

Abstract
Pathogenesis in alcoholic liver disease (ALD) is complicated and multifactorial but clearly involves oxidative stress and inflammation. Currently, conflicting reports exist regarding the role of endoplasmic reticulum (ER) stress in the etiology of ALD. The glucose regulated protein 78 (GRP78) is the ER homologue of HSP70 and plays a critical role in the cellular response to ER stress by serving as a chaperone assisting protein folding and by regulating the signaling of the unfolded protein response (UPR). Comprised of three functional domains, an ATPase, peptide-binding, and lid domain, GRP78 folds nascent polypeptides via the substrate-binding domain. Earlier work has indicated that the ATPase function of GRP78 is intrinsically linked and essential to its chaperone activity. Previous work in our laboratory has indicated that Grp78 and the UPR are not induced in a mouse model of ALD but that Grp78 is adducted by the lipid electrophiles 4-hydroxynonenal (4-HNE) and 4-oxononenal (4-ONE) in vivo. As impairment of Grp78 has the potential to contribute to pathogenesis in ALD, we investigated the functional consequences of aldehyde adduction upon Grp78 function. Identification of 4-HNE and 4-ONE target residues in purified human GRP78 revealed a marked propensity for Lys and His adduction within the ATPase domain and a relative paucity of adduct formation within the peptide-binding domain. Consistent with these findings, we observed a concomitant dose-dependent decrease in ATP-binding and ATPase activity without any discernible impairment of chaperone function. Collectively, our data indicate that ATPase activity is not essential for Grp78 mediated chaperone activity and is consistent with the hypothesis that ER stress does not play a primary initiating role in the early stages of ALD.
Keywords: 4-HNE, 4-ONE, HSP, electrophile, alcoholic liver disease, protein oxidation
Introduction
Oxidative stress has been linked to numerous disease states, including neurodegeneration, diabetes, cancer, and alcoholic liver disease (ALD) (1,2). ALD is thought to stem from a complex triad oxidative stress, ER stress, and inflammation, where each response is capable of propagating the other (3,4). The mechanisms behind these intrinsically linked responses remains unknown, however, the enhanced generation of lipid electrophiles and ensuing protein adduction is thought to be a key mediator in these processes (1,2,5–7). 4-hydroxynonenal (4-HNE) and 4-oxononenal (4-ONE) are among the most prevalent lipid electrophiles generated under the pathologic conditions engendered by the sustained consumption of ethanol (1,5). These electrophiles react with the side-chains of cysteine, histidine, and lysine residues, forming a covalent linkage via either a Michael-type addition or Schiff-base mechanism (6,7), generally leading to impaired protein function. Utilizing rodent models for early-stage ALD, a comprehensive inventory of carbonylated proteins has been generated (8–11). Among the most heavily and consistently adducted proteins are the heat-shock proteins (HSPs), specifically, the endoplasmic reticulum (ER) member of the HSP family, 78kDa glucose-regulated protein (GRP78 or HSPA5) (10). Impaired GRP78 function has been demonstrated to play a major role in the initiation of numerous hepatic disease states through the induction of the ER stress response (12).
HSPs represent a class of critical molecular chaperones that are responsible for maintaining proper protein folding in all cellular compartments (13). HSPs are comprised of three domains, an N-terminal ATPase catalytic site, a substrate-binding domain and a C-terminal lid domain (13). The precise role of these domains in GRP78-mediated protein folding remains unclear; however, current theories suggest that the binding of an unfolded protein is followed by the hydrolysis of ATP to ADP, resulting in a conformational change and a trapping of the nascent peptide (13). Once correct folding is achieved, HSPs bind to a new molecule of ATP, resulting in the release of the correctly folded protein (13). Conversely, efficient chaperone-mediated folding has been observed in an ATP-independent fashion using small HSPs and heat-denatured citrate synthase (14). These conflicting reports suggest independent roles for the ATP-binding and substrate-binding domains of certain molecular chaperones.
Protein folding is arguably the most notable function of the ER, and as a result, a large number of chaperone proteins reside in this compartment (15,16). When the chaperone function of GRP78 and other HSPs become compromised, unfolded proteins accumulate within the ER, resulting in an increase in protein ubiquitylation (17). As the unfolded protein load overwhelms the protein degradation machinery, the cell elicits a signaling cascade referred to as the unfolded protein response (UPR) (18). This response is mediated by GRP78, which is bound to three transmembrane proteins under non-stressed conditions, activating transcription factor 6 (ATF6), inositol-requiring enzyme 1 (IRE-1), and protein kinase RNA-like endoplasmic reticulum kinase (PERK). When the UPR is initiated, GRP78 dissociates from these proteins, initiating the UPR signaling response (17,19). Given its integral role in both protein folding and cellular signaling, maintaining GRP78 function is of critical importance.
Our laboratory has investigated the role of the UPR, oxidative stress, and inflammatory responses in rodent models for early-stage ALD, revealing a minimal role for the UPR (4). These reports also shed light on the pathogenic potential of lipid electrophile adduction on chaperone function (8,9). The impact of 4-HNE and 4-ONE adduction on HSP70 and HSP90 have been investigated, displaying Cys-specific susceptibility to modification (8,9). Recent efforts have identified a 4-ONE adduct on Lys591 of murine GRP78 in a model for ALD; the mechanistic impact of this modification has yet to be assessed (10). In the current study, we demonstrate that GRP78 is susceptible to modification by lipid electrophiles. These modifications were concentrated to the ATPase domain of the protein, revealing a dose-dependent decrease in ATPase activity; contrary to other HSPs, the chaperone activity was preserved. This is likely due to a lack of Cys present in the peptide-binding domain, leaving GRP78 resistant to the deleterious effects of electrophile adduction on chaperone activity.
Experimental Procedures
Animal Model
All procedures involving animals were approved by the Institutional Animal Care and Use Committee of the University of Colorado and were performed in accordance with published National Institutes of Health guidelines. Male, C57BL/6J mice (12 per group) were utilized for the analysis and characterization of ethanol-mediated liver damage. For a detailed description of the ethanol feeding regimen utilized for these studies, see Galligan et. al, 2012 (4). Upon completion of the study, animals were anesthetized via intraperitoneal injection with sodium pentobarbital and euthanized by exsanguination. Livers were excised, weighed, and frozen for biochemical characterization or subjected to differential centrifugation for subcellular fractionation as previously described (11).
