

Abstract
Despite the medical, social, and economic impact of obesity, only a few therapeutic options, focused largely on reducing caloric intake, are currently available and these have limited success rates. A major impediment is that any challenge by caloric restriction is counterbalanced by activation of systems that conserve energy to prevent body weight loss. Therefore, targeting energy-conserving mechanisms to promote energy expenditure is an attractive strategy for obesity treatment. Here, in order to suppress muscle energy efficiency, we target sarcolemmal ATP-sensitive potassium (KATP) channels which have previously been shown to be important in maintaining muscle energy economy. Specifically, we employ intramuscular injections of cell-penetrating vivo-morpholinos to prevent translation of the channel pore-forming subunit. This intervention results in significant reduction of KATP channel expression and function in treated areas, without affecting the channel expression in nontargeted tissues. Furthermore, suppression of KATP channel function in a group of hind limb muscles causes a substantial increase in activity-related energy consumption, with little effect on exercise tolerance. These findings establish a proof-of-principle that selective skeletal muscle targeting of sarcolemmal KATP channel function is possible and that this intervention can alter overall bodily energetics without a disabling impact on muscle mechanical function.
Introduction
Despite the medical, social, and economic impact of obesity,1,2,3 only a few therapeutic options, focused largely on reducing caloric intake or absorption, are currently available and these have limited success rates.1,3,4,5 A major impediment is that any challenge by caloric restriction is counterbalanced by activation of systems that conserve energy to prevent body weight loss.4,6,7 Therefore, targeting energy-conserving mechanisms to promote energy expenditure is an attractive strategy for obesity treatment.
Skeletal muscles account for about 40–50% of body mass and their function relies on the very energy demanding processes of ion homeostasis and actin-myosin cycling, driven by myofiber ATPases.4,8 However, this ATP utilization not only supports active cellular functions, but also results in heat or entropy production, defined as thermogenesis. The ratio of energy consumption contributing toward “useful work” versus thermogenesis defines muscle energy efficiency. Considering the high volume of ATP turnover in skeletal muscles, even small changes in their energy efficiency could have a substantial effect on overall bodily energy consumption, rendering any level of exercise more effective for caloric utilization, and weight loss.4,9 This is particularly important since, although diet and exercise remain the mainstays of body weight control, there are numerous obstacles to increasing exercise activity in overweight or obese individuals.
Recently, we discovered that muscle energy efficiency is regulated by sarcolemmal ATP-sensitive potassium (KATP) channels,9,10,11,12 formed through association of Kir6.2 pore-forming and SUR2A regulatory sulfonylurea receptor subunits.13 These channels have the unique ability to sense changes in cellular energy availability (i.e., alterations in local ATP/ADP concentration) and translate them into constraints on cellular energy utilization through alterations in action potentials that govern calcium dynamics.9,10,11,12,14–20 Disruption of KATP channel function renders muscles less energy efficient9,10,11,12 and, in animals with global Kir6.2 KO, is coupled with lower body weight and reduced adiposity.9
Pharmacologic blockade of skeletal muscle KATP channels to increase cellular energy expenditure would be a logical clinical translation of these findings. Unfortunately, no KATP channel antagonist is selective for skeletal muscle KATP channels.13 Indeed, extensive clinical experience with currently available KATP channel blockers, oral hypoglycemic sulfonylureas, indicates that these drugs promote insulin secretion without significant extrapancreatic effects.21,22 This may be explained by the phenomenon of irreversible block of pancreatic isoforms, while the effect on striated muscle KATP channels disappears promptly after drug washout.21 A further obstacle to pharmacologic inhibition of skeletal muscle KATP channels is that there is no distinction between cardiac and skeletal isoforms13,23,24 and so, even if an antagonist specific to Kir6.2/SUR2A channels were developed, it would be impossible to use in a systemic manner to achieve skeletal muscle blockade without potentially detrimental consequences on cardiac stress resistance.10,12,16,21 Thus, alternative strategies must be sought.
To overcome the systemic limitations of pharmacologic blockade of KATP channels, in the current study, we employ vivo-morpholino technology to target delivery of anti-Kir6.2 oligomers directly to skeletal muscles. We find that intramuscular injection of anti-Kir6.2 vivo-morpholino in mice effectively disrupts skeletal muscle KATP channel expression and function only in treated areas. We further find that a series of anti-Kir6.2 vivo-morpholino injections into selected limb muscles increases metabolic rate and heat generation, compared with controls, while having very limited effect on skeletal muscle mechanical function. Thus, these findings establish a proof-of-principle that selective skeletal muscle targeting of sarcolemmal KATP channel function is possible and that this intervention can alter bodily energetics without a disabling impact on overall exercise tolerance.
