

Abstract
Cryptococcus neoformans, similar to other eukaryotes, undergoes replicative aging. Replicative life spans have been determined for clinical C. neoformans strains, and although they are a reproducible trait, life spans vary considerably among strains. C. neoformans has been proposed as an ideal model organism to investigate the contribution of replicative aging in a fungal pathogen population to emerging phenotypic variation during chronic cryptococcal infections. C. neoformans cells of advanced generational age manifest a distinct phenotype; specifically, a larger cell size, a thicker cell wall, drug resistance, as well as resistance to hydrogen peroxide-mediated killing. Consequently, old cells are selected in the host environment during chronic infection and aging could be an unanticipated mechanism of pathogen adaptation that contributes to persistent disease. Aging as a natural process of phenotypic variation should be further studied as it likely is also relevant for other eukaryotic pathogen populations that undergo asymmetric replicative aging.
Keywords: C. neoformans, aging, pathogen, phenotypic variation
INTRODUCTION
Studies on aging have traditionally focused on fungi, such as Saccharomyces cerevisiae (Kaeberlein, 2010; Kennedy and Guarente, 1996; Smith et al., 2007) and Schizosaccharomyces pombe (Roux et al., 2010b; Roux et al., 2006) with the goal to improve human longevity. However, recent studies in pathogenic fungi, such as Candida albicans (Fu et al., 2008) and Cryptococcus neoformans (Bouklas et al., 2013; Cordero et al., 2011; Jain et al., 2009a) have focused on other aspects and revealed effects of aging on phenotypic variation in these pathogens. Specifically, studies have investigated how age-related phenotypic changes alter virulence-associated traits and change host pathogen interactions. Importantly, this work indicated that selection of older generation cells within the infecting pathogen population occurs throughout chronic infection. The resulting phenotypic variation and biological advantage conferred on old cells could affect the outcome and promote persistent disease. Thus, aging of eukaryotic pathogen populations may contribute to phenotypic variation, which is an unanticipated mechanism of pathoadaptation and should be further studied in relation to virulence and host pathogen interaction.
Phenotypic variation in C. neoformans
Over the years, several studies indicated that during the process of infection, C. neoformans can undergo “microevolution,” which produces phenotypic variants with altered virulence (Guerrero and Fries, 2008; Gupta and Fries, 2010; Jain et al., 2006a; McFadden et al., 2007; Ormerod et al., 2013; Pietrella et al., 2003). Initially, evidence for this phenomenon was derived from investigations with serial isolates from patients with chronic cryptococcosis. Comparison of virulence of these strains demonstrated significant differences in experimental murine infections (Fries and Casadevall, 1998) and also changes in the polysaccharide capsule (Cherniak et al., 1995). Extensive variability of capsule volume and induction was noted among clinical strains and found to correlate with phagocytosis indices (Zaragoza et al., 2003a). In addition, in vivo analysis of infecting fungal population in the murine host demonstrated dynamic capsule sizes and antibody binding patterns (Garcia-Hermoso et al., 2004). Furthermore, more pronounced capsule induction was observed in the lung environment when compared to capsules on C. neoformans cells in the brain environment (Rivera et al., 1998; Vartivarian et al., 1993). Capsular polysaccharide changes were shown to affect many aspects of this anti-opsonic shield. Capsular changes have been, for instance, associated with differing binding patterns of capsular antibodies, complement (Charlier et al., 2005), as well as eliciting different immune responses (Cheng et al., 2009), and importantly altering phagocytosis by macrophages (Zaragoza et al., 2003b). The latter is of particular importance because phagocytosis is important for the transmigration of C. neoformans across the blood-brain barrier, and therefore these studies predicted that capsule changes would affect dissemination (Charlier et al., 2009; Shi et al., 2010). It is important to emphasize that cell size variation can also occur independent of capsule induction. Increasing numbers of cells with larger body sizes were observed in tissue of mice chronically infected with C. neoformans (Feldmesser et al., 2001). Giant C. neoformans cells have been further investigated and are now referred to as titan cells (Okagaki et al., 2010; Zaragoza et al., 2010; Zaragoza and Nielsen, 2013). These cells are 30 – 50 μm large and polyploid. A less dramatic, but nonetheless significant and consistent cell size enlargement, is observed in the course of replicative aging of C. neoformans cells (Bouklas et al., 2013).
Replicative aging creates phenotypic variants within a fungal population
Cell size increase has been a hallmark of generational aging in all eukaryotic yeast cells examined to date (Bilinski and Bartosz, 2006; Yang et al., 2011), but certainly not all cell size differences reflect generational differences. The cell size increase in aging C. neoformans cells is much less impressive (6 – 15 μm) compared to that of titan cells. However, the ensuing size increase is observed consistently in all clinical C. neoformans strains examined to date (Figure 1). Analogous to older S. cerevisiae mother cells, the cell body size of replicating C. neoformans mother cells increases throughout the process of replicative aging (Jain et al., 2009a). C. neoformans strain cell size increase was found to be more gradual than described for Candida albicans (Jain et al., 2009a) and variable among strains (Bouklas et al., 2013). In S. cerevisiae, birth size has been shown to dictate the maximal size that is reached by cells at death (Yang et al., 2011). Specifically, a small birth size predicted a greater number of divisions, and hence a longer replicative life span in a systematic study of S. cerevisae mutants. However, in C. neoformans, birth size and replicative life span were uncorrelated in 360 examined cells derived from clinical and environmental C. neoformans strains (Figure 1). Similarly, in the prokaryotic pathogen, Mycobacterium smegmatis, birth and elongation lengths were uncorrelated (Aldridge et al., 2012). It is conveivable that birth size as a limitation to life span may be lost in pathogens, especially those that also exploit intracellular growth niches. In pathogens, complex selection mechanisms may affect cell size, which appears to be an emerging rather than a predetermined trait. More studies need to be done to shed light on the molecular mechanisms that regulate cell size in C. neoformans.