2-Dimensional Gel Electrophoresis and Western Blotting
Microsomal fractions isolated from the livers of both control and ethanol-fed mice were subjected to two-dimensional gel electrophoresis (2D-PAGE) on IPG strips (pH 3–11) and separated on 7cm gels. Proteins were then transferred to a Hybond-P membrane (GE Healthcare, Buckinghamshire, UK) and then blocked for 30 min with a tris-buffered saline solution containing 1% Tween-20 (TBST) and 5% non-fat dry milk (NFDM). Membranes were probed with primary antibodies directed against either KDEL (which recognizes GRP78 and GRP58) (SPA-827, Stressgen, Ann Arbor, MI), or custom antibodies directed against 4-HNE or 4-ONE modified proteins (Bethyl Laboratories, Montgomery, TX). A horseradish peroxidase conjugated secondary (Jackson Labs, Bar Harbor, ME) was then applied and membranes were developed using ECL-Plus Reagent from GE Healthcare. Chemiluminescence was visualized using a Storm 860 scanner from Molecular Dynamics (Sunnyvale, CA).
Detection of Carbonylated GRP78
Protein collected from microsomal fractions was incubated with 5.0mM biotin hydrazide (Pierce, Rockford, IL) for 2hrs at room temperature in the dark. Samples were reduced with 10.0mM sodium borohydride for 1 h at room temperature. Excess biotin hydrazide was then removed using Zeba Spin Desalting Columns (Pierce, Rockford, IL) and biotin-conjugated proteins were incubated with NutrAvidin Agarose resin (Pierce, Rockford, IL) overnight at 4°C. Avidin beads were washed 5x with PBS containing 0.1% Tween-20 and denatured in loading buffer. Proteins were separated via SDS-PAGE, transferred to PVDF, and blotted with primaries directed against GRP78. As a loading control, input fractions were blotted for GRP78 and actin; carbonylated GRP78 was normalized to the actin corrected input GRP78 expression and is represented as a percent of control.
Synthesis and Purification of Recombinant Human GRP78 in E. Coli
High-level expression of recombinant N-terminal His-tagged human GRP78 was performed in E coli (DE3) cells transformed with a GRP78-pET-28b construct (a generous gift from Professor Richard C. Austin, McMaster University, Canada). Cells were grown in LB media containing 50μg/mL Kanamycin at 37°C until an OD of 0.6 at 600nm was reached. Protein expression was induced by the addition of isopropyl β-D-a-thiogalactopyranoside and proceeded for a further 5 hours at 37°C. Cells were then harvested by centrifugation. The resulting cell pellet was resuspended with lysis buffer containing 15mM Tris-HCl, pH7.4 (4°C), 10mM β-mercaptoethanol, 300mM NaCl, 10mM imidazole, and an appropriate amount of Complete, EDTA-free, Protease Inhibitor Cocktail Tablets from Roche (Indianapolis, Indiana). The cells were lysed by sonication and cell debris and insoluble material were removed by centrifugation for 30 min at 10,000× g at 4°C. The resulting supernatant was applied onto a Ni-NTA affinity chromatography column (Qiagen, Germantown, MD). Briefly, 50% Ni-NTA slurry equilibrated with lysis buffer was incubated with the supernatant for one hour with gentle rocking at 4°C. The resin mixture was loaded onto the column and was then washed with washing buffer containing 15mM Tris-HCl, pH7.4 (4°C), 10mM β-mercaptoethanol, 300mM NaCl, and 40mM imidazole for 10 column volumes. GRP78 was eluted with a linear gradient of 40–400mM imidazole and subsequently concentrated and changed to a buffer containing 25mM Tris-HCl, pH 7.4, 100mM NaCl. Protein concentration was determined by the method of Bradford using bovine serum albumin as a standard and the purity was evaluated by SDS-PAGE.
In Vitro Adduction of GRP78 with 4-HNE and 4-ONE
GRP78 (2.0μg) was treated with increasing molar concentrations of either 4-HNE or 4-ONE in 50mM tricine, pH 7.4 for 1hr. at 37°C. For the chemical reduction of labile aldehyde adducts, samples were treated with 10mM NaBH4 for 1hr at 37°C. Samples were then reduced with standard SDS-PAGE loading buffer and heated at 95°C for 5 min. Proteins were resolved under standard SDS-PAGE and either transferred to PVDF membranes or digested with sequencing grade trypsin for MS analysis as described (10).
GRP78 ATPase Activity Assay
To assess the activity of recombinant GRP78, a phosphate release assay was utilized as described with slight modifications (20). Briefly, 1.0μg of GRP78 was incubated for 1 hour at 37°C with increasing molar concentrations of 4-HNE or 4-ONE in 50mM tricine, pH 7.4. The reaction was then initiated by the addition of 0.1mM ATP, 2mM MgCl and 0.5mM DTT. The reaction was allowed to proceed at 37°C for various times, as indicated. For the calculation of Vmax, the reaction was stopped following 1 hour. Free phosphate was measured by the addition of BIOMOL Green (Enzo Life Sciences, Inc., Farmingdale, NY) at a 1:1 ratio. Following 10 min, samples were read using a microtiter plate reader at 620 nm on a SpectraMax 190 microplate spectrofluorometer (Molecular Devices, Sunnyvale, CA). Values were determined as nanomoles of phosphate released per minute and are presented as a percentage of control reactions.
GRP78 chaperone assay
The chaperone activity of GRP78 was tested analyzing activity to prevent protein aggregation using a previously published method (21). Chaperone activity was monitored via aggregation of citrate synthase (CS) at 1.0μM or malate dehydrogenase (MDH) at 1μM in the absence and presence of 4-HNE and 4-ONE modified GRP78, at increasing concentrations. These samples were incubated for 90 min at 45 °C (CS) and 40°C (MDH) in 40mM HEPES (pH 7.5). Protein aggregation was monitored by light scattering at 320 nm using a temperature controlled SpectraMAX 190 spectrophotometer (Molecular Devices, Sunnyvale, CA). Chaperone activity was determined as a percentage of each substrate in the absence of GRP78 (i.e. 100% aggregation).
LC-MS/MS Identification of GRP78 adducts
Control and modified GRP78 were digested with trypsin using a standard in-gel protocol. Peptide separation was performed by nano-Advance Splitless nano-LC at a flow rate of 500 nL/min with a gradient of 5 to 45% solvent B (90% acetonitrile, 0.1% formic acid) over 60 min on a 0.1mm x 150mm Magic AQ C18 column (Michrome, Auburn, CA). The LC was coupled to an amaZon speed ETD ion trap mass spectrometer with captive spray ion source (Bruker Daltonics, Inc., Billerica, MA). The instrument was operated using data-dependent collision-induced dissociation (CID) and electron transfer dissociation (ETD) MS/MS with a threshold for fragmentation at 100000 counts (TIC)(22). Data analysis was performed using Mascot (v 2.4, www.matrixscience.com) and Proteinscape (Bruker Daltonics). Peptide identifications were accepted if they could be established at greater than 99.0% probability as specified.
