

Abstract
Histone deacetylase inhibitors (HDACi) are novel clinical anticancer drugs that inhibit HDAC gene expression and induce cell apoptosis in human cancers. Nevertheless, the detailed mechanism or the downstream HDAC targets by which HDACi mediates apoptosis in human breast cancer cells remains unclear. Here, we show that HDACi reduce tumorigenesis and induce intrinsic apoptosis of human breast cancer cells through the microRNA miR-125a-5p in vivo and in vitro. Intrinsic apoptosis was activated by the caspase 9/3 signaling pathway. In addition, HDACi mediated the expression of miR-125a-5p by activating RUNX3/p300/HDAC5 complex. Subsequently, miR-125a-5p silenced HDAC5 post-transcriptionally in the cells treated with HDACi. Thus, a regulatory loop may exist in human breast cancer cells involving miR-125a-5p and HDAC5 that is controlled by RUNX3 signaling. Silencing of miR-125a-5p and RUNX3 inhibited cancer progression and activated apoptosis, but silencing of HDAC5 had a converse effect. In conclusion, we demonstrate a possible new mechanism by which HDACi influence tumorigenesis and apoptosis via downregulation of miR-125a-5p expression. This study provides clinical implications in cancer chemotherapy using HDACi.
Introduction
Histone deacetylases (HDACs), which target lysine residues, are epigenetic determinants that are essential for transcriptional regulation because they promote chromatin condensation.1 Cytoplasmic HDACs (nonhistone HDACs) have been shown to play central roles in mediating processes related to human cancers, including promoting tumor progression and metastases and inhibiting apoptosis.2 Consequently, histone deacetylase inhibitors (HDACi) are promising anticancer drugs and several are currently the focus of clinical trials, such as Trichostatin A (TSA) and valproic acid, which decrease HDAC expression.3 TSA and valproic acid stimulate multiple pathways leading to apoptosis, induced cell death, and cell growth arrest in adult patients with solid tumors.4 TSA delays tumor growth and induces apoptosis by mediating expression of Ki-67, matrix metalloprotease 2 (MMP2), caspase 3, and Bcl-xL.5,6 Furthermore, HDACi have been useful as a chemotherapy strategy to restrain proliferation, dedifferentiation, and self-renewal of cancer stem-like cells.7 However, the specific mechanisms by which HDACi affect these cells are largely unknown. We previously showed that HDACs mediate tumorigenesis8 and the epithelial–mesenchymal transition9 in a highly malignant human breast cancer stem-like cells (CSCs) line. These CSCs carry stem-cell markers (CD44+/CD24-, ER+, HER2/neu+) and are capable of self renewal or pluripotent differentiation, making them extremely malignant.10
HDACi can also rapidly alter microRNA (miRNA) levels11 to induce cell death in thyroid cancer cells12 and to inhibit growth via a p53-independent pathway in neuroblastoma SH-SY5Y cells.13 Nevertheless, the exact mechanisms by which HDACi mediates CSCs apoptosis through miRNA remain unknown. MiRNAs are small (~21–23 nucleotides) noncoding RNAs that reduce the post-transcriptional stability of target mRNAs.14 MiRNA hybridization with target mRNAs induces mRNA degradation and inhibits translation, thereby regulating a variety of cellular processes, such as proliferation, apoptosis, senescence, differentiation, and death.15 Several specific miRNAs have been shown to act as tumor suppressors or as oncogenes in cancers.16
In the present work, we demonstrate that HDACi mediate apoptosis through miRNA in human breast cancer cells and we identify possible downstream target genes. This study revealed a novel pathway by which HDACi promotes apoptosis.
Results
HDACi modulate miR-125a-5p expression in breast cancer stem-like cells
To determine whether miRNAs were induced by HDACi in human breast cancer cells, the Human 384 SeraMir miRNA profiler was used to assess two breast cancer cell lines treated with the HDACi TSA, a breast cancer stem cell line (R2N1d) and a metastasis-type breast cancer cell line (MDA-MB-231) (Figure 1a). To compare treatment with control cells, 380 miRNAs were measured and the top miRNA most significantly (>10-fold) increased/decreased miRNAs in the TSA-treated cells relative to control cells are shown in Figure 1b and Supplementary Table S1. miR-125a-5p, miR-150, miR-362-3p, miR-503, miR-133a, let-7c, miR-548b-5p, let-7b, miR-149, miR-512-5p, miR-29c, miR-513c, and miR-187 were induced by TSA, but miR-331-5p, miR-33b, miR-192, miR-195, let-7i, miR-541, miR-200b, miR-146-3p, Hsa-miR-200a, miR-193a-5p, miR-99b, miR-34b, and miR-373 were decreased by TSA in both R2N1d and MDA-MB-231 cells. MiR-125a-5p was the most highly induced by TSA treatment in both human breast cancer lines. We further confirmed that HDACi can induce miR-125a-5p expression with qRT-PCR of breast cancer cells treated with six different HDACi (TSA, valproic acid, sodium butyrate, splitomycin, apicidin, and M344). We found that miR-125a-5p increased in a concentration-dependent manner with each of the HDACi (Figure 1c).
Figure 1.
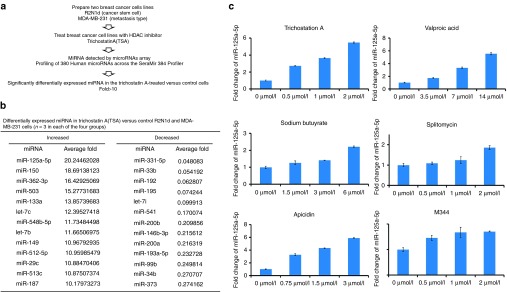
miR-125a-5p is induced by histone deacetylase inhibitors (HDACi). (a) MiRNA profiles were examined in Trichostatin A-treated R2N1d and MDA-MB-231 cells. (b) Differentially expressed miRNAs (>10-fold) in the Trichostatin A (TSA)-treated cells relative to the control cells (n = 3). (c) R2N1d cells were treated with different concentrations of the HDACi Trichostatin A (0, 0.5, 1, and 2 µmol/l), valproic acid (0, 3.5, 7, and 14 µmol/l), sodium butyrate (0, 1.5, 3, and 6 µmol/l), splitomycin (0, 0.5, 1, and 2 µmol/l), apicidin (0, 0.75, 1.5, and 3 µmol/l), or M344 (0, 0.5, 1, and 2 µmol/l), and miR-125a-5p expression was detected with qRT-PCR 48 hours post-treatment. Values are the mean ± standard deviation (SD) of three experiments.