Figure 1. Birth size of clinical C. neoformans cells does not correlate with the strain’s replicative life span (RLS).
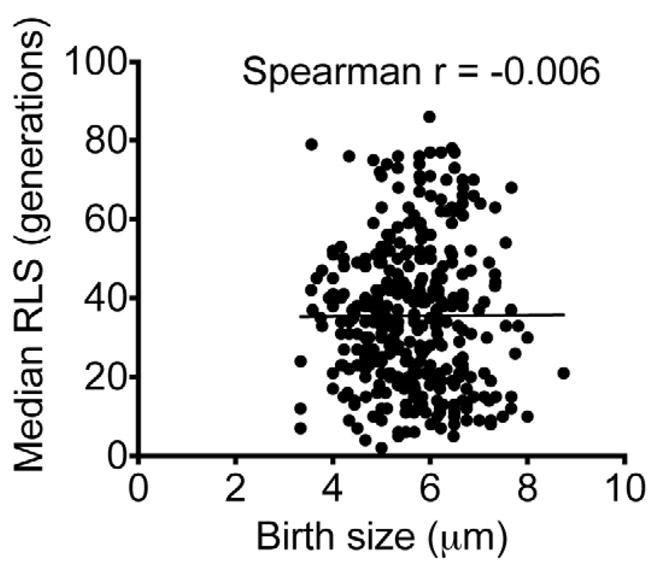
Birth cell body size of C. neoformans cells (n = 360) did not correlate with the median RLS of the strain (Spearman r = −0.006).
Another distinct finding of C. neoformans cells is that their bud scars (Woyke et al., 2002) heal during replication, such as during melanin-defect repairs (Nosanchuk and Casadevall, 2003), or chitooligosaccharide production (Rodrigues et al., 2008). Consequently, the cell wall of older C. neoformans cells does not become weak as in older S. cerevisiae cells, in which it can be successfully stained for bud scars (Powell et al., 2003). Instead, the C. neoformans cell wall grows thicker with age (Figure 2), which likely contributes to the resilience of older cells. Cell size increase of older cells proportionally affects both capsule and cell body, and thus is not the result of an over-induced capsule. With increase of cell size, the ability of older C. neoformans cells to resist phagocytosis by macrophages increases when compared to the younger and smaller cells of that strain; dramatic differences were observed within 10 generations (Bouklas et al., 2013). In addition, more pronounced inhibition of killing by macrophages was observed for older cells, and was most likely the result of enhanced resistance to hydrogen peroxide, which was also documented for older cells. Doubling times only change dramatically in the last third of the life span, and are not greatly affected in very young cells or in middle aged cells.
Figure 2. The cell walls of old C. neoformans cells are not weakened with age unlike those of old S. cerevisiae cells.

(A) Old S. cerevisiae cells can be stained with calcofluor for bud scars, which weaken the cell wall with each division. (B) Old C. neoformans cells cannot be accurately stained for bud scars, and the cell wall appears to thicken with each division.
Phenotypic switching in older cells and its implications for phenotypic variation
C. neoformans strains also undergo phenotypic switching (Fries et al., 1999; Fries et al., 2001; Goldman et al., 1998; Jain et al., 2005), which is defined as the spontaneous emergence of colonies with an altered colony morphology at a rate that is higher than that of somatic mutation (Slutsky et al., 1985). These phenotypic switch variants show enhanced virulence (Fries et al., 2001; Goldman et al., 1998; Guerrero et al., 2010; Jain et al., 2006a; Jain et al., 2009b; Jain et al., 2006b), a finding corroborated by similar observations in other fungal pathogens, such as C. glabrata (Lachke et al., 2002; Srikantha et al., 2005). Interestingly, a recent study (Bouklas et al., 2013) showed that phenotypic switching to a hypervirulent switch variant was also accompanied by a significant loss of replicative life span in the hypervirulent variant compared to the parent, which was at least partially reverted. Thus, life span and phenotypic switching may be regulated by overlapping complex epigenetic regulation, which ultimately could be activated in vivo. Furthermore, studies have also shown that aging of C. neoformans cells is accompanied by an increased phenotypic switching rate, which most likely is the result of age-related genomic instability (Jain et al., 2009a). These findings indicate that the emergence of older cells by itself or in concert with phenotypic switching could increase the heterogeneity of the pathogen population during infection. Sophisticated mathematical modeling has been done (Bouklas et al., 2013; Jain et al., 2009a) to assess the impact of this process.
