces early calcification and changes in aortic valve extracellular matrix composition and structure, rescued differentially by BH4 or peg-SOD.

A, TNFα increased collagen and decreased GAG and elastin (arrows). BH4 maintains GAG and elastin at control levels, but does not block increases in collagen. Peg-SOD significantly reduced collagen compared to TNFα and maintained control levels of GAG and elastin (Blue: glycosaminoglycans, Red: mucins, Yellow: collagen, Black, elastin). B. TNFα increased synthesis of COL1A1 and COL3A1 mRNA, rescued by peg-SOD but not by BH4. COL2A1 was decreased in all treatment conditions. * indicates p < 0.05 versus control, # indicates p < 0.05 versus TNFα. N = 6. One-way ANOVA with Tukey’s post hoc test were used to compare means. C, TNFα increased calcium deposition (Alizarin Red S stain) that is rescued by BH4 or peg-SOD. D, TNFα increased mineralization (von Kossa stain), rescued by BH4 or peg-SOD. E, TNFα increased alkaline phosphatase, decreased Sox9, and increased MMP-9 mRNA, rescued differentially by BH4 or peg-SOD. Representative images from n = 6 leaflets per condition. Scale bar = 200μm. * indicates p < 0.05 versus control, # indicates p < 0.05 versus TNFα, ^ indicates p < 0.05 versus TNFα+BH4. N = 6.
Alizarin red staining showed significant increase in free calcium in leaflets cultured with TNFα, localized to the fibrosa side of the leaflets (Fig 7C, arrows) that was mitigated by BH4 and returned to control levels by peg-SOD. von Kossa staining showed an increase in mineralization, again on the fibrosa, in leaflets cultured with TNFα that was equally blocked by BH4 or peg-SOD (Fig 7D). BH4 caused minimal off-target effects on a range of markers regulating AV function (S7 Fig).
TNFα increased synthesis of mRNA for alkaline phosphatase, an enzyme involved in active bone formation, which was mitigated by co-treatment with BH4 or peg-SOD. TNFα decreased synthesis of mRNA for the anti-osteogenic transcription factor Sox9, indicating a pro-calcific phenotypic shift that was rescued by co-treatment with BH4. TNFα also increased synthesis of mRNA for matrix metalloproteinase-9, mitigated by BH4 or peg-SOD (Fig 7E).
Discussion
Aortic valve disease is a pressing medical issue with few clinically useful pharmacological intervention strategies. New therapies are needed that target specific cellular mechanisms that will guard against valve degeneration. In this study, we have shown the valve endothelium to be a novel source of oxidative stress in the aortic valve when stimulated with the inflammatory cytokine TNFα (Fig 8). We further showed that targeting this mechanism via antioxidants directed at superoxide levels protects against loss of endothelial function, chronic inflammation response, myofibroblastic activation, calcification, and extracellular matrix disorganization.
Fig 8. Valve endothelial oxidative stress contributes directly to aortic valve disease.

In a healthy valve, endothelial cells secrete protective NO, maintaining quiescence of valve interstitial cells. Early AVD is characterized by inflammation-driven side specific increase in VEC oxidative stress, in part via eNOS uncoupling, causing disorganization of the extracellular matrix and loss of protective NO secretion in the fibrosa. In late AVD, oxidative stress from VEC contributes to calcification and mineral deposition, while NO and SOD are decreased in fibrosa VEC.
We showed that regions of the calcified valve endothelium with high VCAM-1 co-localized with regions of high superoxide, linking inflammation with oxidative stress in the diseased valve endothelium. This phenomenon occurs in both mildly and heavily affected regions of the valve, specifically on the fibrosa side of the leaflet. This pervasive and side-specific co-localization of inflammatory activation and oxidative stress in the endothelium suggests that TNFα-driven endothelial oxidative stress occurs throughout mild and advanced stages of AVD. Side-specificity of endothelial oxidative stress points to a distinct involvement of this endothelial-specific mechanism with calcific lesion formation, known to develop preferentially on the fibrosa [36]. Our observation of increased superoxide levels in hVEC but not the adjacent hVIC supports a role for valve endothelial cells as a unique source of oxidative stress in the diseased valve, distinct from oxidative stress previously described in the calcifying regions of the valve [23–24] or as a result of immune cell invasion. We also observed fibrosa-specific loss of SOD1 expression in cHAV, indicating a dearth of SOD1 in the diseased fibrosa endothelium during CAVD, making the valve more susceptible to increases in oxidative stress from the endothelium. Low fibrosa SOD1 compared to the ventricularis in cHAV can also explain the preferential accumulation of superoxide that we observed in the fibrosa endothelium of calcified valves.