Computational-based molecular modeling of human GRP78
All molecular modeling studies were conducted using Accelrys Discovery Studio 3.5 (Accelrys Software, Inc., San Diego, CA; (http://accelrys.com) and all crystal structure coordinates were obtained from the protein data bank (http://www.pdb.org). The ATPase domain of human GRP78 (residues 26–407) has been crystallized previously (PDB ID: 3LDO) (23). This structure was combined with a protein homology model of the C-terminal substrate binding domain (residues 408–631) generated with the MODELLER protocol (24) utilizing the crystal structure templates of bovine heat shock cognate 71 kDa protein (PDB ID: 1YUW) (25) (63.4% identity, 82.0% similarity) and E. coli Hsp70 (PDB ID: 2KHO) (26) (49.2% identity, 70.8% similarity). No suitable templates were available for residues 1–26 or 633–654 and were therefore omitted from the model. The resulting structure was then subjected to energy minimization utilizing the conjugate gradient minimization protocol (10,000 iterations) with a CHARMm forcefield and the Generalized Born implicit solvent model with simple switching (27,28). Schiff base and/or Michael adducts of 4-HNE or 4-ONE were built onto the respective specific residues identified by MS/MS analysis and both the native and adducted models subjected to a further round of minimization as described above. Figures were generated using Lightwave 11.5 (NewTek, Inc., San Antonio, TX; www.lightwave3d.com).
ATP Affinity Assay
To examine the effects of ATP binding to GRP78, a Kinase Enrichment Kit was used (Pierce, Rockford, IL). Briefly, 10μg of GRP78 was incubated in triplicate with increasing molar ratios (1:1, 5:1 10:1, 25:1 and 50:1) of 4-HNE or 4-ONE for 30 min at RT. Reactions were then treated with 10μM desthiobiotin ATP in 20mM MgCl for 10 min. ATP bound GRP78 or HSP72 was denatured in 8M urea and purified by incubation with streptavidin agarose beads (25μl/sample) for 60 minutes. Samples were then analyzed using SDS PAGE and Western blotting using anti-KDEL antibodies.
Statistical Analysis
Statistical analysis and generation of graphs was performed using GraphPad Prism 4.02 (GraphPad Software, San Diego, CA). Differences between control and ethanol-fed mice were assessed using a paired Student’s t-test. To determine the impact of aldehyde treatments on GRP78 function, a one-way ANOVA with a Tukey’s post-hoc was utilized. Differences were considered significant if P < 0.05.
Results
GRP78 is a target for modification in ALD
We recently identified GRP78 as a target for adduction by 4-ONE on K592 in a murine model for ALD (29). Given its pivotal role in protein folding and cellular signaling pathways, we investigated the potential deleterious impact of this oxidative modification on protein function. As shown in Figure 1, immunostaining of microsomal proteins reveals an accumulation of 4-HNE (A) and 4-ONE (B) modified proteins in liver sections from ethanol-fed mice. To identify GRP78 as a potential target for modification, immunostaining was confirmed with antibodies directed against the C-terminus of GRP78 (anti-KDEL) (C). The spot corresponding to GRP78 (arrow) was also positively identified as GRP78 utilizing MALDI-MS/MS (data not shown).
Figure 1. GRP78 is a target for modification by 4-HNE and 4-ONE in a murine model for early-stage ALD.

Positive immunostaining was observed following western blotting with antibodies directed against 4-HNE-modified proteins (A) and 4-ONE-modified proteins (B). As a reference, immunostaining was also conducted with antibodies directed against the C-terminal KDEL sequence of GRP78 (C) Coomassie blue staining reveals GRP78, which was positively identified utilizing MALDI-MS (D). GRP78 is indicated by the arrow in each figure.
While the immunostaining of GRP78 demonstrated an increase in the adducted species in ethanol-fed mice (Figure 1), we sought to further validate this observation. Utilizing carbonyl-reactive biotin hydrazide, adducted microsomal protein species were selectively captured via streptavidin pull-down. As shown in Figure 2A, immunoblotting revealed an increase in carbonylated GRP78 following sustained ethanol consumption. Immunoblotting was also conducted to determine the effects of ethanol feeding on the expression of GRP78. While no change in GRP78 expression was observed, blot densitometry reveals an approximate 40% increase in carbonylated GRP78 when normalized to total protein expression (Figure 2B, (P < 0.05).
Figure 2. Carbonylation of GRP78 is increased following sustained ethanol consumption.

(A) Carbonylated GRP78 is visualized through biotin hydrazide conjugation, streptavidin pulldown and GRP78 western blotting. While the expression of GRP78 does not change following ethanol consumption, carbonylated protein is increased. (B) Blot densitometry of (A) reveals a statistically significant increase in modified protein.
Electrophile modification of GRP78 results in protein-protein cross-linking in vitro
To further elucidate the impact of aldehyde modification on GRP78 function, purified recombinant human GRP78 was treated with increasing molar concentrations of either 4-HNE or 4-ONE and subjected to SDS-PAGE. Immunoblotting for either 4-HNE- or 4-ONE-modified proteins demonstrated a dose-dependent increase in adducted species (Figure 3A). Treatment of recombinant GRP78 with pathophysiologically relevant concentrations of 4-HNE (~28μM) induced roughly a 5-fold increase in positive immunostaining (blot densitometry not shown). Given the highly reactive nature of 4-ONE, it is not surprising that treatment with the same concentration (~28μM) resulted in more extensive adduction, leading to roughly a 10-fold increase over control levels.
Figure 3. Adduction by 4-HNE and 4-ONE leads to proteins-protein cross-linking.

A.) Western blotting of purified GRP78 following adduction with either 4-HNE or 4-ONE. Adduction induces high molecular weight protein-protein cross-linking with purified GRP78 at physiologically relevant doses of either 4-HNE (B) or 4-ONE (C). Chemical stabilization of these adducts using NaBH4, displays the high susceptibility of this lysine cross-linking with concentrations as low as 3μM (1x).
4-HNE and 4-ONE have been reported to generate protein-protein cross-links via a Michael addition at Cys, His, or Lys, followed by a Schiff base reaction at a Lys residue (30). Human GRP78 contains 61 Lys residues, leaving it highly susceptible to inter- and intramolecular cross-linking. As shown in Figure 3B, GRP78 displayed a concentration-dependent increase in protein-protein crosslinking, resulting in the formation of higher molecular weight species (~150kD and ~210kD) with as little as 5X molar excess 4-HNE (~14μM aldehyde) (left). Following reduction with 10mM NaBH4 to stabilize labile Schiff-base adducts, cross-linking of GRP78 is observed with as little as 2.8μM 4-HNE (right). Similar results were observed in response to incubation with 4-ONE, with GRP78 cross-linking apparent following treatment with 2.8μM (Figure 3C, left) and formation of very high molecular weight aggregates incapable of penetrating the stacking gel following treatment with doses of ~14μM (Figure 3C, right). In contrast to 4-HNE, incubation with 4-ONE lead to the progressive, concentration-dependent loss of monomeric GRP78, as evidenced by the disappearance of the 78kD band. These cross-links were hypothesized to greatly impact the activity of GRP78 via molecular aggregation.