Drug resistance in older cells and its implications for phenotypic variation
Time killing curves of young and old cells with different drug concentrations have shown that old C. neoformans cells resist killing by antifungals, such as amphotericin B (AMB) and fluconazole, better than young cells (Jain et al., 2009a). These data have also been confirmed in more recent studies (Bouklas et al., 2013) with clinical C. neoformans strains that were directly isolated from cerebrospinal fluid (CSF) of patients. Dramatic differences in killing were observed even with cells that were aged for 10 generations (0 – 50% killing of old cells compared to 100% killing of young cells with sub-therapeutic levels of AMB). Especially, resistance to AMB could be clinically relevant as this drug has poor central nervous system (CNS) penetration, and the fungus is potentially exposed to sub-therapeutic concentrations. Future studies will need to determine whether increased resistance of old C. neoformans cells to antifungal agents is the result of phenotypic drug tolerance, genetic drug resistance, or both. Electron microscopy images of old C. neoformans cells have documented a thickened cell wall in older cells (Figure 2), which could lead to insufficient drug penetration, a possible mechanism by which increased resistance is conferred upon older cells. It would also be reasonable to study the cell wall composition of older cells, which could have an altered sugar composition (Reese et al., 2007) or even lipid deposition (Rodrigues et al., 2007), and potentially serve as a trapping ground for antifungals before they can reach the cell membrane. Increased age could also lead to genomic instability (Jain et al., 2009a; McMurray and Gottschling, 2003), which in return may promote heteroresistance (Sionov et al., 2010) and chromosomal loss (Hu et al., 2011; Li et al., 2012; Ormerod and Fraser, 2013).
Comparison of “old cells” and titan cells
Of the described morphological forms of C. neoformans, old cells could be mistaken for titan cells. Titan cells, however, occur as early as 24 hours after inoculation in murine pulmonary infection, are 5 to 10 times larger than infecting C. neoformans cells, and are therefore not phagocytosed by macrophages (Okagaki and Nielsen, 2012). In contrast, old cells, which rise to a size approximately 30 – 63% larger than infecting cells are smaller than titan cells. They emerge late after weeks of infection in spinal fluids of rats because they are rare and require selection. Titan cells manifest an altered capsule, cell wall, ploidy, and resistance to nitrosative and oxidative stress (Zaragoza and Nielsen, 2013). Old cells are larger, but their size still permits uptake by macrophages, even though less easily with increasing age (Bouklas et al., 2013). Similar to titan cells, they demonstrate an altered cell wall (Figure 2), and resistance to oxidative stress (Bouklas et al., 2013). Whether these cells have increased ploidy or nitrosative resistance needs to be determined. Genome duplication, for instance, can occur in the absence of cell division during endoreplication in S. cerevisiae, where large cells with ploidy have also been observed (Kondorosi et al., 2000). Both titan and old cell morphologies may demonstrate a strategy for C. neoformans to generate phenotypic variation at different points and sites of infection. Titan cells, which account for 20% of cryptococcal cell population during pulmonary infection may be more relevant to earlier infection; whereas old cells, for which proportion is modeled to be high in meningitis may be more relevant to later CNS infection.
Selection of phenotypic variants within a cell population
Phenotypic variation can affect the entire fungal population, a subset of the fungal population, or only individual cells; the latter two require selection in order to dominate within a fungal population. Phenotypic changes, such as universal capsule enlargement commonly constitute a global response to an environmental signal like low iron, glucose, or carbon dioxide levels, and therefore this change manifests in all cells of the fungal population. As long as the environmental signal is operative, selection is not required. In contrast, epigenetically-regulated changes of the polysaccharide capsule that occur in the setting of phenotypic switching affect a smaller proportion of the fungal population. However, these capsular changes are stable and inherited by the progenies, and because they confer a biological advantage in vivo, the switch variants with a large mucoid capsule are selected and accumulate. Although significant differences in doubling times of C. neoformans variants are observed (Jain et al., 2006a; Jain et al., 2009b), host selection still contributes to their emergence because the slower growing mucoid variants persist and outgrow the faster growing parent smooth cells (Fries et al., 2001; Jain et al., 2009a).
One aspect that is unique about emerging older cells within a cryptococcal pathogen population is the fact that the trait being of “old age” is a global natural change that occurs eventually in all cells, but is not inherited by the next generation (Figure 3). Inheritance of “old age” trait occurs in S. cerevisiae cells only in the last 30% of their life span (Kennedy et al., 1994), or in long lived mutants (Delaney et al., 2011), and likely does not matter for the middle aged C. neoformans population that persists in the host during chronic infection (Figure 4). Therefore, the probability of emerging “old cells” is actually low unless significant selection occurs and young generations are preferentially killed, whereas older generations survive. Hence, high selection pressures that include the host immune cells and antifungal treatment contribute to the persistence of old cells, and shift the generational pyramid of pathogen population. In other fungi, there appears to be a trade-off between reproduction and longevity, as evidenced by S. cerevisiae mutants that extend life span, but reduce reproduction and fitness (Delaney et al., 2011). Mathematical modeling was used to show that these mutants had significant defects in their fitness that resulted from reduced maximal growth rate and a cell-cycle delay. Whether this is true in C. neoformans will need to be examined.