Recently, TNFα signaling has been implicated as a key inflammatory pathway in stenotic aortic valves [37–39]. Mouse models of hypercholesterolemia show inflammatory activation of the endothelium, increased superoxide [40], and endothelial dysfunction [41]. Here, we have demonstrated that TNFα increases endothelial oxidative stress, decreases NO secretion, and lowers VE-cadherin and eNOS protein expression in valve endothelial cells, both in vitro and ex vivo. This finding provides a mechanistic link between inflammation, endothelial dysfunction, and oxidative stress. TNFα-derived oxidative stress effects were blocked by co-treatment with eNOS co-factor BH4, demonstrating that eNOS uncoupling is a primary mechanism of increased endothelial oxidative stress and a significant cause of endothelial dysfunction that leads to increased AV calcification via degradation of endothelial protection.
Co-treatment of VEC with TNFα+catalase did not protect the endothelium, but led to increased cellular apoptosis and elevated inflammatory activation. This agrees with previous studies that have shown that in certain cell types, H2O2 actually protects against TNFα –induced apoptosis and that increased levels of catalase has a neutral or negative effect [42–44]. The ability of apocynin to mitigate TNFα-driven endothelial oxidative stress levels as indicated by DCF staining shows that NADPH oxidases also have a role to play in this phenomenon, though it appears to be less important than the uncoupling mechanism, as demonstrated by the more potent effects of BH4. These findings define two unique mechanisms of endothelial oxidative stress contribution to AVD: via changes in eNOS activity due to uncoupling and disruption of native SOD1 and H2O2 levels. TNFα driven oxidative stress, in part due to eNOS uncoupling, degrades the endothelial protection that would normally mitigate valve degeneration [15, 17] and increases oxidative stress in the valve. This establishes a novel mechanism of valve oxidative stress based on endothelial inflammatory activation, distinct from existing descriptions of oxidative stress that have focused on valve interstitial cells [25, 45–47].
We further established the ability of BH4 and peg-SOD to mitigate increases in VCAM-1 that occurred in VEC in response to TNFα. This dovetails with recent studies that implicate side-specific VCAM-1 expression in the progression and regulation of AVD [48–49]. Thus, mitigating the cellular stress imposed on VEC by TNFα-driven eNOS uncoupling may also decrease the need to activate downstream inflammatory signaling pathways. Mitigation of the symptoms of chronic inflammation could prove important to aortic valve calcification, as the TNFα signaling pathway is known to promote osteogenesis [50] and to be an active regulator of lesion formation in the vasculature [51].
Translation of our cellular-level findings into an ex vivo evaluation of AV leaflets allowed us to assess the specific effects of TNFα and antioxidants on the complex, multicellular aortic valve leaflet. We found that superoxide levels in TNFα-treated valve leaflets was specifically elevated in VEC, with highest elevation on the fibrosa side, confirming that endothelial superoxide in cHAV is linked to inflammation of the valve endothelium. Interestingly, co-treatment of ex vivo AV leaflets with TNFα+BH4 reduced oxidative stress only in the fibrosa endothelium compared to TNFα alone, whereas peg-SOD reduced oxidative stress on both sides of the valve leaflet. Examination of mitochondrial ROS provided an explanation for this difference, as peg-SOD but not BH4 lowered mtROS levels within the ventricularis of ex vivo leaflets. We conclude that BH4 treatment mitigates valve oxidative stress via direct targeting of fibrosa-specific eNOS uncoupling, whereas peg-SOD mitigates both fibrosa-specific eNOS uncoupling and mtROS within ventricularis VEC. A limitation of this study is the lack of direct experimental interrogation of mtROS as a propagator of AVD; however, the evidence presented here provides a foundation for future investigation of mitochondrial contribution to increased oxidative stress in the aortic valve in the early inflammatory stages of AVD.