Aldehyde adduction of GRP78 leads disrupted ATPase activity
As a member of the HSP family of proteins, GRP78 contains three domains, an ATPase domain, a substrate-binding domain, and a C-terminal lid domain (13,16,23). Given that GRP78 function requires the binding of substrate, followed by the hydrolysis of ATP for efficient chaperone activity (13,16), we sought to independently evaluate the functional impact of aldehyde adduction on both the ATPase and substrate-binding domains. To determine the impact of adduction on the ATPase activity of GRP78, we monitored the hydrolysis of ATP through the release of phosphate (31,32). As shown in Figure 4, incubation of GRP78 with increasing concentrations of either 4-HNE (open bars) or 4-ONE (closed bars) resulted in a concentration-dependent decrease in enzymatic ATPase activity. These effects were more pronounced following incubation with 4-ONE, where ~50% of activity was abolished following incubation with a 25x molar excess (~70μM) of aldehyde (P < 0.001). Efforts were made to measure ATPase activity in the microsomes collected from both control- and ethanol-fed mouse livers; however, due to the high level of residual phosphates present in the sample preparations, no reliable measurements could be made.
Figure 4. 4-HNE and 4-ONE adduction of GRP78 results in a concentration-dependent decrease in ATP hydrolysis.
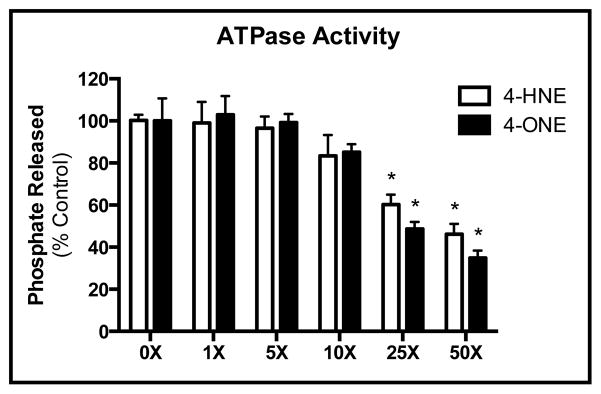
Treatment of GRP78 with 4-HNE (open bars) or 4-ONE (closed bars) reveals a corresponding decrease in ATPase activity. Estimated IC50 values were calculated revealing a Ki of 51.8μM for 4-HNE and a Ki of 43.9μM for 4-ONE.
The chaperone activity of GRP78 is preserved following adduction by lipid electrophiles
Although much work has analyzed the function of the ATPase domain of GRP78, few studies have evaluated the enzymatic activity of the peptide-binding domain (23,33). We sought to assess the impact of modification on the chaperone function of GRP78. Using either heat-denatured malate dehydrogenase (MDH) or citrate synthase (CS) as a substrate, the chaperone properties of GRP78 were monitored. As shown in Figure 5A, incubation of MDH at 40°C resulted in protein aggregation (presented as a % of aggregation). The addition of native GRP78 resulted in approximately 75% refolding of aggregated MDH. Following incubation of GRP78 with either 4-HNE (closed bars) or 4-ONE (open bars), a minor impact on refolding was observed, with approximately 25% of the protein activity being impeded with higher doses of 4-ONE. Conversely, using heat denatured CS as a substrate (B), no change in GRP78 activity was observed, regardless of the treatment.
Figure 5. Chaperone-mediated refolding of CS and MDH is preserved following adduction.

GRP78-mediated refolding of (A) MDH or (B) CS is unaffected by electrophile adduction with either 4-HNE (closed bars) or 4-ONE (open bars). (C) Sequence alignment using ClustalW demonstrates a conserved Cys residue in electrophile-susceptible HSPs. HSPs lacking this Cys residue are presented in green font. Mean is plotted with error bars representing SEM.
Previous works have demonstrated a critical role for Cys residues within the substrate-binding domain of other HSP family members (9). A sequence alignment of the HSP70 family of proteins highlights the conserved Cys-containing domain within the backbone of the ATPase domain (Figure 5C). Adduction of this residue on HSP72 was found to drastically disrupt the chaperone-mediated refolding of firefly luciferase; this residue was found to be the major site of adduction on HSP72 (9). Conversely, DnaK was resistant to adduction by 4-HNE due to the lack of a reactive Cys residue in its substrate-binding domain. Similar to DnaK, GRP78 lacks this Cys residue across all species, demonstrating a potential mechanism by which GRP78 is resistant to the deleterious impact of modification by Cys reactive electrophiles (9).
MS analysis reveals the ATPase domain as a susceptible region for adduction
Recently, our laboratory has identified K592 as a specific target for modification by 4-ONE in the ethanol-feeding model utilized for these studies (29). Various strategies were used to identify additional sites of modification in vivo, however no other sites of modification were identified. We therefore sought to characterize potential sites of modification in vitro utilizing recombinant human GRP78. This technique provides a model for other less stable or otherwise unidentifiable in vivo adducts of GRP78. Following treatment with physiologically relevant concentrations (27.7μM) of either 4-HNE or 4-ONE, 23 different adducts on 12 different residues (8 Lys, 4 His) were identified using MS (Table 1). Consistent with the observed inhibition of ATPase activity presented in Figure 4A and B, 16 of these adducts were identified in the ATPase domain of GRP78. In contrast, only two adducts were identified in the peptide-binding domain of GRP78, further suggesting that the major mechanism of aldehyde-mediated inhibition of GRP78 function is due to the greater susceptibility of the ATPase domain to adduction.
Table 1.