miR-125a-5p positively regulates cell apoptosis through caspase 9/3 in vitro
Previous studies have reported that HDACi induce apoptosis through the intrinsic pathway in cancer cells. Therefore, we examined whether miR-125a-5p regulates apoptosis in the two breast cancer cell lines. First, we examined miR-125a-5p expression with a dose-dependent manner of miR-125a-5p plasmid by q-PCR. We found that miR-125a-5p expression was positively correlated with indicated doses in both R2N1d (Figure 2a) and MDA-MB-231 (Supplementary Figure S1a) cells. In addition, we investigated cell cycle progression by flow cytometry and found that overexpression of miR-125a-5p caused a significant increase in cell cycle arrest at the sub-G1 phase and a decrease in the proportion of cells in G1 phase in both R2N1d (Figure 2b) and MDA-MB-231 (Supplementary Figure S1b) cells. Cell cycle arrest at the sub-G1 phase frequently leads to cell death through apoptosis.17 Consistent with this, TUNEL assays showed that miR-125a-5p overexpression significantly enhanced apoptosis in both R2N1d (Figure 2c) and MDA-MB-231 (Supplementary Figure S1c) cells compared with the control cells.
Figure 2.

miR-125a-5p mediates the apoptosis signaling pathway. The control miRNA (5 μg/ml), miR-125a-5p (5 μg/ml), anti-miR-125a-5p (150 nmol/l), or anti-miR-125a-5p (150 nmol/l) + miR-125a-5p (5 μg/ml) were transfected into R2N1d cells and the cells assessed at 48 hours post-transfection. (a) MiR-125a-5p expression was analyzed by q-PCR with a dose-dependent manner of miR-125a-5p plasmid. (b) The cell cycle stage of the transfected cells was evaluated with propidium iodide staining and flow cytometry. (c) Apoptosis was evaluated with the TUNEL assay. (d) The apoptosis pathway was evaluated with a caspase activity assay, and (e,f) the apoptosis markers caspase 3 and Bcl-xL were evaluated by western blotting with β-actin as a loading control. Values are the mean ± SD of three experiments. *P < 0.05 versus untreated control; two-tailed Student's t-test.
Next, a caspase activity assay showed that miR-125a-5p overexpression activated caspase 3 and 9 activity in both R2N1d (Figure 2d) and MDA-MB-231 (Supplementary Figure S2a) cells. We used specific inhibitors of caspases 2, 3, 8, and 9 to further analyze the apoptosis signaling pathway. The caspase 9 inhibitor blocked the miR-125a-5p–mediated increase in caspase 3 activation (Supplementary Figure S2c), but the caspase 3 inhibitor did not affect caspase 9 activation (Supplementary Figure S2e). Furthermore, caspase 2 and 8 were not activated by overexpression of miR-125a-5p in the breast cancer cell lines (Supplementary Figure S2b,d). To further confirm that miR-125a-5p promotes apoptosis, we examined the levels of caspase 3, and Bcl-xL by immunoblotting. We found that active caspase 3 was induced, but Bcl-xL decreased in R2N1d (Figure 2e,f) and MDA-MB-231 (Supplementary Figure S2f,g) cells overexpressing miR-125a-5p. These findings suggested that miR-125a-5p overexpression functions in the same way as HDACi to induce the intrinsic apoptosis pathway through caspases 9 and 3.
HDAC5 is the direct target of miR-125a-5p and mediates its cellular function
We next used RepTar and RNAhybrid,18,19 a computational motif prediction method, to identify miR-125a-5p target genes in the human genome (Supplementary Table S2). RNAhybrid calculates the binding thermodynamic between miRNA sequences and the 3′-UTRs of putative target genes. RepTar and RNAhybrid identified HDAC5 as a possible target, with two free energy value for binding in the 3′-UTR of −22.3 (Supplementary Figure S3a) and −32.9 kcal/mol (Figure 3a). RepTar prediction software found that 3′UTR of HDAC5 has two miR-125a-5p targeting site located at ~425 and 710 bp downstream of the HDAC5 3′ start (Supplementary Figure S3a). Therefore, the full length, 427~449 bp and 708~734 bp fragment of 3′-UTR was cloned into a luciferase reporter vector and transfecting into HEK-293T cells. The results found that full length and 708~734 bp fragment of HDAC5 3′-UTR was decreased by miR-125a-5p, but 427~449 bp signal was not (Supplementary Figure S3b). We next examined whether miR-125a-5p could directly silence the biological function of HDAC5 through binding the 3′-UTR (708~734 bp fragment) by cloning the wild-type (WT) and mutated (MT) 3′-UTR into a luciferase reporter vector and transfecting these plasmids, along with different concentrations of the miR-125a-5p expression plasmid into HEK-293T cells. We found that the HDAC5 WT luciferase signal was decreased by miR-125a-5p in a concentration-dependent manner, but the HDAC5 MT signal was not (Figure 3b). To verify these results, we investigated whether miR-125a-5p affected HDAC5 protein levels. The levels of HDAC5 were lower in miR-125a-5p–overexpressing cells compared with control cells, whereas the levels of other HDACs (HDAC7, HDAC10) were not affected by miR-125a-5p (Figure 3c).
Figure 3.

HDAC5 is the direct target for miR-125a-5p. (a) RNAhybrid predicted miR-125a-5p binding sites and free energy values (−32.9 kcal/mol) in the 3′-UTR of HDAC5. The wild-type (WT, black letters) and mutated (MT, red letters) 3′-UTR of the HDAC5 sequence were cloned into a luciferase reporter plasmid. (b) HEK-293T cells were cotransfected with different ratios of miR-125a-5p and the plasmids containing the wild-type (WT) or mutant (MT) 3′-UTR of HDAC5. Luciferase activity was analyzed with the dual luciferase system. Activity of the WT and MT constructs was normalized to firefly/Renilla luciferase activity. (c) Cells were transfected as in Figure 2 and HDAC5, 7, and 10 protein expression was examined by western blotting 72 hours later. β-actin served as a loading control. (d) Cells were transfected with control-siRNA or HDAC5-siRNA-1/2 (5 μg/ml) and HDAC5, Ki-67, MMP2, caspase 3 and Bcl-xL levels were examined by western blotting 72 hours later. R2N1d cells were transfected as in (d) and 48 hours after transfection, the cell growth (e), apoptosis rates (f), wound healing (HDAC5-siRNA-1) (g), and invasion (HDAC5-siRNA-1) (h) were evaluated. Values are the mean ± SD of three experiments. *P < 0.05 versus untreated control; two-tailed Student's t-test.