Immunofluorescent staining confirmed the side-specific effect of BH4. CD31, VE-cadherin, and eNOS were more robustly restored on the fibrosa with co-treatment of TNFα+BH4, compared to TNFα alone. TNFα caused increased αSMA in the fibrosa, potentially due to mesenchymal transition of valve endothelial cells [34], in addition to direct myofibroblastic activation of valvular interstitial cells [31]. TNFα-induced increases in αSMA were blocked by co-treatment of valve leaflets with TNFα+BH4 or TNFα+peg-SOD, demonstrating a role for both eNOS uncoupling and superoxide from other sources such as NADPH oxidases and mtROS in driving myofibroblastic activation during AVD.
TNFα exposure caused significant disruption of the extracellular matrix organization and content of the aortic valve. TNFα increased collagen and decreased GAG and elastin, agreeing with studies that find differential GAG localization [52] and reduced elastin [53] in diseased aortic valves. GAG and elastin were preserved by BH4 or peg-SOD. Increases in collagen were only mitigated by peg-SOD, not by BH4. mRNA analysis showed that increased collagen was caused by increased expression of COL1A1 and COL3A1 isoforms in TNFα and TNFα+BH4 conditions, but rescued in TNFα+peg-SOD condition. Interestingly, COL2A1 was uniformly decreased across TNFα, TNFα+BH4, and TNFα+peg-SOD conditions, pointing to TNFα as a driver of early chondrocyte differentiation in valve cells and as an important contributor to valve calcification via a mechanism separate from the oxidative stress pathways examined here [54]. These findings indicate that protection against excess valve endothelial oxidative stress can guard against ECM changes known to feature in AVD [55] via antioxidant mitigation of inflammation, endothelial dysfunction, and oxidative stress.
TNFα accelerates calcification of aortic valve interstitial cells in vitro [32–33]. However, the contribution of TNFα to AVD within the complex multi-cellular environment of the native aortic valve is unknown. Here we used TNFα with and without BH4 treatment to show that stimulation of endothelial eNOS uncoupling by TNFα contributes to valve calcification, elucidating a novel mechanism of TNFα contribution to AVD. Increases in alkaline phosphatase point to the mechanism behind these changes, indicating that TNFα stimulates early osteogenic differentiation of valve interstitial cells, as has been observed in vitro under TGF-β stimulation [56], that can be protected against by preservation of endothelial function and reductions in oxidative stress.
Antioxidant therapy for AVD must be founded on specific mechanisms generating oxidative stress in order to be efficacious [57]. The evidence presented here establishes inflammatory activation of endothelial oxidative stress as a unique integrated pathobiological mechanism effecting early to late stages of aortic valve disease. Our findings support that specific targeting of eNOS uncoupling and the resulting excess superoxide may be key elements of managing AVD via antioxidant treatments.
Supporting Information
A, Intracellular superoxide in VEC+TNFα at 30 minutes after treatment. Presented using colorimetric scale to show relative DHE intensity, indicated in inset triangle. B, Quantification of superoxide production at 30 minutes using microplate assay for DHE fluorescence. * indicates p < 0.05 versus control. N > 6 for each condition. DHE fluorescence was measured using integrated pixel density in three different fields of view (250μm2) on each sample. The three measurements were averaged within each sample, with each average corresponding to N of 1. Means were compared using unpaired Student’s t-test, assuming unequal variance.
(TIFF)
PAVEC cultured 48 hrs on 3D hydrogels with control, +30 ng/mL TNFα, or 1μM H2O2. Boxed regions indicate bands shown in Fig 3.
(TIFF)
A. TNFα+catalase treatment caused a significant increase in apoptosis. B. TNFα+catalase treatment caused increased loss of VE-cadherin, increased nuclear translocation of NFkB, and increased VCAM-1 expression compared to both control and TNFα alone. N = 3, * indicates p < 0.05 vs CTL, # indicates p < 0.05 vs TNFα. Scale bar is 20μm.
(TIFF)
PAVEC cultured 48 hrs on 3D hydrogels with control, +30 ng/mL TNFα, +30 ng/mL TNFα+10μM BH4, or +30 ng/mL TNFα+20U/mL peg-SOD. Boxes indicate bands shown in Fig 4. *Analysis of NFκB p65 protein expression was not used in this study.
(TIFF)
There was no significant apoptosis in any of AV leaflets, as measured by the TUNEL assay. N = 6 for all sample groups. Means were compared using one-way ANOVA with Tukey’s post hoc test.