Identification of 4-HNE and 4-ONE Modified Peptides on Human GRP78
| Range | Sequence | Modified Residue | Modification | NaBH4 | Type | Scores | Domain |
|---|---|---|---|---|---|---|---|
|
| |||||||
| 75 – 96 | R.LIGDAAK*NQLTSNPENTVFDAK.R | K81 | 4-HNE M.A. | X | CID | 35.6 (M:35.6) | ATPase |
| 75 – 96 | R.LIGDAAK*NQLTSNPENTVFDAK.R | K81 | 4-ONE S.B | CID | 25.5 (M:25.5) | ATPase | |
| 164 – 181 | K.K*VTH*AVVTVPAYFNDAQR.Q | K164; H167 | 4-ONE M.A.; 4-ONE M.A. | CID | 42.5 (M:42.5) | ATPase | |
| 164 – 181 | K.K*VTHAVVTVPAYFNDAQR.Q | K164 | 4-HNE M.A. | X | CID | 38.8 (M:38.8) | ATPase |
| 164 – 181 | K.K*VTHAVVTVPAYFNDAQR.Q | K164 | 4-HNE S.B | CID | 48.7 (M:48.7) | ATPase | |
| 165 – 181 | K.VTH*AVVTVPAYFNDAQR.Q | H167 | 4-HNE M.A. | X | ETD | 76.0 (M:76.0) | ATPase |
| 165 – 181 | K.VTH*AVVTVPAYFNDAQR.Q | H167 | 4-HNE M.A. | CID | 65.7 (M:65.7) | ATPase | |
| 165 – 181 | K.VTH*AVVTVPAYFNDAQR.Q | H167 | 4-HNE M.A. | X | ETD | 71.2 (M:71.2) | ATPase |
| 198 – 214 | R.IINEPTAAAIAYGLDK*R.E | K213 | 4-HNE S.B | ETD | 20.1 (M:20.1) | ATPase | |
| 198 – 214 | R.IINEPTAAAIAYGLDK*R.E | K213 | 4-ONE S.B | CID | 48.0 (M:48.0) | ATPase | |
| 298 – 306 | R.ALSSQH*QAR.I | H303 | 4-HNE M.A. | X | CID | 56.2 (M:56.2) | ATPase |
| 325 – 336 | R.AK*FEELNmDLFR.S | K326 | 4-HNE S.B | ETD | 24.9 (M:24.9) | ATPase | |
| 353 – 367 | K.K*SDIDEIVLVGGSTR.I | K353 | 4-HNE S.B | ETD | 30.9 (M:30.9) | ATPase | |
| 368 – 376 | R.IPK*IQQLVK.E | K370 | 4-HNE S.B | ETD | 27.2 (M:27.2) | ATPase | |
| 368 – 376 | R.IPK*IQQLVK.E | K370 | 4-ONE M.A. | CID | 25.6 (M:25.6) | ATPase | |
| 475 – 492 | K.DNH*LLGTFDLTGIPPAPR.G | H477 | 4-HNE M.A. | X | ETD | 37.2 (M:37.2) | Peptide-Binding |
| 475 – 492 | K.DNH*LLGTFDLTGIPPAPR.G | H477 | 4-HNE M.A. | CID | 26.0 (M:26.0) | Peptide-Binding | |
| 582 – 596 | K.LGGK*LSSEDKETmEK.A | K585 | 4-ONE S.B | ETD | 43.9 (M:43.9) | Lid | |
| 602 – 617 | K.IEWLESH*QDADIEDFK.A | H608 | 4-HNE M.A. | X | ETD | 42.1 (M:42.1) | Lid |
| 602 – 617 | K.IEWLESH*QDADIEDFK.A | H608 | 4-HNE M.A. | CID | 32.4 (M:32.4) | Lid | |
| 602 – 617 | K.IEWLESH*QDADIEDFK.A | H608 | 4-HNE M.A. | X | CID | 21.5 (M:21.5) | Lid |
| 621 – 633 | K.K*ELEEIVQPIISK.L | K621 | 4-HNE M.A. | ETD | 53.4 (M:53.4) | Lid | |
Structural analysis of GRP78 adducts
Our results have indicated that aldehyde adduction is concentrated in the ATPase domain of GRP78, resulting in the inhibition of ATPase activity without significant effect on chaperone activity. Adduction of the residues comprising the ATPase domain have the potential to interfere with protein function due to alterations in protein conformation, flexibility, cofactor (ATP) binding, and/or ATPase capacity. In order to gain a greater mechanistic insight as to the potential functional and structural consequences of aldehyde adduction on GRP78, we generated a protein homology model of human GRP78, built 4-ONE adducts onto the residues listed in Table 1, and both the native and adducted structures subjected to extensive solvent-based energy minimization calculations. Additional minimizations were conducted using 4-HNE, revealing similar results (data not shown). A molecular overlay of the native and 4-ONE adducted structures of GRP78 (Figure 6) revealed minimal conformational changes to the protein backbone of the GRP78 structure in response to 4-ONE adduction, likely due to the peripheral location of the majority of the adducted residues. However, the side chain of K81 (Figure 6, indicated) lays within the ATP binding site of GRP78, suggesting that modification of this residue has the potential to directly interfere with ATP binding into the pocket, hydrolysis of ATP to ADP, and/or release of ADP after cleavage. Closer examination of the active site of the native vs. adducted models indicates potential alterations in side chain conformations due to the introduction of the 4-ONE modification at K81 (Figure 6 inset). E293 and R297 are involved in direct interactions with the ATP molecule as well as with each other. Adduction at K81 is predicted to induce the formation of hydrogen bonds between R297 and the carbonyl of 4-ONE, resulting in a predicted shift in the position of R297 that disrupts both its pi bond interactions with ATP and hydrogen bond interactions with E293, as well as altering the hydrogen bond interactions between E293 and ATP. Overall, these results suggest that adduction of K81 would lead to decreased GRP78 ATPase activity via an inhibition of ATP binding, rather than an impaired capacity to effectively hydrolyze ATP to ADP. Of the adducted residues, H477, K585, H608, and K621 are contained within the peptide-binding or lid domains of GRP78 (Figure 6). These adducts are localized on the periphery, distal to the peptide binding site, and do not appear likely to directly interfere with protein binding. Consistent with this observation, although adduction of GRP78 inhibited ATPase activity, we did not observe any effect on chaperone activity, further suggesting that these residues may not be critical to this function, or that adduction of these residues is not sufficient to impair or prevent these necessary protein-protein interactions from occurring.
Figure 6. Computational-based molecular modeling of 4-ONE adduction reveals potential conformational changes in the ATPase domain of GRP78.

Multiple viewpoints of the overlay of energy-minimized native (blue) and 4-ONE adducted (magenta) human GRP78. 4-ONE adducts are depicted as yellow sticks and the specific identities of the modified residues indicated. Key conformational changes in the ATP binding site between the native (blue, left inset) and 4-ONE adducted (magenta, right inset) are shown. Solid orange lines indicate predicted pi bond interactions and dashed green lines indicate predicted hydrogen bond interactions.
Lys adduction in the nucleotide-binding domain decreases the affinity of GRP78 for ATP
Based on the in situ simulations, we sought to investigate the ATP-binding efficiency of GRP78 as a potential mechanism for the alterations in ATPase activity. As shown in Figure 7, GRP78 bound efficiently to ATP-coated agarose beads. This binding was disrupted however, following pre-treatment with either 4-HNE (top) or 4-ONE (bottom). Consistent with the ATPase activity presented in Figure 4, a significant decrease in ATP-binding was found following lower concentrations of 4-ONE (~14μM) compared to 4-HNE (Figure 7B). These data further support the hypothesis that Lys modification to the ATP-binding pocket of GRP78 blocks access to the ATPase domain, selectively disrupting ATPase activity via impaired ATP binding.
Figure 7. Electrophilic modification of GRP78 decreases its affinity for ATP.

(A) Treatment of GRP78 with 4-HNE (top) or 4-ONE (bottom) results in a concentration-dependent decrease in ATP binding (B) Quantification of (A); mean is plotted from an N of 3; error bars represent SEM.