Figure 3. Schemata demonstrating the rare probability of finding old cells.
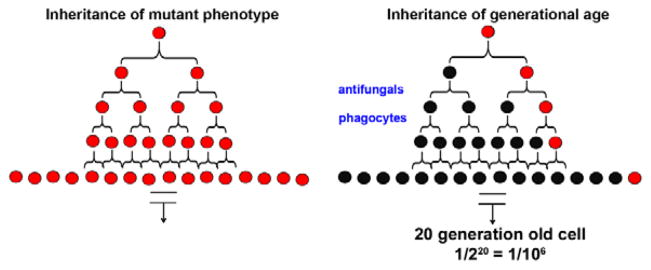
The probability of finding a mutant phenotype (red) in a clonally expanding population is 1/1 regardless of the number of replications because the phenotype is inherited by all progeny. However, the probability of finding a cell with a specific age (20-generation-old cell in red) is rare, and is 1/106 in a population that has replicated 20 times.
Figure 4. A model that depicts the relationship between replicative age and intracellular proliferation rate of C. neoformans.
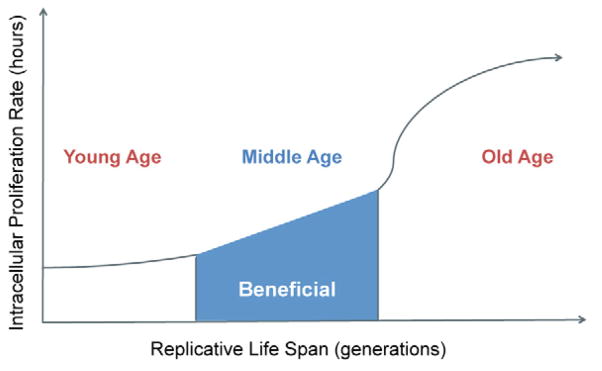
The pathogen is most likely to benefit during middle age, when it has acquired the old age phenotype and still proliferates at a rate comparable to that of young cells.
Phenotypic variation of C. neoformans in the host environment
Ideally relevance of phenotypic variations observed in vitro should be investigated in the human host environment. In patients with chronic cryptococcosis, high cryptococcal burden, neurocognitive impairment, and poor fungal clearance are markers of death and adverse outcome (Bicanic et al., 2009; Dromer et al., 2007). The factors responsible for these surrogate markers are still poorly understood. It is conceivable that the ability of C. neoformans to undergo phenotypic variation contributes to differences in clinical outcomes. The capsule has been established to be an important determinant of phenotypic variation (McFadden et al., 2007). Raised intracranial pressure (ICP) during chronic infection has been attributed to capsule shedding and/or the capsule on the organism itself (Fries et al., 2005; Robertson et al., 2014). Recent investigations have examined phenotype variation for the first time in C. neoformans cells derived directly from the spinal fluid (ex vivo) of infected patients enrolled in a standardized treatment trial in an attempt to correlate capsule size with ICP, fungal clearance, and CSF inflammation (Robertson et al., 2014). These data confirmed that the C. neoformans population in the spinal fluid is more heterogeneous than in vitro propagated C. neoformans populations. Thus variation of cell and capsule size is more consistent with an evolved population exposed to diverse selection pressures during chronic infection. Noteworthy, despite extensive size variation, titan cells (> 30 μm) were only observed in a small percentage (3.6%) of samples and seemed to occur in a subfraction of patients. Larger ex vivo capsule size always remained associated with ICP and decreased inflammatory response. In contrast, the degree of capsular polysaccharide shedding did not correlate with ICP, and most importantly ex vivo and in vitro capsule size did not correlate.
Another highly relevant finding with respect to phenotypic variation in clinical strains is that high fungal uptake into macrophages in vitro was associated with high CSF fungal burden in the patient who was infected with that strain and ultimately the patient’s survival (Sabiiti et al., 2014). As predicted by in vitro data (Zaragoza et al., 2008), high fungal uptake strains in this study had smaller capsules. In clinical strains “high fungal uptake strains” also manifested enhanced laccase activity, which would likely confer a higher degree of melanization. To date, laccase activity in old C. neoformans cells has not been compared to that in young cells. Interestingly, intracellular proliferation rate (IPR) was inversely correlated with uptake. This finding may also explain why younger smaller cells are more easily phagocytosed compared to older cells, but do not have a higher IPR, unless compared to extremely old cells. In the host, high fungal uptake may be important to improve transmigration across the BBB or dissemination to various organs, and the low IPR may result in a lower inflammatory response. Last, it is noteworthy that phenotypic switching has been observed in serotype A, D, and C strains, and in all cases was shown to result in variants with altered virulence (Guerrero et al., 2010; Jain and Fries, 2008). Recent analysis of clinical strains (Robertson et al., 2014) yielded predominantly smooth colony morphologies and established switched colonies were not detected on the plates (n = 200 to 1000 colonies plated) (T. Bicanic, personal communication).