(TIFF)
Porcine AV leaflets cultured 21 days in control, +30 ng/mL TNFα, +30 ng/mL TNFα+10μM BH4, or +30 ng/mL TNFα+20U/mL peg-SOD. Boxes indicate bands shown in Fig 6. *Analysis of NFκB p65 protein expression was not used in this study.
(TIFF)
A. Complex regulation of transcription factors Runx2 and Msx2 by TNFα and BH4 or SOD. N = 6, * indicates p < 0.05 vs CTL, # indicates p < 0.05 vs TNFα. B. BH4 co-treatment consistently causes either neutral or beneficial effects compared to TNFα alone. N = 6, * indicates p < 0.05 vs TNFα.
(TIFF)
(DOCX)
Acknowledgments
The authors thank Dr. Sanjay Samy, Robert Packer Hospital, Sayre, PA, for generous donation of calcified aortic valve leaflets, Shirk Meats in Dundee, NY for donation of porcine aortic valve tissue, Dr. Jonathan Chen, Seattle Children’s Hospital, for generous donation of non-diseased aortic valve leaflets, Kevin S. Hsu for assistance with data acquisition and analysis, and Jennifer M. Richards for assistance in the procurement and processing of diseased aortic valve tissue.
Data Availability
All relevant data are within the paper and its Supporting Information files.
Funding Statement
This work was supported by the National Science Foundation (nsf.gov, CBET-0955172 and MRSEC DMR-1120296 to JTB), and by the National Institutes of Health (nih.gov, HL110328 and HL118672 to JTB). EJF was supported by a National Science Foundation graduate research fellowship. GDH was supported by the National Science Foundation Research Experience for Undergraduates Site program (DMR-1063059). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Go AS, Mozaffarian D, Roger VL, Benjamin EJ, Berry JD, Borden WB, et al. Heart disease and stroke statistics—2013 update: A report from the American Heart Association. Circulation. 2013;127: e6–e245. 10.1161/CIR.0b013e31828124ad [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Rajamannan NM, Evans FJ, Aikawa E, Grande-Allen KJ, Demer LL, Heistad DD, et al. Calcific aortic valve disease: Not simply a degenerative process. Circulation. 2011;124: 1783–91. 10.1161/CIRCULATIONAHA.110.006767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Teo KK, Corsi DJ, Tam JW, Dumesnil JG, Chan KL. Lipid lowering on progression of mild to moderate aortic stenosis: meta-analysis of the randomized placebo-controlled clinical trials on 2344 patients. Can J Cardiol. 2011;27: 800–8. 10.1016/j.cjca.2011.03.012 [DOI] [PubMed] [Google Scholar]
- 4. Shavelle DM, Takasu J, Budoff MJ, Mao S, Zhao X-Q, O'Brien KD. HMG CoA reductase inhibitor (statin) and aortic valve calcium. Lancet. 2002;359: 1125–6. [DOI] [PubMed] [Google Scholar]
- 5. Rossebø AB, Pedersen TR, Boman K, Brudi P, Chambers JB, Gerdts E, et al. Intensive lipid lowering with Simvastatin and Ezetimibe in aortic stenosis. New Engl J Med. 2008;359: 1343–56. 10.1056/NEJMoa0804602 [DOI] [PubMed] [Google Scholar]
- 6. Butcher JT, Penrod AM, Garcia AJ, Nerem RM. Unique morphology and focal adhesion development of valvular endothelial cells in static and fluid flow environments. Arterioscler Thromb Vasc Biol. 2004;24: 1429–34. [DOI] [PubMed] [Google Scholar]
- 7. Simmons CA, Grant GR, Manduchi E, Davies PF. Spatial heterogeneity of endothelial phenotypes correlates with side-specific vulnerability to calcification in normal porcine aortic valves. Circ Res. 2005;96: 792–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Bischoff J, Aikawa E. Progenitor cells confer plasticity to cardiac valve endothelium. J Cardiovasc Trans Res. 2011;4: 710–9. 10.1007/s12265-011-9312-0 [DOI] [PubMed] [Google Scholar]