We also analyzed the biological functions of HDAC5 in R2N1d and MDA-MB-231 cells. Silencing of HDAC5 with siRNA-1/2 resulted in decreased cell growth, wound healing and invasion (Figure 3e,g,h and Supplemetary Figure S4a,c,d), but increased apoptosis (Figure 3f and Supplementary Figure S4b). Immunoblotting also showed that Ki-67, active MMP2, and Bcl-xL levels decreased, but active caspase 3 levels increased when HDAC5 was silenced in human breast cancer cells (Figure 3d). Hence, these data indicate that HDAC5 levels are controlled by miR-125a-5p and that silencing of HDAC5 has the same biological effect as increasing miR-125a-5p in human breast cancer cells.
HDACi induce miR-125a-5p expression via the RUNX3/p300/HDAC5 complex
To further analyze the underlying mechanism by which HDACi affect miR-125a-5p, the sequence of the miR-125a-5p promoter region was examined with the TFSEARCH program for potential transcription factor binding sites. We identified a potential p300 binding motif (TGACTCCCTCTTATT) in the proximal region of the promoter (Figure 4a). Pre-miR-125a was located at chr19: 51693254-339 and has two clustered miRNAs in the upstream sequence including pre-mir-99b (chr19: 51692612-81) and pre-let-7e (chr19: 51692786-864). We found that pre-miR-125a and pre-mir-99b-let-7e may have independent promoters and only one P300 putative binding site located at ~160 bp upstream of the pre-miR-125a 5′ end (Supplementary Figure S5). Interestingly, a previous study demonstrated that HDACi increase acetylated RUNX3 through inhibition of HDAC5, which in turn enhances binding of the RUNX3-p300 complex to target promoters.20 Therefore, we hypothesized that HDACi induce miR-125a-5p expression by decreasing HDAC5 expression and acetylating RUNX3, leading to enhanced binding of the RUNX3/p300 complex to the miR-125a-5p promoter in human breast cancer. To test this hypothesis, RUNX3, p300, and HDAC5 were silenced with siRNA in the presence and absence of TSA, and the association of the RUNX3/p300/HDAC5 complex with miR-125a-5p was examined. MiR-125a-5p expression was essentially blocked by different concentrations of RUNX3 siRNA (Supplementary Figure S6a) and p300 siRNA (Supplementary Figure S6b). On the other hand, HDAC5 siRNA (Supplementary Figure S6c) promoted miR-125a-5p expression when cells were treated with TSA (Figure 4b). Immunoprecipitation assays showed that acetylated RUNX3 increased in the presence of TSA and HDAC5 siRNA (Figure 4c). Additionally, ChIP showed that RUNX3, but not HDAC5, was required for p300 to bind the miR-125a-5p promoter (Figure 4d). These results suggested that RUNX3 may play an important role in tumor suppression through miR-125a-5p. Indeed, upon RUNX3 silencing, levels of Ki-67, active MMP2, and Bcl-xL increased, whereas active caspase 3 decreased in both R2N1d and MDA-MB-231 cells (Figure 4e), and this was accompanied by increased cell growth, wound healing, and invasion (Figure 4f–h).
Figure 4.

Histone deacetylase inhibitors (HDACi) induce synthesis of miR-125a-5p via the RUNX3/p300/HDAC5 complex. (a) Schematic diagram of the miR-125a-5p promoter showing the one predicted p300 binding site (red). (b) R2N1d cells were treated with Trichostatin A (TSA) or transfected with HDAC5 siRNA-1 at the indicated doses, and HDAC5 expression was analyzed by immunoblotting (IB) with β-actin as a loading control. Protein–protein interactions with RUNX3 and acetyl-lysine were analyzed with immunoprecipitation. (c) R2N1d cells were transfected with RUNX3 siRNA-1 (5 μg/ml), p300 siRNA (5 μg/ml), or HDAC5 siRNA-1 (5 μg/ml) then treated with TSA, and miR-125a-5p expression was analyzed with qRT-PCR 48 hours after treatment. (d) Binding of the transcription factor p300 to miR-125a-5p was analyzed with anti-p300 ChIP. (e–h) R2N1d and MDA-MB-231 cells were transfected with control siRNA (5 μg/ml) or RUNX3 siRNA-1/2 (5 μg/ml). At 48 hours post-transfection, RUNX3, Ki-67, MMP2, caspase 3, and Bcl-xL proteins expression was analyzed by western blotting (e) with β-actin as a loading control, and the cell growth (f), invasion (RUNX3-siRNA-1) (g), and wound healing (RUNX3-siRNA-1) (h) rates were evaluated. Values are the mean ± SD of three experiments. *P < 0.05 versus untreated control; two-tailed Student's t-test.
HDAC5 participates in a positive feedback mechanism through miR-125a-5p
HDAC5 appears to act upstream of miR-125a-5p in the apoptosis-induction pathway and may mediate miR-125a-5p expression in human breast cancer. We hypothesized that HDAC5 may participate in a positive feedback loop mediated through miR-125a-5p. To test this hypothesis, we analyzed miR-125a-5p expression following overexpression of HDAC5 in R2N1d cells by qRT-PCR. We found that miR-125a-5p expression decreased in a HDAC5 concentration-dependent manner (Figure 5a,b). Converse, miR-125a-5p expression was induced by silencing of HDAC5 in a dose-dependent manner (Figure 5c,d). Additionally, cell growth increased with overexpression of HDAC5 in both cell lines (Figure 5e). We also found that overexpression of HDAC5 abolished miR-125a-5p-induced apoptosis (Figure 5f) and increased migration (Figure 5g) in R2N1d. Thus, knocking down HDAC5 will mimic the effect of TSA on miR-125a expression. HDAC5 and miR-125a-5p appear to operate through a positive feedback mechanism via the RUNX3/p300 complex to affect tumor growth, metastasis and apoptosis in human breast cancer.