Discussion
The HSP family of proteins represents a class of highly abundant and critical chaperones (13). In this report, we identify an ER-resident member of the HSP family, GRP78, which is susceptible to electrophile modification in the alcoholic liver but also highly resistant to functional inhibition by these modifications. Previous studies have evaluated the impact of electrophile modification to other members of the HSP family, including HSP72 and HSP90 (8,9). These studies indicated Cys residues present in the substrate-binding domain as the major sites of adduction, highlighting a potential mechanism for the marked decrease in the chaperone activity following modification by 4-HNE (8,9,34). Additionally, Connor et al. have demonstrated His residues on HSP90 as highly sensitive to electrophile modification; however, the functional effects of these modifications were not evaluated (34).
We have previously identified Lys591 of murine GRP78 as a target for 4-ONE-mediated adduction in a chronic ethanol-feeding model (10). Lys591 is located on the solvent-accessible lid domain of GRP78, leaving it susceptible to modification by electrophiles. Although Lys591 was not identified in the current study, Lys585 of human GRP78 was identified as a target for adduction by 4-ONE and is also located in the lid domain. The precise role of this domain has yet to be elucidated, and literature surrounding its role remains conflicted. Current theories propose that ATP binding results in a conformational change in the backbone of the protein, resulting in a displacement of the lid domain and release of the newly refolded client protein from the peptide-binding domain (13). Our results from the current study, however, suggest that these domains may act independently and that chaperone activity is, in fact, uncoupled from ATPase activity. Adduction of purified GRP78 by either 4-HNE or 4-ONE was found to alter the ATPase activity of the protein, while these same concentrations were found to have little effect on the chaperone-mediated refolding of denatured substrates in the absence of ATP. These data suggest that pathophysiological concentrations of electrophile are sufficient to impede the ATPase activity of the protein, which may leave a functional population of GRP78 capable of executing the critical function of chaperone-mediated folding of unfolded substrates. If ATP hydrolysis was indeed the rate-limiting step in protein folding, the chaperone assays conducted here would fail to correctly refold denatured CS due to a trapping of the client protein in the substrate-binding domain.
GRP78 is heavily susceptible to electrophile modification, with the majority of these adducts (7) being found on Lys residues in the ATPase domain. Utilizing a combination of MS, in silico molecular modeling, and biochemical approaches, the impact of electrophile modification on GRP78 function was determined. Adducts were modeled onto identified Lys residues and structural alterations were monitored; adduction to the ATPase domain of GRP78 was not found to alter the overall conformation of either the substrate-binding or lid domains of the protein, consistent with the observed preservation of chaperone activity. Modification of Lys81 within the ATPase domain of the protein, however, suggests a potential stearic hindrance of ATP-binding to the ATPase domain of the protein. Consistent with this hypothesis, adduction of GRP78 by both 4-HNE and 4-ONE were found to decrease the binding of GRP78 to ATP, with 4-ONE having a greater effect. Collectively, these data support the hypothesis that Lys modification in the ATP-binding domain of GRP78 selectively disrupts ATPase activity, while maintaining the critical chaperone function of the protein (Figure 8).
Figure 8.

Although ATPase activity is inhibited, the chaperone function of GRP78 is resistant to the deleterious impact of electrophile adduction.
Lipid electrophiles have been shown to impact protein function in a multitude of ways, including alterations in enzymatic activity, post-translational modifications, and protein folding (35–38). Chaperones are particularly susceptible to modification by lipid electrophiles, often resulting in altered activity (8,9). MS characterization of in vitro adduction sites revealed a propensity for aldehyde modification in the ATPase domain that can likely attributed to the prevalence of Lys residues within this domain. Conversely, the substrate-binding domain of GRP78 was found to be relatively resistant to functionally detrimental modification. Although adducts were identified in both the substrate-binding domain (His477) and the lid domain (Lys585, His608 and Lys621), these adducts do not impact the chaperone activity of GRP78. This is likely a result of their location on the periphery of the protein and do not appear to be involved in the protein-protein interactions between GRP78 and its client proteins. This resistance to electrophile-mediated disruption in chaperone activity may be the result of an evolutionary divergence from other HSP members. An assessment of the primary structure of HSP70 family members reveals a conserved Cys residue on proteins that were found to be susceptible to electrophile-induced alterations in chaperone activity. Although GRP78 exhibits a high degree of sequence homology with HSP72 (80%), this Cys is lacking in both human GRP78 and the E. coli HSP (DnaK), perhaps conferring their resistance to functionally relevant adduction in this domain (9). This Cys-specific resistance to electrophile-induced inhibition is perhaps key to maintaining chaperone viability and efficient protein folding.
As the master regulator for the ER stress response and UPR signaling, alterations in GRP78 function have been outlined in numerous diseases, most notably ALD (12,39–42). Our previous works, however, have defined a minimal role for GRP78 in the pathogenesis of ALD (4). Conversely, Ji et al. have utilized GRP78 knockout mice to demonstrate increased hepatotoxicity following sustained intragastric ethanol consumption compared to wild-type mice (12). This is likely due to the major differences in the carbohydrate content of the diet utilized; Ji et al. have utilized very high concentrations of sucrose (360mM) as their carbohydrate source, while our studies used a more physiologically relevant concentration of carbohydrate (~90mM) while increasing the fat-derived calories (4,12). Gentile et al. have highlighted the pathogenic effects of these high sucrose-containing diets, displaying a pro-ER stress environment compared to their starch-fed counterparts (42). These effects were largely mediated through the induction of the UPR via the release of ATF6, PERK, and IRE1; the binding sites for these proteins, however, have not been mapped, although the large substrate-binding domain is thought to contain this function (43). Modification to this domain would likely result in a disruption in ATF6, IRE1 and PERK binding and initiation of the UPR. An extensive evaluation of these stress signaling pathways has been conducted with this model for ALD and no significant alterations in these pathways was observed (4). The studies presented here do have their limitations, however, as measuring the functional effects of only the electrophile-adducted pool of proteins in vivo is a limitation in technology. Additionally, it is conceivable that adduction to GRP78 may alter the functional pool of protein, leading to enhanced proteasomal degradation. While this possibility cannot be ruled out, it does offer an additional mechanism by which electrophile adduction to ER chaperone proteins may be enhancing the deleterious effects of ethanol toxicity. Collectively, these data demonstrate a high propensity for lipid electrophile adduction of the ATPase domain of GRP78. Despite these modifications, however, the chaperone and substrate/protein-binding domain of this protein are shielded from deleterious effects, preventing protein aggregation and unnecessary UPR signaling.
Supplementary Material
Highlights.
Grp78 is a target for adduction by lipid electrophiles in the alcoholic liver.
The ATPase domain of GRP78 heavily modified, impacting ATP-binding and activity.
Substrate binding is resistant to adduction, preserving chaperone activity.
Unlike HSP70, GRP78 lacks Cys residues in its substrate-binding domain.