- 9. Farivar RS, Cohn LH, Soltesz EG, Mihaljevic T, Rawn JD, Byrne JD. Transcriptional profiling and growth kinetics of endothelium reveals differences between cells derived from porcine aorta versus aortic valve. Eur J Cardiothorac Surg. 2003;24: 527–34. [DOI] [PubMed] [Google Scholar]
- 10. Butcher JT, Nerem RM. Valvular endothelial cells and the mechanoregulation of valvular pathology. Philos Trans R Soc Lond B. 2007;362: 1445–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Pompilio G, Rossoni G, Sala A, Polvani GL, Berti F, Dainese L, et al. Endothelial-dependent dynamic and antithrombotic properties of porcine aortic and pulmonary valves. Ann Thorac Surg. 1998;65: 986–92. [DOI] [PubMed] [Google Scholar]
- 12. Müller AM, Cronen C, Kupferwasser LI, Oelert H, Müller K-M, Kirkpatrick CJ. Expression of endothelial cell adhesion molecules on heart valves: up‐regulation in degeneration as well as acute endocarditis. J Pathol. 2000;191: 54–60. [DOI] [PubMed] [Google Scholar]
- 13. Poggianti E, Venneri L, Chubuchny V, Jambrik Z, Baroncini LA, Picano E. Aortic valve sclerosis is associated with systemic endothelial dysfunction. J Am Coll Cardiol. 2003;41: 136–41. [DOI] [PubMed] [Google Scholar]
- 14. Gould ST, Srigunapalan S, Simmons CA, Anseth KS. Hemodynamic and cellular response feedback in calcific aortic valve disease. Circ Res. 2013;113:186–97. 10.1161/CIRCRESAHA.112.300154 [DOI] [PubMed] [Google Scholar]
- 15. Richards J, El-Hamamsy I, Chen S, Sarang Z, Sarathchandra P, Yacoub MH, et al. Side-specific endothelial-dependent regulation of aortic valve calcification. Am J Pathol. 2013;182: 1922–31. 10.1016/j.ajpath.2013.01.037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Rajamannan NM, Subramaniam M, Stock SR, Stone NJ, Springett M, Ignatiev KI, et al. Atorvastatin inhibits calcification and enhances nitric oxide synthase production in the hypercholesterolaemic aortic valve. Heart. 2005;91: 806–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. El-Hamamsy I, Balachandran K, Yacoub MH, Stevens LM, Sarathchandra P, Taylor PM, et al. Endothelium-dependent regulation of the mechanical properties of aortic valve cusps. J Am Coll Cardiol. 2009;53: 1448–55. 10.1016/j.jacc.2008.11.056 [DOI] [PubMed] [Google Scholar]
- 18. Bosse K, Hans CP, Zhao N, Koenig SN, Huang N, Guggilam A, et al. Endothelial nitric oxide signaling regulates Notch1 in aortic valve disease. J Mol Cell Cardiol. 2013;60: 27–35. 10.1016/j.yjmcc.2013.04.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ciprian AB, Cheng B, Sharma S, Sharma S, Yao L, Caldwell RW, et al. Increased Superoxide and eNOS uncoupling in blood vessels of Bmal1-KO mice. Circ Res. 2012;111: 1157–65. 10.1161/CIRCRESAHA.111.261750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Wever RMF, van Dam T, van Rijn HJM, de Groot F, Rabelink TJ. Tetrahydrobiopterin regulates superoxide and nitric oxide generation by recombinant endothelial nitric oxide synthase. Biochem Biophys Res Comm. 1997;237: 340–4. [DOI] [PubMed] [Google Scholar]
- 21. Kar S, Bhandar B, Kavdia M. Impact of SOD in eNOS uncoupling: a two-edged sword between hydrogen peroxide and peroxynitrite. Free Radic Res. 2012;46: 1496–513. 10.3109/10715762.2012.731052 [DOI] [PubMed] [Google Scholar]
- 22. Li J-M, Shah AM. Endothelial cell superoxide generation: regulation and relevance for cardiovascular pathophysiology. Am J Physiol Regul Integr Comp Physiol. 2004;287: R1014–30. [DOI] [PubMed] [Google Scholar]
- 23. Miller JD, Chu Y, Brooks RM, Richenbacher WE, Pena-Silva R, Heistad DD. Dysregulation of antioxidant mechanisms contributes to increased oxidative stress in calcific aortic valvular stenosis in humans. J Am Coll Cardiol. 2008;52: 843–50. 10.1016/j.jacc.2008.05.043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Liberman M, Bassi E, Martinatti MK, Lario FC, Wosniak J, Pomerantzeff PMA, et al. Oxidant generation predominates around calcifying foci and enhances progression of aortic valve calcification. Arterioscler Thromb Vasc Biol. 2008;28: 463–70. [DOI] [PubMed] [Google Scholar]