Figure 5.

miR-125a-5p is repressed by HDAC5. R2N1d cells were transfected with the indicated amounts of an HDAC5 expression or silencing plasmid (μg/ml). At 24 hours post-transfection, miR-125a-5p expression was detected using western blot and qRT-PCR (a–d), and the cell growth rate was evaluated by determining XTT assay (e). The control miRNA (5 μg/ml), miR-125a-5p (5 μg/ml), HDAC5 (5 μg/ml), or miR-125a-5p (5 μg/ml) + HDAC5 (5 μg/ml) were transfected into R2N1d cells and the cells assessed at 48 hours post-transfection. (f) Apoptosis was evaluated with the TUNEL assay and the wound healing rates were evaluated (g). Values are the mean ± SD of three experiments. *P < 0.05 versus untreated control; Two-tailed Student's t-test.
HDACi inhibit cancer stem-like cell tumorigenesis through miR-125a-5p
We next examined whether HDACi inhibit cancer stem-like cell tumorigenesis via miR-125a-5p targeting of HDAC5 in vivo. R2N1d-YFP and R2N1d-GFP-mir-125a-5p cells were implanted into immunodeficient nude mice and tumor growth was monitored, respectively. After 1 week, the mice were intratumorally injected with TSA (500 μg/kg) or normal saline once every 2 days for 30 days. Mir-125a-5p and TSA inhibited R2N1d cell tumorigenesis as indicated by a decrease in the fluorescence signal by in vivo imaging (Figure 6a) and tumor photon flux (Figure 6b).
Figure 6.

Histone deacetylase inhibitors (HDACi) induce miR-125a-5p to inhibit tumorigenesis. R2N1d-YFP and R2N1d-GFP-mir-125a-5p cells were injected into the right flank of immunodeficient mice (n = 6 per group), and 1 week later, Trichostatin A (TSA) (500 μg/kg body weight) or normal saline was injected into the tumor once every 2 days for 30 days. (a) Tumor images were captured by in vivo imaging and (b) photon flux was measured with the AxioVision Software. (c) Tumor thin sections were analyzed with fluorescence microscopy, hematoxylin and eosin (H&E) staining, TUNEL, in situ hybridization with miR-125a-5p and immunohistochemistry with anti-HDAC5. (d) A model is reported in which TSA determine RUNX3/p300 complex to the miR-125a-5p promoter through acetylating of RUNX3 and accordingly miR-125a-5p upregulation, gives rise to HDAC5 downregulation and subsequent apoptosis activation.
In addition, fluorescence microscopy, TUNEL assay, in situ miR-125a-5p hybridization, and HDAC5 immunochemistry of the tumor tissues, showed similar results (Figure 6c) to those observed in vitro. Specifically, the R2N1d-YFP fluorescence signal in xenograft tumor sections remained constant, whereas the apoptosis-inducing ability or the expression of miR-125a-5p increased, but expression of HDAC5 decreased with TSA treatment.
Discussion
In this study, we used a commercial human miRNA profiler, to identify miRNAs associated with HDACi treatment in human breast cancer stem-like cells. We identified and validated the induction of miR-125a-5p by HDACi, which activated the RUNX3/p300/HDAC5 complex and mediated cell apoptosis through a feedback loop with the target gene HDAC5 (Figure 6d). We further showed that miR-125a-5p and HDAC5 modulate the tumorigenesis of cancer stem-like cells in vivo/vitro and may be appropriate clinical indicators to monitor the success of TSA treatment in breast cancer patients.
Among the differentially expressed miRNAs identified by the profiler, miR-125a-5p, miR-150,21 miR-362-3p,22 miR-503,23 miR-133a,24 let-7c,25 miR-548b-5p, let-7b,25 miR-149,26 miR-512-5p, miR-29c,27 miR-513c, and miR-187 emerged as the most consistently increased, suggesting that these miRNA may potentially act as tumor suppressors.
HDACi have been evaluated in clinical trials as potential anticancer drugs,28 and TSA has shown great promise as a clinical therapy for human breast cancer.29,30 HDACi decrease tumorigenesis and induce apoptosis through the intrinsic apoptosis pathway in different cancer types.31 Interestingly, we found that miR-125a-5p mediated the intrinsic apoptosis pathway through caspases 9 and 3, providing a potential mechanism for the induction of the intrinsic apoptosis pathway by HDACi and identifying miR-125a-5p as a potential therapeutic target for HDACi. Previous studies also have showed that class I HDAC inhibitor, entinostat induced apoptosis through miR-125a, miR-125b, and miR-205 in erbB2-overexpressing breast cancer cells.32 Therefore, HDACi positively have the ability to induce miR-125a-5p expression and mediate apoptosis. MiR-125a-5p has been reported to be downregulated in non-small cell lung cancer tissues, and to decrease migration and invasion of human lung cancer cell lines.33 MiR-125a-5p interacts with hepatitis B virus surface antigen and directly suppresses its activity.34 In addition, miR-125a-5p decreases cell growth more potently when combined with trastuzumab in the treatment of gastric cancer,35 and mediates apoptosis of human lung cancer cells through a p53-dependent pathway.36 These previous findings are similar to our observation that miR-125a-5p promotes apoptosis in human breast cancer stem cells. Therefore, miR-125a-5p appears to play an important role in promoting cell apoptosis by targeting apoptosis-related genes in multiple cancer types. Furthermore, an early report found that miR-125a-5p targets proapoptotic protein, BAK1 to suppress apoptosis in immature hematopoietic stem cells37 and thus revealing that miR-125a-5p biology can assume varied roles.
HDAC5 is a member of the class II HDAC family (HDAC4, 5, 6, 9, and 11) and localizes in both the nucleus and cytoplasm. HDAC4 and HDAC5 are highly similar enzymes, with an overall sequence identity of ~70%.38 Early studies indicated that miR-2861 represses HDAC5 expression to enhance bone morphogenetic protein 2-induced osteoblastogenesis.39 Interesting, we found that in addition to directly targeting HDAC5, miR-125a-5p can also directly target HDAC4 and inhibit its protein expression (data not shown). This result reveals that, not only do HDACs regulate miRNA expression, but miRNAs can also reciprocally control HDAC activity. In addition, high levels of HDAC5 are significantly associated with poor survival in human brain cancer patients and knockdown of HDAC5 enhances apoptosis through caspase 3 (ref. 40). Consistent with these results, we found that silencing of HDAC5 inhibits cell growth, migration, and invasion and increases apoptosis in human breast cancer stem-like cells.