Acknowledgments
Studies were supported by the National Institutes of Health/National Institutes of Alcoholism and Alcohol Abuse under grant numbers R37AA09300 (DRP), R01DK074487-01 (DRP) and F31AA018606-01 (JJG); MS analysis was carried out by the Proteomic Mass Spectrometry Facility and is supported in part by the Colorado Clinical Translational Science Institute (UL1 RR025780) and the University of Colorado Cancer Center (P30 CA046934). The authors would also like to thank the Computational Chemistry and Biology Core Facility at the University of Colorado Anschutz Medical Campus, which is supported in part by NIH/NCATS Colorado CTSI Grant UL1 TR001082, for their contributions to this manuscript.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Albano E. Alcohol, oxidative stress and free radical damage. The Proceedings of the Nutrition Society. 2006;65:278–290. doi: 10.1079/pns2006496. [DOI] [PubMed] [Google Scholar]
- 2.Dalle-Donne I, Giustarini D, Colombo R, Rossi R, Milzani A. Protein carbonylation in human diseases. Trends in molecular medicine. 2003;9:169–176. doi: 10.1016/s1471-4914(03)00031-5. [DOI] [PubMed] [Google Scholar]
- 3.Malhotra JD, Kaufman RJ. Endoplasmic reticulum stress and oxidative stress: a vicious cycle or a double-edged sword? Antioxidants & redox signaling. 2007;9:2277–2293. doi: 10.1089/ars.2007.1782. [DOI] [PubMed] [Google Scholar]
- 4.Galligan JJ, Smathers RL, Shearn CT, Fritz KS, Backos DS, Jiang H, Franklin CC, Orlicky DJ, Maclean KN, Petersen DR. Oxidative Stress and the ER Stress Response in a Murine Model for Early-Stage Alcoholic Liver Disease. Journal of toxicology. 2012;2012:207594. doi: 10.1155/2012/207594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mottaran E, Stewart SF, Rolla R, Vay D, Cipriani V, Moretti M, Vidali M, Sartori M, Rigamonti C, Day CP, Albano E. Lipid peroxidation contributes to immune reactions associated with alcoholic liver disease. Free radical biology & medicine. 2002;32:38–45. doi: 10.1016/s0891-5849(01)00757-2. [DOI] [PubMed] [Google Scholar]
- 6.Poli G, Schaur RJ, Siems WG, Leonarduzzi G. 4-hydroxynonenal: a membrane lipid oxidation product of medicinal interest. Medicinal research reviews. 2008;28:569–631. doi: 10.1002/med.20117. [DOI] [PubMed] [Google Scholar]
- 7.Esterbauer H, Schaur RJ, Zollner H. Chemistry and biochemistry of 4-hydroxynonenal, malonaldehyde and related aldehydes. Free radical biology & medicine. 1991;11:81–128. doi: 10.1016/0891-5849(91)90192-6. [DOI] [PubMed] [Google Scholar]
- 8.Carbone DL, Doorn JA, Kiebler Z, Ickes BR, Petersen DR. Modification of heat shock protein 90 by 4-hydroxynonenal in a rat model of chronic alcoholic liver disease. The Journal of pharmacology and experimental therapeutics. 2005;315:8–15. doi: 10.1124/jpet.105.088088. [DOI] [PubMed] [Google Scholar]
- 9.Carbone DL, Doorn JA, Kiebler Z, Sampey BP, Petersen DR. Inhibition of Hsp72-mediated protein refolding by 4-hydroxy-2-nonenal. Chemical research in toxicology. 2004;17:1459–1467. doi: 10.1021/tx049838g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Galligan JJ, Smathers RL, Fritz KS, Epperson LE, Hunter LE, Petersen DR. Protein carbonylation in a murine model for early alcoholic liver disease. Chemical research in toxicology. 2012;25:1012–1021. doi: 10.1021/tx300002q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Roede JR, Stewart BJ, Petersen DR. Decreased expression of peroxiredoxin 6 in a mouse model of ethanol consumption. Free radical biology & medicine. 2008;45:1551–1558. doi: 10.1016/j.freeradbiomed.2008.08.032. [DOI] [PubMed] [Google Scholar]
- 12.Ji C, Kaplowitz N, Lau MY, Kao E, Petrovic LM, Lee AS. Liver-specific loss of glucose-regulated protein 78 perturbs the unfolded protein response and exacerbates a spectrum of liver diseases in mice. Hepatology. 2011;54:229–239. doi: 10.1002/hep.24368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kampinga HH, Craig EA. The HSP70 chaperone machinery: J proteins as drivers of functional specificity. Nature reviews Molecular cell biology. 2010;11:579–592. doi: 10.1038/nrm2941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jakob U, Gaestel M, Engel K, Buchner J. Small heat shock proteins are molecular chaperones. The Journal of biological chemistry. 1993;268:1517–1520. [PubMed] [Google Scholar]
- 15.Gorlach A, Klappa P, Kietzmann T. The endoplasmic reticulum: folding, calcium homeostasis, signaling, and redox control. Antioxidants & redox signaling. 2006;8:1391–1418. doi: 10.1089/ars.2006.8.1391. [DOI] [PubMed] [Google Scholar]
- 16.Maattanen P, Gehring K, Bergeron JJ, Thomas DY. Protein quality control in the ER: the recognition of misfolded proteins. Seminars in cell & developmental biology. 2010;21:500–511. doi: 10.1016/j.semcdb.2010.03.006. [DOI] [PubMed] [Google Scholar]
- 17.Walter P, Ron D. The unfolded protein response: from stress pathway to homeostatic regulation. Science. 2011;334:1081–1086. doi: 10.1126/science.1209038. [DOI] [PubMed] [Google Scholar]
- 18.Malhi H, Kaufman RJ. Endoplasmic reticulum stress in liver disease. J Hepatol. 2011;54:795–809. doi: 10.1016/j.jhep.2010.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Malhotra JD, Kaufman RJ. The endoplasmic reticulum and the unfolded protein response. Seminars in cell & developmental biology. 2007;18:716–731. doi: 10.1016/j.semcdb.2007.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shearn CT, Smathers RL, Stewart BJ, Fritz KS, Galligan JJ, Hail N, Jr, Petersen DR. Phosphatase and tensin homolog deleted on chromosome 10 (PTEN) inhibition by 4-hydroxynonenal leads to increased Akt activation in hepatocytes. Molecular pharmacology. 2011;79:941–952. doi: 10.1124/mol.110.069534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Silva KP, Seraphim TV, Borges JC. Structural and functional studies of Leishmania braziliensis Hsp90. Biochimica et biophysica acta. 2013;1834:351–361. doi: 10.1016/j.bbapap.2012.08.004. [DOI] [PubMed] [Google Scholar]