Experimental challenges
Unfortunately to this date, technical methods are not available to establish the precise generational age of C. neoformans cells in human specimens. However recent data from our laboratory (Bouklas et al., 2013) addressed that question by determining the age of individual C. neoformans cells recovered from the spinal fluid and brains of chronically-infected rats. As discussed, generational age of C. neoformans cells cannot be determined by bud scar count alone as done in S. cerevisiae (Bouklas and Fries, 2013; Powell et al., 2003), but has to be determined indirectly through correlation of remaining life span with the overall life span potential of an individual C. neoformans strain. In addition many traits, such as mating and melanization are investigated in growing populations, which by default generates young cells and creates a generationally mixed yeast population. Consequently a causative relation cannot be established between the investigated trait and old phenotype. The above study established that ex vivo fungal populations derived from rats with chronic CNS cryptococcosis contain old cells as they accumulated through selection over the course of infection. Such a shift in generational distribution could also be demonstrated in two C. neoformans populations derived from spinal fluid of treated and untreated humans. While the study controlled for in vivo stresses that may result in adaptation to a shortened RLS, we acknowledge that epigenetic or genetic re-programming could have lead to a shortened RLS. An independent mechanism to identify older cells would be most beneficial. Additionally, demonstrating this in other pathogens, as well as further investigations on the transcriptional signature of old cells may shed some light on this issue, and help circumvent the challenge of more precise markers for the generational age of C. neoformans cells.
Relevance of replicative aging for phenotypic variation in other pathogens
Aging research in C. neoformans (Bouklas et al., 2013; Jain et al., 2009a) has demonstrated considerable variation in replicative life span among clinical strains and highlighted the importance of aging for the outcome of infection. Phenotypic variation is commonly described in encapsulated organisms, including Klebsiella, Neisseria, and Staphylococcus, which are easy to detect as they commonly change colony morphologies (Poolman et al., 1985; Proctor et al., 2006; Randall, 1939); however, this is certainly not limited to encapsulated organisms. Phenotypic changes as a consequence of aging have also been described for other eukaryotic organisms that are not encapsulated, such as Candida albicans (Fu et al., 2008) and Schizosaccharomyces pombe (Roux et al., 2010a). Now the relevance for pathogenesis has to be further investigated in other pathogenic fungi, such as Candida spp. It is conceivable that this unanticipated mechanism of generating phenotypic variation may even be relevant for prokaryotes, and may be exclusive of capsular or cell wall changes. It appears that cells of Mycobacterium species undergo acentric division and asymmetrical polar growth to give rise to unequal daughters (Singh et al., 2013), leading to differences in elongation lengths and growth rates (Joyce et al., 2012) that differentially affect the antibiotic susceptibilities, at least for M. smegmatis (Aldridge et al., 2012). The relevance of asymmetric division to other bacteria remains to be studied, but has been suggested in Caulobacter crescentus, where each division leads to one flagellated “swarmer” cell and one immotile “stalked” cell (Tsokos and Laub, 2012). Thus, in both eukaryotic and prokaryotic pathogens that undergo some form of asymmetric division, there is a potential for generation of phenotypic variants.
CONCLUSIONS
Fungal pathogens, such as C. neoformans, continue to evade the host immune system by successfully undergoing phenotypic variation during chronic infection. Hence, despite the use of widespread antifungal therapy, they continue to persist. In order to treat such persistent infections, it may be time to reassess our approaches to studying the pathogenesis of this fungal pathogen. The ability of the pathogen to replicate and age in the host poses a new challenge and adds another layer of complexity to the pathogenesis of C. neoformans. Thus, a reductionist approach to study variations that are inherited by the progeny may not be sufficient in light of the growing evidence, which indicates that not all variations are inherited. The distinct phenotype of old cells could confer an unanticipated virulence trait that could be relevant to the pathogenesis of other eukaryotic, and even some prokaryotic pathogens. Thus, aging should be further investigated in relation to host pathogen interactions.
Highlights.
Cryptococcus neoformans is capable of generating phenotypic variants during chronic infection.
C. neoformans undergoes replicative aging, which results in old cells with a distinct and conceivably advantageous phenotype.
Old cells have been shown to be selected in vivo.
Aging could generate phenotypic variation and contribute to cryptococcal persistence.