RUNX3 is a tumor suppressor41 that controls gene expression by interacting with p300 and HDAC5 (ref. 20). RUNX3 promotes apoptosis in K-Ras-activated lung cancer cells42 and transcriptionally activates the proapoptotic gene Bim in transforming growth factor-β-induced apoptosis.43 Similarly, we found that RUNX3 inhibits cell invasion and migration, and induces apoptosis in human breast cancer stem cells. Acetylating of RUNX3 enhanced binding of the RUNX3/p300 complex to the miR-125a-5p promoter and silencing of RUNX3 has the same biological effect as decreasing miR-125a-5p in human breast cancer cells. Therefore, we believe that RUNX3 is an important role for transcription regulations and biological effect of miR-125a-5p. Our data further indicated that HDAC5 silencing in breast cancer cells is due to increased miR-125a-5p and its inhibitory effect on HDAC5. Thus, RUNX3 may mediate apoptosis through a regulatory loop involving miR-125a-5p and HDAC5.
In summary, our study identified a critical regulatory RUNX3/p300/HDAC5/miR-125a-5p loop network that modulates HDAC5 levels when HDACi are used to treat breast cancer cells. Furthermore, miR-125a-5p and RUNX3 appear to play similar biological roles in mediating apoptosis and tumorigenesis in human breast cancer stem cells. Thus, the present study provides valuable insight that will be of great use in clinical applications of HDACi.
Materials and Methods
Cell lines and drug treatment. The MDA-MB-231 human breast cancer cell line was purchased from the American Type Culture Collection (Manassas, VA) and maintained in Dulbecco's modified Eagle's medium/F12 (Life Technologies, Grand Island, NY) supplemented with 10% fetal bovine serum, and 5% penicillin, streptomycin, and amphotericin B (equal parts). The R2N1d human breast cancer stem cells (a kind gift from Professor Chang, Michigan State University) were grown in MSU-1 medium and cultured as described.9 All cells were grown at 37 °C in 5% CO2. For drug treatment, the cells were seeded at a concentration of 1 × 105 cells per six-well plate 24 hours before treatment with HDACi (TSA, valproic acid, sodium butyrate, splitomycin, apicidin, and M344, BioVision, Mountain View, CA) for 1 day.
miRNA profiling using multiplex quantitative reverse transcription (qRT)-PCR. Expression profiles of MiRNA were obtained with the Human 384 SeraMir miRNA Profiler (System Biosciences, Mountain View, CA). Briefly, miRNA was converted to cDNA using a SeraMir Exosome RNA Amplification Kit (System Biosciences). The kit contains 380 mature human miRNA qRT-PCR primers that were used in the SYBRGreen 7900 HT fast real-time PCR system (Applied Biosystems, Foster City, CA).
miRNA and mRNA quantification. Total RNA was extracted from cell lines using TRIzol Reagent (Invitrogen, Paisley, UK) and the miRNA was amplified with the TaqMan MicroRNA Reverse Transcription kit (Applied Biosystems). The RT primers for miR-125a-5p (mature miRNA Sequence: UCCCUGAGACCCUUUAACCUGUG) and miR-16 (refs. 44,45) (for normalization; mature miRNA Sequence: UAGCAGCACGUAAAUAUUGGCG) were obtained from Applied Biosystems. mRNA levels were quantified with the ABI Prism 7000 Sequence Detection System (Applied Biosystems) using the comparative CT method (2^-ΔΔCT).
Flow cytometry. Cells were transfected with the miR-125a-5p plasmid with or without the miR-125a-5p inhibitor (anti-miR-125a-5b) using the TurboFect Transfection Reagent (Fermentas, Hanover, MD). After transfection (24 hours), the cells were resuspended by 0.25% trypsin-EDTA (Gibco, Grand Island, NY) and stained with propidium iodide (Sigma, St Louis, MO) followed by cell cycle analysis with an EPICS XL-MCL cytometer (Beckman Coulter, Fullerton, CA).
Caspase activity and terminal deoxynucleotidyl transferase dUTP nick-end labeling (TUNEL) assays. Caspase activity was analyzed with ApoAlert Caspase assay plates (Clontech, Palo Alto, CA). Briefly, the cells were transfected with the miR-125a-5p plasmid and then incubated for 24 hours in the presence or absence of individual caspase-specific inhibitors (Clontech). The cells were harvested with 0.25% trypsin-EDTA (Gibco) and the caspase activity was detected in the ApoAlert plate with an ELISA fluorescent plate reader (Multiskan EX, excitation: 380 nm; emission: 460 nm). For the TUNEL assay, apoptosis was detected using the Apo-BrdU-red DNA Fragmentation Assay Kit by following the manufacturer's instructions (BioVision). The image of nuclei was captured by fluorescence microscopy and the quantity was obtained by Flow cytometry.
Immunoblot analysis and immunoprecipitation. For immunoblotting, cells were lysed with Radio-Immunoprecipitation assay buffer on ice for 1 h. Proteins were resolved by SDS-PAGE and transferred to Hybond nitrocellulose membranes (GE Healthcare Europe GmbH, Pollards Wood, UK). The membranes were blocked in 5% non-fat milk in PBS/Tween-20(5%) and hybridized with antibodies against Bcl-xL (1:1,000, Cell Signaling Technology, Beverly, MA), caspase 3 (1:1,000, Cell Signaling Technology), HDAC family members (HDAC5, 7, and 10; 1:1,000, BioVision), Ki-67 (1:1,000, Sigma), MMP2 (1:1,000, Cell Signaling Technology), RUNX3 (1:1,000, Epitomics, Burlingame, CA), and acetyl-lysine (1:1,000, Abcam, Cambridge, MA). For immunoprecipitation, the lysates were incubated with protein-G beads (Roche Applied Science, Indianapolis, IN), anti-IgG beads (Santa Cruz Biotechnology, Santa Cruz, CA), or anti-p300 beads (1:1,000, Epitomics) for 24 hours at 4 °C with rocking. The immunoprecipitated complexes were washed and used for immunoblotting.