- 25. Branchetti E, Sainger R, Poggio P, Grau JB. Antioxidant enzymes reduce DNA damage and early activation of valvular interstitial cells in aortic valve sclerosis. Arterioscler Thromb Vasc Biol. 2013;33: e66–74. 10.1161/ATVBAHA.112.300177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Marchesi C, Ebrahimian T, Angulo O, Paradis P, Schiffrin EL. Endothelial nitric oxide synthase uncoupling and perivascular adipose oxidative stress and inflammation contribute to vascular dysfunction in a rodent model of metabolic syndrome. Hypertension. 2009;54: 1384–92. 10.1161/HYPERTENSIONAHA.109.138305 [DOI] [PubMed] [Google Scholar]
- 27. Karbach S, Wenzel P, Waisman A, Münzel T, Daiber A. eNOS uncoupling in cardiovascular diseases—the role of oxidative stress and inflammation. Curr Pharm Des. 2014;20: 3579–94. [DOI] [PubMed] [Google Scholar]
- 28. New SEP, Aikawa E. Molecular imaging insights into early inflammatory stages of arterial and aortic valve calcification. Circ Res. 2010;108: 1381–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Hjortnaes J, Butcher J, Figueiredo JL, Riccio M, Kohler RH, Kozloff KM, et al. Arterial and aortic valve calcification inversely correlates with osteoporotic bone remodeling: a role for inflammation. Eur Heart J. 2010;31: 1975–84. 10.1093/eurheartj/ehq237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Aikawa E, Nahrendorf M, Sosnovik D, Lok VM, Jaffer FA, Aikawa M, et al. Multimodality molecular imaging identifies proteolytic and osteogenic activities in early aortic valve disease. Circulation. 2007;115: 377–86. [DOI] [PubMed] [Google Scholar]
- 31. Kaden JJ, Dempfle C-E, Grobholz R, Fischer CS, Vocke DC, Kiliç R, et al. Inflammatory regulation of extracellular matrix remodeling in calcific aortic valve stenosis. Cardiovasc Pathol. 2005;14: 80–7. [DOI] [PubMed] [Google Scholar]
- 32. Yu Z, Seya K, Daitoku K, Motomura S, Fukuda I, Furukawa KI. Tumor necrosis factor-α accelerates the calcification of human aortic valve interstitial cells obtained from patients with calcific aortic valve stenosis via the BMP2-Dlx5 pathway. J Pharmacol Exp Ther. 2011;337: 16–23. 10.1124/jpet.110.177915 [DOI] [PubMed] [Google Scholar]
- 33. Galeone A, Brunetti G, Oranger A, Greco G, Di Benedetto A, Mori G, et al. Aortic valvular interstitial cells apoptosis and calcification are mediated by TNF-related apoptosis-inducing ligand. Int J Cardiol. 2013;169: 296–304. 10.1016/j.ijcard.2013.09.012 [DOI] [PubMed] [Google Scholar]
- 34. Mahler GJ, Farrar EJ, Butcher JT. Inflammatory cytokines promote mesenchymal transformation in embryonic and adult valve endothelial cells. Arterioscler Thromb Vasc Biol. 2013;33: 121–30. 10.1161/ATVBAHA.112.300504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Farrar EJ, Butcher JT. Heterogeneous susceptibility of valve endothelial cells to mesenchymal transformation in response to TNFα. Ann Biomed Eng. 2013;42: 149–61. 10.1007/s10439-013-0894-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Chen JH, Simmons CA. Cell-matrix interactions in the pathobiology of calcific aortic valve disease: Critical roles for matricellular, matricrine, and matrix mechanics cues. Circ Res. 2011;108: 1510–24. 10.1161/CIRCRESAHA.110.234237 [DOI] [PubMed] [Google Scholar]
- 37.Mahler GJ, Butcher JT. Inflammatory regulation of valvular remodeling: The good(?), the bad, and the ugly. Int J Inflam. 2011;721419. [DOI] [PMC free article] [PubMed]