- 22.Fritz KS, Kellersberger KA, Gomez JD, Petersen DR. 4-HNE adduct stability characterized by collision-induced dissociation and electron transfer dissociation mass spectrometry. Chemical research in toxicology. 2012;25:965–970. doi: 10.1021/tx300100w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Macias AT, Williamson DS, Allen N, Borgognoni J, Clay A, Daniels Z, Dokurno P, Drysdale MJ, Francis GL, Graham CJ, Howes R, Matassova N, Murray JB, Parsons R, Shaw T, Surgenor AE, Terry L, Wang Y, Wood M, Massey AJ. Adenosine-derived inhibitors of 78 kDa glucose regulated protein (Grp78) ATPase: insights into isoform selectivity. Journal of medicinal chemistry. 2011;54:4034–4041. doi: 10.1021/jm101625x. [DOI] [PubMed] [Google Scholar]
- 24.Eswar N, Eramian D, Webb B, Shen MY, Sali A. Protein structure modeling with MODELLER. Methods Mol Biol. 2008;426:145–159. doi: 10.1007/978-1-60327-058-8_8. [DOI] [PubMed] [Google Scholar]
- 25.Jiang J, Prasad K, Lafer EM, Sousa R. Structural basis of interdomain communication in the Hsc70 chaperone. Mol Cell. 2005;20:513–524. doi: 10.1016/j.molcel.2005.09.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bertelsen EB, Chang L, Gestwicki JE, Zuiderweg ER. Solution conformation of wild-type E. coli Hsp70 (DnaK) chaperone complexed with ADP and substrate. Proc Natl Acad Sci U S A. 2009;106:8471–8476. doi: 10.1073/pnas.0903503106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Feig M, Onufriev A, Lee MS, Im W, Case DA, Brooks CL., 3rd Performance comparison of generalized born and Poisson methods in the calculation of electrostatic solvation energies for protein structures. J Comput Chem. 2004;25:265–284. doi: 10.1002/jcc.10378. [DOI] [PubMed] [Google Scholar]
- 28.Brooks BR, Brooks CL, 3rd, Mackerell AD, Jr, Nilsson L, Petrella RJ, Roux B, Won Y, Archontis G, Bartels C, Boresch S, Caflisch A, Caves L, Cui Q, Dinner AR, Feig M, Fischer S, Gao J, Hodoscek M, Im W, Kuczera K, Lazaridis T, Ma J, Ovchinnikov V, Paci E, Pastor RW, Post CB, Pu JZ, Schaefer M, Tidor B, Venable RM, Woodcock HL, Wu X, Yang W, York DM, Karplus M. CHARMM: the biomolecular simulation program. Journal of computational chemistry. 2009;30:1545–1614. doi: 10.1002/jcc.21287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Galligan JJ, Smathers RL, Fritz KS, Epperson E, Hunter LE, Petersen DR. Protein Carbonylation in a Murine Model for Early Alcoholic Liver Disease. Chem Res Toxicol. 2012 doi: 10.1021/tx300002q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sayre LM, Lin D, Yuan Q, Zhu X, Tang X. Protein adducts generated from products of lipid oxidation: focus on HNE and one. Drug metabolism reviews. 2006;38:651–675. doi: 10.1080/03602530600959508. [DOI] [PubMed] [Google Scholar]
- 31.Gaut JR, Hendershot LM. Mutations within the nucleotide binding site of immunoglobulin-binding protein inhibit ATPase activity and interfere with release of immunoglobulin heavy chain. J Biol Chem. 1993;268:7248–7255. [PubMed] [Google Scholar]
- 32.Kassenbrock CK, Kelly RB. Interaction of heavy chain binding protein (BiP/GRP78) with adenine nucleotides. EMBO J. 1989;8:1461–1467. doi: 10.1002/j.1460-2075.1989.tb03529.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.King LS, Berg M, Chevalier M, Carey A, Elguindi EC, Blond SY. Isolation, expression, and characterization of fully functional nontoxic BiP/GRP78 mutants. Protein expression and purification. 2001;22:148–158. doi: 10.1006/prep.2001.1424. [DOI] [PubMed] [Google Scholar]
- 34.Connor RE, Marnett LJ, Liebler DC. Protein-selective capture to analyze electrophile adduction of hsp90 by 4-hydroxynonenal. Chemical research in toxicology. 2011;24:1275–1282. doi: 10.1021/tx200157t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Smathers RL, Fritz KS, Galligan JJ, Shearn CT, Reigan P, Marks MJ, Petersen DR. Characterization of 4-HNE modified L-FABP reveals alterations in structural and functional dynamics. PloS one. 2012;7:e38459. doi: 10.1371/journal.pone.0038459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Roede JR, Carbone DL, Doorn JA, Kirichenko OV, Reigan P, Petersen DR. In vitro and in silico characterization of peroxiredoxin 6 modified by 4-hydroxynonenal and 4-oxononenal. Chemical research in toxicology. 2008;21:2289–2299. doi: 10.1021/tx800244u. [DOI] [PubMed] [Google Scholar]
- 37.Fritz KS, Petersen DR. An overview of the chemistry and biology of reactive aldehydes. Free radical biology & medicine. 2013;59:85–91. doi: 10.1016/j.freeradbiomed.2012.06.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Backos DS, Fritz KS, Roede JR, Petersen DR, Franklin CC. Posttranslational modification and regulation of glutamate-cysteine ligase by the alpha, beta-unsaturated aldehyde 4-hydroxy-2-nonenal. Free radical biology & medicine. 2011;50:14–26. doi: 10.1016/j.freeradbiomed.2010.10.694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yoshida H. ER stress and diseases. The FEBS journal. 2007;274:630–658. doi: 10.1111/j.1742-4658.2007.05639.x. [DOI] [PubMed] [Google Scholar]
- 40.Kaplowitz N, Than TA, Shinohara M, Ji C. Endoplasmic reticulum stress and liver injury. Seminars in liver disease. 2007;27:367–377. doi: 10.1055/s-2007-991513. [DOI] [PubMed] [Google Scholar]
- 41.Nuss JE, Choksi KB, DeFord JH, Papaconstantinou J. Decreased enzyme activities of chaperones PDI and BiP in aged mouse livers. Biochemical and biophysical research communications. 2008;365:355–361. doi: 10.1016/j.bbrc.2007.10.194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gentile CL, Nivala AM, Gonzales JC, Pfaffenbach KT, Wang D, Wei Y, Jiang H, Orlicky DJ, Petersen DR, Pagliassotti MJ, Maclean KN. Experimental evidence for therapeutic potential of taurine in the treatment of nonalcoholic fatty liver disease. American journal of physiology Regulatory, integrative and comparative physiology. 2011;301:R1710–1722. doi: 10.1152/ajpregu.00677.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Shen J, Snapp EL, Lippincott-Schwartz J, Prywes R. Stable binding of ATF6 to BiP in the endoplasmic reticulum stress response. Molecular and cellular biology. 2005;25:921–932. doi: 10.1128/MCB.25.3.921-932.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.