Luciferase assay. The luciferase reporter plasmid carrying the wild-type or mutated HDAC5 3′-untranslated region (UTR) (pGL3-HDAC5-WT-3′-UTR and pGL3-HDAC5-MT-3′-UTR, respectively) was transfected into HEK-293T cells along with the miR-125a-5p plasmid with the TurboFect Transfection Reagent (Fermentas, Vilnius, Lithuania). After transfection (24 hours), cells were lysed and luciferase activity was measured with the Dual-Luciferase Reporter Assay system (Promega, Madison, WI). The sequences of HDAC5-WT-3′-UTR and HDAC5-MT-3′-UTR are showed in Figure 3a.
Cell growth, invasion, and wound-healing assays. The cell growth, invasion, and wound-healing assays were performed as described.46
Chromatin immunoprecipitation. Chromatin immunoprecipitation (ChIP) assays were performed with the EZ-ChIP kit (Millipore) using anti-P300 (1:500, Santa Cruz Biotechnology), and the following miR-125a-5p promoter primers: forward: 5′-CTGTGTCTCTTTCACAGTGG-3′ and reverse: 5′-CGGGAGGGGCAGAGACCTAG-3′. Non-immune IgG was used for negative control and the positive control of input chromatin was amplified before immunoprecipitation and mock immunoprecipitated chromatin. The PCR products quantitative were separated by 1% agarose gels.
Lentiviral infection. To produce yellow fluorescent protein (YFP) and Green fluorescent protein (GFP)-mir-125a-5p-expressing R2N1d cells, pLKO.1 lentiviral supernatants were produced using a standard RNAi Core Facility protocol (Academia Sinica, Taipei, Taiwan). R2N1d cells were infected for 48 hours with the lentiviral supernatants and then the stable clone cell lines (R2N1d-YFP and R2N1d-GFP-mir-125a-5p) were selected with puromycin.
Tumor cell xenograft study. The experimental procedures complied with the Kaohsiung Medical University animal care and use standards and the study was approved by the Institutional Animal Care and Use Committee (approval number: 101036). Female BALB/cAnN.Cg-Foxn1nu/Crl-Narl mice (~4–6 weeks old) from the National Laboratory Animal Center (Taipei, Taiwan) were used for xenograft experiments. R2N1d-YFP cells were suspended in 200 μl of medium (MSU-1) and injected subcutaneously into the flanks of 12 immunodeficient nude mice. After 1 week, half of the mice were injected with TSA and the other half with normal saline once every 2 days for 30 days. Fluorescence was measured 30 days after injection using an in vivo imaging system (Xenogen, Alameda, CA).
In situ hybridization and immunohistochemistry. In situ hybridization in cryosections of the xenografted human breast cancer tissue was performed using a 5′-digoxygenin–labeled miR-125a-5p miRCURY LNA detection probe (5′-TCACAGGTTAAAGGGTCTCAGGGA-3′; Exiqon, Vedbaek, Denmark) and an IsHyb In Situ Hybridization kit (BioChain, Hayward, CA). Briefly, the human miR-125a-5p detection probe was hybridized to tissues sections mounted on glass slides and images were captured on a DM16000 B microscope (Leica, Bannockburn, IL). For immunohistochemistry, the optimal cutting temperature compound–embedded cryopreserved tissues were cut into 5-μm-thick sections and the immunostaining with HDAC5 antibodies (1:1,000, BioVision) and counterstaining of nuclei with hematoxylin and eosin were performed with the Dako LSAB kit (Dako, Carpinteria, CA).
Transfection assay, oligonucleotides, and anti-miR miRNA. The TurboFect Transfection Reagent (Fermentas, Hanover, MD) was used to transfect the following overexpression plasmid (µg/ml) and small hairpin RNAs (shRNAs) (µg/ml) into R2N1d and MDA-MB-231 cells by following the manufacturer's instructions: pLKO.TRC-miR-125a-5p: 5′-UCCCUGAGACCCUUUAACCUGUG-3′, pLKO.1-HDAC5 shRNA-1: 5′-GCTAGAGAAAGTCATCGAGAT-3′, pLKO.1-HDAC5 shRNA-2: 5′-GCCGGGTTTGATGCTGTTGAA-3′, pLKO.1-RUNX3 shRNA-1: 5′-ACCACCTCTACTACGGGACAT-3′, and pLKO.1-RUNX3 shRNA-2: 5′-GTTCAACGACCTTCGCTTCGT-3′. Anti-miR miRNA inhibitors (Ambion, Austin, TX) are chemically modified oligonucleotides which were single-stranded nucleic acids synthesized to inhibit specifically endogenous miRNAs. The cells were transfected with anti-miR inhibitors (anti-mir-125a-5p, 150 nmol/l) based on the manufacturer's instructions.
SUPPLEMENTARY MATERIAL Figure S1. miR-125a-5p mediates the apoptosis of MDA-MB-231 Figure S2. miR-125a-5p Induces Apoptosis Through the Caspase9/3 Signaling Pathway Figure S3. miR-125a-5p post-transcriptionally downregulates HDAC5 expression by targeting its 3′-UTR. Figure S4. HDAC5 Regulate Biological Function in MDA-MB-231 Breast Cancer Cells Figure S5. The separate transcriptional map of three-miRNA cluster Figure S6. The RUNX3/P300/HDAC5 Complex Mediates miR-125a-5p Expression Table S1. Differentially expressed miRNA in Trichostatin A (TSA) vs. control R2N1d and MDA-MB-231 cells (n = 3 in each of the four groups) Table S2. A total of 3,974 genes as candidates for miR-125a-5p indirect targets
Acknowledgments
This work was supported by the Ministry of Science and Technology of Taiwan (grant numbers 102-2628-B-037-011-MY3 and 102-2632-B-037-001-MY3) and the Kaohsiung Medical University (Hospital) Research Fund “Aim for the Top Universities Grant, grant No. KMU-TP103G01, KMU-TP103G04, KMU-TP103G05, KMU-TP103A15, KMUH103-3R26). We thank the Center for Research Resources and Development of Kaohsiung Medical University for lentivirus infection technical support, the Laboratory Animal Center of Kaohsiung Medical University for assistance with animal experiments, and the National RNAi Core Facility for providing the shRNAs. The authors declared no conflict of interest.