- 38. Ghaisas N, Foley J, O'Briain D, Crean P, Kelleher D, Walsh M. Adhesion molecules in nonrheumatic aortic valve disease: Endothelial expression, serum levels and effects of valve replacement. J Am Coll Cardiol. 2000;36:2257–62. [DOI] [PubMed] [Google Scholar]
- 39. Bosse Y, Miqdad A, Fournier D, Pepin A, Pibarot P, Mathieu P. Refining Molecular pathways leading to calcific aortic valve stenosis by studying gene expression profile of normal and calcified stenotic human aortic valves. Circ Cardiovasc Genet. 2009;2: 489–98. 10.1161/CIRCGENETICS.108.820795 [DOI] [PubMed] [Google Scholar]
- 40. Miller JD, Weiss RM, Serrano KM, Castaneda LE, Brooks RM, Zimmerman K, et al. Evidence for active regulation of pro-osteogenic signaling in advanced aortic valve disease. Arterioscler Thromb Vasc Biol. 2010;30: 2482–6. 10.1161/ATVBAHA.110.211029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Stapleton PA, Goodwill AG, James ME, Brock RW, Frisbee JC. Hypercholesterolemia and microvascular dysfunction: interventional strategies. J Inflam. 2010;7: 54 10.1186/1476-9255-7-54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Dasgupta J, Subbaram S, and Connor K M. Manganese superoxide dismutase protects from TNF-α-induced apoptosis by increasing the steady-state production of H2O2 . Antiox Redox Signaling. 2006;8: 1295–305. [DOI] [PubMed] [Google Scholar]
- 43. Mele J, Van Remmen H, Vijg J, Richardson A. Characterization of transgenic mice that overexpress both copper zinc superoxide dismutase and catalase. Antiox Redox Signaling. 2006;8: 628–38. [DOI] [PubMed] [Google Scholar]
- 44. Bai J, Cederbaum AI. Overexpression of catalase in the mitochondrial or cytosolic compartment increases sensitivity of HepG2 cells to tumor necrosis factor-α-Induced apoptosis. J Biol Chem. 2000;275: 19241–9. [DOI] [PubMed] [Google Scholar]
- 45. Rajamannan NM. Bicuspid aortic valve disease: the role of oxidative stress in Lrp5 bone formation. Cardiovasc Pathol. 2011;20: 168–76. 10.1016/j.carpath.2010.11.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Rajamannan N. Role of oxidative stress in calcific aortic valve disease: From bench to bedside—The role of a stem cell niche In: Morales-Gonzalez JA, ed. Oxidative Stress and Chronic Degenerative Diseases—A Role for Antioxidants. Rijeka, Croatia: InTech. 2013. pp. 265–87. [Google Scholar]
- 47. Heistad DD, Wakisaka Y, Miller J, Chu Y, Pena-Silva R. Novel aspects of oxidative stress in cardiovascular diseases. Circ J. 2009;73: 201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Sucosky P, Balachandran K, Elhammali A, Jo H, Yoganathan AP. Altered shear stress stimulates upregulation of endothelial VCAM-1 and ICAM-1 in a BMP-4–and TGF-β1–dependent pathway. Arterioscler Thromb Vasc Biol. 2009;29: 254–60. 10.1161/ATVBAHA.108.176347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Guerraty MA, Grant GR, Karanian JW, Chiesa OA, Pritchard WF, Davies PF. Hypercholesterolemia induces side-Specific phenotypic changes and peroxisome proliferator-activated receptor-pathway activation in swine aortic valve endothelium. Arterioscler Thromb Vasc Biol. 2010;30: 225–31. 10.1161/ATVBAHA.109.198549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Hess K, Ushmorov A, Fiedler J, Brenner RE, Wirth T. TNFα promotes osteogenic differentiation of human mesenchymal stem cells by triggering the NF-κB signaling pathway. Bone. 2009;45: 367–76. 10.1016/j.bone.2009.04.252 [DOI] [PubMed] [Google Scholar]
- 51. Gareus R, Kotsaki E, Xanthoulea S, van der Made I, Gijbels MJJ, Kardakarus R, et al. Endothelial cell-specific NF-κB inhibition protects mice from atherosclerosis. Cell Metab. 2008;8: 372–83. 10.1016/j.cmet.2008.08.016 [DOI] [PubMed] [Google Scholar]