Acknowledgments
We thank Emily Cook and Neena Jain for their technical expertise. B.C.F. is supported by NIH award R01 AI059681.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aldridge BB, et al. Asymmetry and aging of mycobacterial cells lead to variable growth and antibiotic susceptibility. Science. 2012;335:100–4. doi: 10.1126/science.1216166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bicanic T, et al. Relationship of cerebrospinal fluid pressure, fungal burden and outcome in patients with cryptococcal meningitis undergoing serial lumbar punctures. AIDS. 2009;23:701–6. doi: 10.1097/QAD.0b013e32832605fe. [DOI] [PubMed] [Google Scholar]
- Bilinski T, Bartosz G. Hypothesis: cell volume limits cell divisions. Acta Biochim Pol. 2006;53:833–5. [PubMed] [Google Scholar]
- Bouklas T, Fries BC. Cryptococcus neoformans constitutes an ideal model organism to unravel the contribution of cellular aging to the virulence of chronic infections. Curr Opin Microbiol. 2013;16:391–7. doi: 10.1016/j.mib.2013.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouklas T, et al. Old Cryptococcus neoformans cells contribute to virulence in chronic cryptococcosis. M Bio. 2013;4 doi: 10.1128/mBio.00455-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charlier C, et al. Capsule structure changes associated with Cryptococcus neoformans crossing of the blood-brain barrier. Am J Pathol. 2005;166:421–32. doi: 10.1016/S0002-9440(10)62265-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charlier C, et al. Evidence of a role for monocytes in dissemination and brain invasion by Cryptococcus neoformans. Infect Immun. 2009;77:120–7. doi: 10.1128/IAI.01065-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng PY, et al. Cryptococcus gattii isolates from the British Columbia cryptococcosis outbreak induce less protective inflammation in a murine model of infection than Cryptococcus neoformans. Infect Immun. 2009;77:4284–94. doi: 10.1128/IAI.00628-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherniak R, et al. Variation in the structure of glucuronoxylomannan in isolates from patients with recurrent cryptococcal meningitis. Infect Immun. 1995;63:1899–905. doi: 10.1128/iai.63.5.1899-1905.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cordero RJ, et al. Chronological aging is associated with biophysical and chemical changes in the capsule of Cryptococcus neoformans. Infect Immun. 2011;79:4990–5000. doi: 10.1128/IAI.05789-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delaney JR, et al. Quantitative evidence for early life fitness defects from 32 longevity-associated alleles in yeast. Cell Cycle. 2011;10:156–65. doi: 10.4161/cc.10.1.14457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dromer F, et al. Determinants of disease presentation and outcome during cryptococcosis: the CryptoA/D study. PLoS Med. 2007;4:e21. doi: 10.1371/journal.pmed.0040021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feldmesser M, et al. Dynamic changes in the morphology of Cryptococcus neoformans during murine pulmonary infection. Microbiology. 2001;147:2355–65. doi: 10.1099/00221287-147-8-2355. [DOI] [PubMed] [Google Scholar]
- Fries BC, Casadevall A. Serial isolates of Cryptococcus neoformans from patients with AIDS differ in virulence for mice. J Infect Dis. 1998;178:1761–6. doi: 10.1086/314521. [DOI] [PubMed] [Google Scholar]
- Fries BC, et al. Phenotypic switching in Cryptococcus neoformans results in changes in cellular morphology and glucuronoxylomannan structure. Infect Immun. 1999;67:6076–83. doi: 10.1128/iai.67.11.6076-6083.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fries BC, et al. Phenotypic switching of Cryptococcus neoformans can produce variants that elicit increased intracranial pressure in a rat model of cryptococcal meningoencephalitis. Infect Immun. 2005;73:1779–87. doi: 10.1128/IAI.73.3.1779-1787.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fries BC, et al. Phenotypic switching of Cryptococcus neoformans occurs in vivo and influences the outcome of infection. J Clin Invest. 2001;108:1639–48. doi: 10.1172/JCI13407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu XH, et al. Candida albicans, a distinctive fungal model for cellular aging study. Aging cell. 2008;7:746–57. doi: 10.1111/j.1474-9726.2008.00424.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Hermoso D, et al. Cryptococcus neoformans capsule structure evolution in vitro and during murine infection. Infect Immun. 2004;72:3359–65. doi: 10.1128/IAI.72.6.3359-3365.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldman DL, et al. Phenotypic switching in the human pathogenic fungus Cryptococcus neoformans is associated with changes in virulence and pulmonary inflammatory response in rodents. Proc Natl Acad Sci U S A. 1998;95:14967–72. doi: 10.1073/pnas.95.25.14967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guerrero A, Fries BC. Phenotypic switching in Cryptococcus neoformans contributes to virulence by changing the immunological host response. Infect Immun. 2008;76:4322–31. doi: 10.1128/IAI.00529-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guerrero A, et al. Cryptococcus neoformans variants generated by phenotypic switching differ in virulence through effects on macrophage activation. Infect Immun. 2010;78:1049–57. doi: 10.1128/IAI.01049-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta G, Fries BC. Variability of phenotypic traits in Cryptococcus varieties and species and the resulting implications for pathogenesis. Future Microbiol. 