Supplementary Material
References
- Strahl BD, Allis CD. The language of covalent histone modifications. Nature. 2000;403:41–45. doi: 10.1038/47412. [DOI] [PubMed] [Google Scholar]
- Glozak MA, Sengupta N, Zhang X, Seto E. Acetylation and deacetylation of non-histone proteins. Gene. 2005;363:15–23. doi: 10.1016/j.gene.2005.09.010. [DOI] [PubMed] [Google Scholar]
- Johnstone RW. Histone-deacetylase inhibitors: novel drugs for the treatment of cancer. Nat Rev Drug Discov. 2002;1:287–299. doi: 10.1038/nrd772. [DOI] [PubMed] [Google Scholar]
- Dokmanovic M, Clarke C, Marks PA. Histone deacetylase inhibitors: overview and perspectives. Mol Cancer Res. 2007;5:981–989. doi: 10.1158/1541-7786.MCR-07-0324. [DOI] [PubMed] [Google Scholar]
- El-Zawahry A, Lu P, White SJ, Voelkel-Johnson C. In vitro efficacy of AdTRAIL gene therapy of bladder cancer is enhanced by trichostatin A-mediated restoration of CAR expression and downregulation of cFLIP and Bcl-XL. Cancer Gene Ther. 2006;13:281–289. doi: 10.1038/sj.cgt.7700905. [DOI] [PubMed] [Google Scholar]
- Medina V, Edmonds B, Young GP, James R, Appleton S, Zalewski PD. Induction of caspase-3 protease activity and apoptosis by butyrate and trichostatin A (inhibitors of histone deacetylase): dependence on protein synthesis and synergy with a mitochondrial/cytochrome c-dependent pathway. Cancer Res. 1997;57:3697–3707. [PubMed] [Google Scholar]
- Jones PA, Baylin SB. The epigenomics of cancer. Cell. 2007;128:683–692. doi: 10.1016/j.cell.2007.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsieh TH, Tsai CF, Hsu CY, Kuo PL, Hsi E, Suen JL.et al. (2012n-Butyl benzyl phthalate promotes breast cancer progression by inducing expression of lymphoid enhancer factor 1 PLoS One 7e42750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsieh TH, Tsai CF, Hsu CY, Kuo PL, Lee JN, Chai CY.et al. (2012Phthalates stimulate the epithelial to mesenchymal transition through an HDAC6-dependent mechanism in human breast epithelial stem cells Toxicol Sci 128365–376. [DOI] [PubMed] [Google Scholar]
- Wang KH, Kao AP, Chang CC, Lee JN, Hou MF, Long CY.et al. (2010Increasing CD44+/CD24(-) tumor stem cells, and upregulation of COX-2 and HDAC6, as major functions of HER2 in breast tumorigenesis Mol Cancer 9288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott GK, Mattie MD, Berger CE, Benz SC, Benz CC. Rapid alteration of microRNA levels by histone deacetylase inhibition. Cancer Res. 2006;66:1277–1281. doi: 10.1158/0008-5472.CAN-05-3632. [DOI] [PubMed] [Google Scholar]
- Brest P, Lassalle S, Hofman V, Bordone O, Gavric Tanga V, Bonnetaud C.et al. (2011MiR-129-5p is required for histone deacetylase inhibitor-induced cell death in thyroid cancer cells Endocr Relat Cancer 18711–719. [DOI] [PubMed] [Google Scholar]
- Jin H, Liang L, Liu L, Deng W, Liu J. HDAC inhibitor DWP0016 activates p53 transcription and acetylation to inhibit cell growth in U251 glioblastoma cells. J Cell Biochem. 2013;114:1498–1509. doi: 10.1002/jcb.24491. [DOI] [PubMed] [Google Scholar]
- Bartel DP. MicroRNAs: target recognition and regulatory functions. Cell. 2009;136:215–233. doi: 10.1016/j.cell.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang TC, Mendell JT. microRNAs in vertebrate physiology and human disease. Annu Rev Genomics Hum Genet. 2007;8:215–239. doi: 10.1146/annurev.genom.8.080706.092351. [DOI] [PubMed] [Google Scholar]
- Gandellini P, Profumo V, Folini M, Zaffaroni N. MicroRNAs as new therapeutic targets and tools in cancer. Expert Opin Ther Targets. 2011;15:265–279. doi: 10.1517/14728222.2011.550878. [DOI] [PubMed] [Google Scholar]
- Darzynkiewicz Z, Juan G, Li X, Gorczyca W, Murakami T, Traganos F. Cytometry in cell necrobiology: analysis of apoptosis and accidental cell death (necrosis) Cytometry. 1997;27:1–20. [PubMed] [Google Scholar]
- Elefant N, Berger A, Shein H, Hofree M, Margalit H, Altuvia Y. RepTar: a database of predicted cellular targets of host and viral miRNAs. Nucleic Acids Res. 2011;39 Database issue:D188–D194. doi: 10.1093/nar/gkq1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krüger J, Rehmsmeier M. RNAhybrid: microRNA target prediction easy, fast and flexible. Nucleic Acids Res. 2006;34 Web Server issue:W451–W454. doi: 10.1093/nar/gkl243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin YH, Jeon EJ, Li QL, Lee YH, Choi JK, Kim WJ.et al. (2004Transforming growth factor-beta stimulates p300-dependent RUNX3 acetylation, which inhibits ubiquitination-mediated degradation J Biol Chem 27929409–29417. [DOI] [PubMed] [Google Scholar]
- Ma Y, Zhang P, Wang F, Zhang H, Yang J, Peng J.et al. (2012miR-150 as a potential biomarker associated with prognosis and therapeutic outcome in colorectal cancer Gut 611447–1453. [DOI] [PubMed] [Google Scholar]
- Christensen LL, Tobiasen H, Holm A, Schepeler T, Ostenfeld MS, Thorsen K.et al.; COLOFOL steering group 2013MiRNA-362-3p induces cell cycle arrest through targeting of E2F1, USF2 and PTPN1 and is associated with recurrence of colorectal cancer Int J Cancer 13367–78. [DOI] [PubMed] [Google Scholar]
- Forrest AR, Kanamori-Katayama M, Tomaru Y, Lassmann T, Ninomiya N, Takahashi Y.et al. (2010Induction of microRNAs, mir-155, mir-222, mir-424 and mir-503, promotes monocytic differentiation through combinatorial regulation Leukemia 24460–466. [DOI] [PubMed] [Google Scholar]