- 52. Stephens EH, Saltarrelli JG, Baggett LS, Nandi I, Kuo JJ, Olmsted-Davis EA, et al. Differential proteoglycan and hyaluronan distribution in calcified aortic valves. Cardiovasc Pathol. 2011;20: 334–42. 10.1016/j.carpath.2010.10.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Hinton RB, Lincoln J, Deutsch GH, Osinska H, Manning PB, Benson DW, et al. Extracellular matrix remodeling and organization in developing and diseased aortic valves. Circ Res. 2006;98: 1431–8. [DOI] [PubMed] [Google Scholar]
- 54. Acharya A, Hans CP, Koenig SN, Nichols HA, Galindo CL. Inhibitory role of Notch1 in calcific aortic valve disease. PLoS ONE. 2011;6: e27743 10.1371/journal.pone.0027743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Fondard O, Detaint D, Lung B, Choqueux C, Adle-Biassette H, Jarraya M, et al. Extracellular matrix remodelling in human aortic valve disease: the role of matrix metalloproteinases and their tissue inhibitors. Eur Heart J. 2005;26: 1333–41. [DOI] [PubMed] [Google Scholar]
- 56. Clark-Greuel JN, Connolly JM, Sorichillo E, Narula NR, Rapoport HS, Mohler ER, et al. Transforming growth factor-β1 mechanisms in aortic valve calcification: Increased alkaline phosphatase and related events. Ann Thorac Surg. 2005;83: 946–53. [DOI] [PubMed] [Google Scholar]
- 57.Pashkow FJ. Oxidative stress and inflammation in heart disease: Do antioxidants have a role in treatment and/or prevention? Int J Inflamm. 2011; 514623. [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
A, Intracellular superoxide in VEC+TNFα at 30 minutes after treatment. Presented using colorimetric scale to show relative DHE intensity, indicated in inset triangle. B, Quantification of superoxide production at 30 minutes using microplate assay for DHE fluorescence. * indicates p < 0.05 versus control. N > 6 for each condition. DHE fluorescence was measured using integrated pixel density in three different fields of view (250μm2) on each sample. The three measurements were averaged within each sample, with each average corresponding to N of 1. Means were compared using unpaired Student’s t-test, assuming unequal variance.
(TIFF)
PAVEC cultured 48 hrs on 3D hydrogels with control, +30 ng/mL TNFα, or 1μM H2O2. Boxed regions indicate bands shown in Fig 3.
(TIFF)
A. TNFα+catalase treatment caused a significant increase in apoptosis. B. TNFα+catalase treatment caused increased loss of VE-cadherin, increased nuclear translocation of NFkB, and increased VCAM-1 expression compared to both control and TNFα alone. N = 3, * indicates p < 0.05 vs CTL, # indicates p < 0.05 vs TNFα. Scale bar is 20μm.
(TIFF)
PAVEC cultured 48 hrs on 3D hydrogels with control, +30 ng/mL TNFα, +30 ng/mL TNFα+10μM BH4, or +30 ng/mL TNFα+20U/mL peg-SOD. Boxes indicate bands shown in Fig 4. *Analysis of NFκB p65 protein expression was not used in this study.
(TIFF)
There was no significant apoptosis in any of AV leaflets, as measured by the TUNEL assay. N = 6 for all sample groups. Means were compared using one-way ANOVA with Tukey’s post hoc test.
(TIFF)
Porcine AV leaflets cultured 21 days in control, +30 ng/mL TNFα, +30 ng/mL TNFα+10μM BH4, or +30 ng/mL TNFα+20U/mL peg-SOD. Boxes indicate bands shown in Fig 6. *Analysis of NFκB p65 protein expression was not used in this study.
(TIFF)
A. Complex regulation of transcription factors Runx2 and Msx2 by TNFα and BH4 or SOD. N = 6, * indicates p < 0.05 vs CTL, # indicates p < 0.05 vs TNFα. B. BH4 co-treatment consistently causes either neutral or beneficial effects compared to TNFα alone. N = 6, * indicates p < 0.05 vs TNFα.
(TIFF)
(DOCX)
Data Availability Statement
All relevant data are within the paper and its Supporting Information files.