2010;5:775–87. doi: 10.2217/fmb.10.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu G, et al. Variation in chromosome copy number influences the virulence of Cryptococcus neoformans and occurs in isolates from AIDS patients. BMC Genomics. 2011;12:526. doi: 10.1186/1471-2164-12-526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain N, et al. Isolation and characterization of senescent Cryptococcus neoformans and implications for phenotypic switching and pathogenesis in chronic cryptococcosis. Eukaryot Cell. 2009a;8:858–66. doi: 10.1128/EC.00017-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain N, Fries BC. Phenotypic switching of Cryptococcus neoformans and Cryptococcus gattii. Mycopathologia. 2008;166:181–8. doi: 10.1007/s11046-008-9137-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain N, et al. Phenotypic switching and its implications for the pathogenesis of Cryptococcus neoformans. FEMS yeast research. 2006a;6:480–8. doi: 10.1111/j.1567-1364.2006.00039.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain N, et al. Loss of allergen 1 confers a hypervirulent phenotype that resembles mucoid switch variants of Cryptococcus neoformans. Infect Immun. 2009b;77:128–40. doi: 10.1128/IAI.01079-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain N, et al. Phenotypic switching in a Cryptococcus neoformans variety gattii strain is associated with changes in virulence and promotes dissemination to the central nervous system. Infect Immun. 2006b;74:896–903. doi: 10.1128/IAI.74.2.896-903.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain N, et al. Molecular epidemiology of clinical Cryptococcus neoformans strains from India. J Clin Microbiol. 2005;43:5733–42. doi: 10.1128/JCM.43.11.5733-5742.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joyce G, et al. Cell division site placement and asymmetric growth in mycobacteria. PLoS One. 2012;7:e44582. doi: 10.1371/journal.pone.0044582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaeberlein M. Lessons on longevity from budding yeast. Nature. 2010;464:513–9. doi: 10.1038/nature08981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennedy BK, et al. Daughter cells of Saccharomyces cerevisiae from old mothers display a reduced life span. J Cell Biol. 1994;127:1985–93. doi: 10.1083/jcb.127.6.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennedy BK, Guarente L. Genetic analysis of aging in Saccharomyces cerevisiae. Trends Genet. 1996;12:355–9. [PubMed] [Google Scholar]
- Kondorosi E, et al. Plant cell-size control: growing by ploidy? Curr Opin Plant Biol. 2000;3:488–92. doi: 10.1016/s1369-5266(00)00118-7. [DOI] [PubMed] [Google Scholar]
- Lachke SA, et al. Phenotypic switching and filamentation in Candida glabrata. Microbiology. 2002;148:2661–74. doi: 10.1099/00221287-148-9-2661. [DOI] [PubMed] [Google Scholar]
- Li W, et al. Genetic Diversity and Genomic Plasticity of Cryptococcus neoformans AD Hybrid Strains. G3 (Bethesda) 2012;2:83–97. doi: 10.1534/g3.111.001255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McFadden DC, et al. Capsule structural heterogeneity and antigenic variation in Cryptococcus neoformans. Eukaryot Cell. 2007;6:1464–73. doi: 10.1128/EC.00162-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMurray MA, Gottschling DE. An age-induced switch to a hyper-recombinational state. Science. 2003;301:1908–11. doi: 10.1126/science.1087706. [DOI] [PubMed] [Google Scholar]
- Nosanchuk JD, Casadevall A. Budding of melanized Cryptococcus neoformans in the presence or absence of L-dopa. Microbiology. 2003;149:1945–51. doi: 10.1099/mic.0.26333-0. [DOI] [PubMed] [Google Scholar]
- Okagaki LH, Nielsen K. Titan cells confer protection from phagocytosis in Cryptococcus neoformans infections. Eukaryot Cell. 2012;11:820–6. doi: 10.1128/EC.00121-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okagaki LH, et al. Cryptococcal cell morphology affects host cell interactions and pathogenicity. PLoS Pathog. 2010;6:e1000953. doi: 10.1371/journal.ppat.1000953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ormerod KL, Fraser JA. Balancing stability and flexibility within the genome of the pathogen Cryptococcus neoformans. PLoS Pathog. 2013;9:e1003764. doi: 10.1371/journal.ppat.1003764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ormerod KL, et al. Comparative Genomics of Serial Isolates of Cryptococcus neoformansReveals Gene Associated with Carbon Utilization and Virulence. G3 (Bethesda) 2013 doi: 10.1534/g3.113.005660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pietrella D, et al. Phenotypic switching of Cryptococcus neoformans can influence the outcome of the human immune response. Cell Microbiol. 2003;5:513–22. doi: 10.1046/j.1462-5822.2003.00297.x. [DOI] [PubMed] [Google Scholar]
- Poolman JT, et al. Colony variants of Neisseria meningitidis strain 2996 (B:2b:P1.2): influence of class-5 outer membrane proteins and lipopolysaccharides. J Med Microbiol. 1985;19:203–9. doi: 10.1099/00222615-19-2-203. [DOI] [PubMed] [Google Scholar]
- Powell CD, et al. Chitin scar breaks in aged Saccharomyces cerevisiae. Microbiology. 2003;149:3129–37. doi: 10.1099/mic.0.25940-0. [DOI] [PubMed] [Google Scholar]
- Proctor RA, et al. Small colony variants: a pathogenic form of bacteria that facilitates persistent and recurrent infections. Nat Rev Microbiol. 2006;4:295–305. doi: 10.1038/nrmicro1384. [DOI] [PubMed] [Google Scholar]