- Kano M, Seki N, Kikkawa N, Fujimura L, Hoshino I, Akutsu Y.et al. (2010miR-145, miR-133a and miR-133b: Tumor-suppressive miRNAs target FSCN1 in esophageal squamous cell carcinoma Int J Cancer 1272804–2814. [DOI] [PubMed] [Google Scholar]
- Esquela-Kerscher A, Trang P, Wiggins JF, Patrawala L, Cheng A, Ford L.et al. (2008The let-7 microRNA reduces tumor growth in mouse models of lung cancer Cell Cycle 7759–764. [DOI] [PubMed] [Google Scholar]
- Lin RJ, Lin YC, Yu AL. miR-149* induces apoptosis by inhibiting Akt1 and E2F1 in human cancer cells. Mol Carcinog. 2010;49:719–727. doi: 10.1002/mc.20647. [DOI] [PubMed] [Google Scholar]
- Pass HI, Goparaju C, Ivanov S, Donington J, Carbone M, Hoshen M.et al. (2010hsa-miR-29c* is linked to the prognosis of malignant pleural mesothelioma Cancer Res 701916–1924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim HJ, Bae SC. Histone deacetylase inhibitors: molecular mechanisms of action and clinical trials as anti-cancer drugs. Am J Transl Res. 2011;3:166–179. [PMC free article] [PubMed] [Google Scholar]
- Vigushin DM, Ali S, Pace PE, Mirsaidi N, Ito K, Adcock I.et al. (2001Trichostatin A is a histone deacetylase inhibitor with potent antitumor activity against breast cancer in vivo Clin Cancer Res 7971–976. [PubMed] [Google Scholar]
- Alao JP, Stavropoulou AV, Lam EW, Coombes RC, Vigushin DM. Histone deacetylase inhibitor, trichostatin A induces ubiquitin-dependent cyclin D1 degradation in MCF-7 breast cancer cells. Mol Cancer. 2006;5:8. doi: 10.1186/1476-4598-5-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolden JE, Peart MJ, Johnstone RW. Anticancer activities of histone deacetylase inhibitors. Nat Rev Drug Discov. 2006;5:769–784. doi: 10.1038/nrd2133. [DOI] [PubMed] [Google Scholar]
- Wang S, Huang J, Lyu H, Lee CK, Tan J, Wang J.et al. (2013Functional cooperation of miR-125a, miR-125b, and miR-205 in entinostat-induced downregulation of erbB2/erbB3 and apoptosis in breast cancer cells Cell Death Dis 4e556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang L, Huang Q, Zhang S, Zhang Q, Chang J, Qiu X.et al. (2010Hsa-miR-125a-3p and hsa-miR-125a-5p are downregulated in non-small cell lung cancer and have inverse effects on invasion and migration of lung cancer cells BMC Cancer 10318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potenza N, Papa U, Mosca N, Zerbini F, Nobile V, Russo A. Human microRNA hsa-miR-125a-5p interferes with expression of hepatitis B virus surface antigen. Nucleic Acids Res. 2011;39:5157–5163. doi: 10.1093/nar/gkr067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishida N, Mimori K, Fabbri M, Yokobori T, Sudo T, Tanaka F.et al. (2011MicroRNA-125a-5p is an independent prognostic factor in gastric cancer and inhibits the proliferation of human gastric cancer cells in combination with trastuzumab Clin Cancer Res 172725–2733. [DOI] [PubMed] [Google Scholar]
- Jiang L, Huang Q, Chang J, Wang E, Qiu X. MicroRNA HSA-miR-125a-5p induces apoptosis by activating p53 in lung cancer cells. Exp Lung Res. 2011;37:387–398. doi: 10.3109/01902148.2010.492068. [DOI] [PubMed] [Google Scholar]
- Guo S, Lu J, Schlanger R, Zhang H, Wang JY, Fox MC.et al. (2010MicroRNA miR-125a controls hematopoietic stem cell number Proc Natl Acad Sci USA 10714229–14234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Ruijter AJ, van Gennip AH, Caron HN, Kemp S, van Kuilenburg AB. Histone deacetylases (HDACs): characterization of the classical HDAC family. Biochem J. 2003;370 Pt 3:737–749. doi: 10.1042/BJ20021321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Xie H, Liu W, Hu R, Huang B, Tan YF.et al. (2009A novel microRNA targeting HDAC5 regulates osteoblast differentiation in mice and contributes to primary osteoporosis in humans J Clin Invest 1193666–3677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milde T, Oehme I, Korshunov A, Kopp-Schneider A, Remke M, Northcott P.et al. (2010HDAC5 and HDAC9 in medulloblastoma: novel markers for risk stratification and role in tumor cell growth Clin Cancer Res 163240–3252. [DOI] [PubMed] [Google Scholar]
- Bae SC, Choi JK. Tumor suppressor activity of RUNX3. Oncogene. 2004;23:4336–4340. doi: 10.1038/sj.onc.1207286. [DOI] [PubMed] [Google Scholar]
- Lee YS, Lee JW, Jang JW, Chi XZ, Kim JH, Li YH.et al. (2013Runx3 inactivation is a crucial early event in the development of lung adenocarcinoma Cancer Cell 24603–616. [DOI] [PubMed] [Google Scholar]
- Yano T, Ito K, Fukamachi H, Chi XZ, Wee HJ, Inoue K.et al. (2006The RUNX3 tumor suppressor upregulates Bim in gastric epithelial cells undergoing transforming growth factor beta-induced apoptosis Mol Cell Biol 264474–4488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wittmann J, Jäck HM. Serum microRNAs as powerful cancer biomarkers. Biochim Biophys Acta. 2010;1806:200–207. doi: 10.1016/j.bbcan.2010.07.002. [DOI] [PubMed] [Google Scholar]
- Mitchell PS, Parkin RK, Kroh EM, Fritz BR, Wyman SK, Pogosova-Agadjanyan EL.et al. (2008Circulating microRNAs as stable blood-based markers for cancer detection Proc Natl Acad Sci USA 10510513–10518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsieh TH, Tsai CF, Hsu CY, Kuo PL, Lee JN, Chai CY.et al. (2012Phthalates induce proliferation and invasiveness of estrogen receptor-negative breast cancer through the AhR/HDAC6/c-Myc signaling pathway FASEB J 26778–787. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.