- Randall WA. Colony and Antigenic Variation in Klebsiella pneumoniae Types A, B and C. J Bacteriol. 1939;38:461–77. doi: 10.1128/jb.38.4.461-477.1939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reese AJ, et al. Loss of cell wall alpha(1-3) glucan affects Cryptococcus neoformans from ultrastructure to virulence. Mol Microbiol. 2007;63:1385–98. doi: 10.1111/j.1365-2958.2006.05551.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivera J, et al. Organ-dependent variation of capsule thickness in Cryptococcus neoformans during experimental murine infection. Infect Immun. 1998;66:5027–30. doi: 10.1128/iai.66.10.5027-5030.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robertson EJ, et al. Cryptococcus neoformans ex vivo capsule size is associated with intracranial pressure and host immune response in HIV-associated cryptococcal meningitis. J Infect Dis. 2014;209:74–82. doi: 10.1093/infdis/jit435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodrigues ML, et al. Binding of the wheat germ lectin to Cryptococcus neoformans suggests an association of chitinlike structures with yeast budding and capsular glucuronoxylomannan. Eukaryot Cell. 2008;7:602–9. doi: 10.1128/EC.00307-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodrigues ML, et al. Vesicular polysaccharide export in Cryptococcus neoformans is a eukaryotic solution to the problem of fungal trans-cell wall transport. Eukaryot Cell. 2007;6:48–59. doi: 10.1128/EC.00318-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roux AE, et al. A screen for genes involved in respiration control and longevity in Schizosaccharomyces pombe. Ann N Y Acad Sci. 2010a;1197:19–27. doi: 10.1111/j.1749-6632.2010.05198.x. [DOI] [PubMed] [Google Scholar]
- Roux AE, et al. Fission yeast and other yeasts as emergent models to unravel cellular aging in eukaryotes. J Gerontol A Biol Sci Med Sci. 2010b;65:1–8. doi: 10.1093/gerona/glp152. [DOI] [PubMed] [Google Scholar]
- Roux AE, et al. Regulation of chronological aging in Schizosaccharomyces pombe by the protein kinases Pka1 and Sck2. Aging cell. 2006;5:345–57. doi: 10.1111/j.1474-9726.2006.00225.x. [DOI] [PubMed] [Google Scholar]
- Sabiiti W, et al. Efficient phagocytosis and laccase activity affect the outcome of HIV-associated cryptococcosis. J Clin Invest. 2014;124:2000–8. doi: 10.1172/JCI72950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi M, et al. Real-time imaging of trapping and urease-dependent transmigration of Cryptococcus neoformans in mouse brain. J Clin Invest. 2010;120:1683–93. doi: 10.1172/JCI41963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh B, et al. Asymmetric growth and division in Mycobacterium spp.: compensatory mechanisms for non-medial septa. Mol Microbiol. 2013;88:64–76. doi: 10.1111/mmi.12169. [DOI] [PubMed] [Google Scholar]
- Sionov E, et al. Cryptococcus neoformans overcomes stress of azole drugs by formation of disomy in specific multiple chromosomes. PLoS pathogens. 2010;6:e1000848. doi: 10.1371/journal.ppat.1000848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slutsky B, et al. High-frequency switching of colony morphology in Candida albicans. Science. 1985;230:666–9. doi: 10.1126/science.3901258. [DOI] [PubMed] [Google Scholar]
- Smith ED, et al. Genome-wide identification of conserved longevity genes in yeast and worms. Mech Ageing Dev. 2007;128:106–11. doi: 10.1016/j.mad.2006.11.017. [DOI] [PubMed] [Google Scholar]
- Srikantha T, et al. Phenotypic switching in Candida glabrata accompanied by changes in expression of genes with deduced functions in copper detoxification and stress. Eukaryot Cell. 2005;4:1434–45. doi: 10.1128/EC.4.8.1434-1445.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsokos CG, Laub MT. Polarity and cell fate asymmetry in Caulobacter crescentus. Curr Opin Microbiol. 2012;15:744–50. doi: 10.1016/j.mib.2012.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vartivarian SE, et al. Regulation of cryptococcal capsular polysaccharide by iron. J Infect Dis. 1993;167:186–90. doi: 10.1093/infdis/167.1.186. [DOI] [PubMed] [Google Scholar]
- Woyke T, et al. Effect of auristatin PHE on microtubule integrity and nuclear localization in Cryptococcus neoformans. Antimicrob Agents Chemother. 2002;46:3802–8. doi: 10.1128/AAC.46.12.3802-3808.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, et al. Cell size and growth rate are major determinants of replicative lifespan. Cell Cycle. 2011;10:144–55. doi: 10.4161/cc.10.1.14455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaragoza O, et al. Capsule enlargement in Cryptococcus neoformans confers resistance to oxidative stress suggesting a mechanism for intracellular survival. Cell Microbiol. 2008;10:2043–57. doi: 10.1111/j.1462-5822.2008.01186.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaragoza O, et al. Induction of capsule growth in Cryptococcus neoformans by mammalian serum and CO(2) Infect Immun. 2003a;71:6155–64. doi: 10.1128/IAI.71.11.6155-6164.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaragoza O, et al. Fungal cell gigantism during mammalian infection. PLoS Pathog. 2010;6:e1000945. doi: 10.1371/journal.ppat.1000945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaragoza O, Nielsen K. Titan cells in Cryptococcus neoformans: cells with a giant impact. Curr Opin Microbiol. 2013;16:409–13. doi: 10.1016/j.mib.2013.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaragoza O, et al. The efficacy of complement-mediated phagocytosis of Cryptococcus neoformans is dependent on the location of C3 in the polysaccharide capsule and involves both direct and indirect C3-mediated interactions. Eur J Immunol. 2003b;33:1957–67. doi: 10.1002/eji.200323848. [DOI] [PubMed] [Google Scholar]