Results
Anti-Kir6.2 vivo-morpholino suppresses KATP channel expression only in treated skeletal muscles
To assess the effect of local intramuscular injection of anti-Kir6.2 vivo-morpholino on expression of KATP channels, the bilateral gastrocnemius (GCN) muscles of mice were injected with anti-Kir6.2 or negative control (control) vivo-morpholinos. One week after the third injection, the GCN, the untreated nearby tibialis anterior muscle (TA), and hearts were harvested and Kir6.2 expression assessed by western blot (Figure 1a,b). Kir6.2 expression, normalized to glyceraldehyde 3-phosphate dehydrogenase (GAPDH) expression, was reduced in GCN of anti-Kir6.2 vivo-morpholino injected mice by 70% compared to control vivo-morpholino injected mice, while Kir6.2 expression in the heart and in the untreated TA muscle was no different between the groups (Figure 1b). Specifically, the Kir6.2 fractional expression in GCN normalized to average control was 0.3 ± 0.1 in the anti-Kir6.2 group versus 1.0 ± 0.1 in the control group (n = 9 each, P < 0.01), however expression was no different between the groups for noninjected TA muscle where fractional Kir6.2 expression normalized to average control was 0.9 ± 0.3 in the anti-Kir6.2 group versus 1.0 ± 0.2 in the control group (n = 3 each, P = NS (not significant)) or heart where fractional expression normalized to average control was 1.0 ± 0.1 in the anti-Kir6.2 group versus 1.0 ± 0.1 in the control group (n = 3 each, P = NS).
Figure 1.

KATP channel expression in response to anti-Kir6.2 vivo-morpholino treatment of skeletal muscles. (a) Representative western blots of Kir6.2 and GAPDH expression from anti-Kir6.2 (Tx: anti-Kir6.2) and control (Tx: control) vivo-morpholino treated gastrocnemius (GCN—treated), untreated tibialis anterior (TA—untreated) and hearts from the same animals (Heart—untreated). (b) Summary statistics of Kir6.2 expression normalized to GAPDH for control (solid bars) and anti-Kir6.2 (hatched bars) vivo-morpholino treatment in each of the following tissues: the treated gastrocnemius (GCN, n = 9 each), and the untreated TA (n = 3 each) and heart (n = 3 each) from the same animals (*P < 0.01 for GCN anti-Kir6.2 versus GCN control). (c) Representative tracings of single channel activity in cell-attached myofiber patches from bilateral flexor digitorum brevis (FDB) muscles of mice after treatment with control versus anti-Kir6.2 vivo-morpholino. KATP channel activation is induced by pinacidil (100 µmol/l) and 2,4-dinitrophenol (DNP, 200 µmol/l). (d) Summary statistics of the number of KATP channels activated by pinacidil (100 µmol/l) and DNP (200 µmol/l) per cell-attached patch in bilateral FDB isolated myofibers after control (n = 116 cells from eight mice) versus anti-Kir6.2 (n = 99 cells from 14 mice) vivo-morpholino treatment (*P < 0.05 for anti-Kir6.2 versus control).
Anti-Kir6.2 vivo-morpholino suppression of KATP channel expression and function was further confirmed by patch clamp survey of isolated skeletal myofibers. The left flexor digitorum brevis muscle (FDB) of mice was injected with anti-Kir6.2 vivo-morpholino while the FDB of the contralateral limb was injected with control vivo-morpholino. One week after the final injection, the bilateral FDB were harvested and single myofibers enzymatically dissociated and assessed for KATP channel activation in the cell-attached mode (Figure 1c) following application of a metabolic inhibitor (2,4-dinitrophenol (DNP)) and a specific KATP channel opener (pinacidil). This revealed a nearly 60% reduction in the number of KATP channels/patch in the anti-Kir6.2 versus contralateral control vivo-morpholino injected FDB (3.0 ± 0.2, n = 99 cells from 14 mice versus 7.4 ± 0.4, n = 116 cells from eight mice, respectively, P < 0.05, Figure 1d). Overall, these data indicate that intramuscularly injected anti-Kir6.2 vivo-morpholino effectively suppresses KATP channel expression and current only within the treated muscle, without systemic spread or effect on nearby muscles.
Effects of vivo-morpholino-induced disruption of KATP channel expression on skeletal muscle transmembrane potentials and force in situ
To establish the electrophysiological effects of the anti-Kir6.2 vivo-morpholino-induced reduction in KATP channel expression, transmembrane action potentials from anti-Kir6.2 and control vivo-morpholino-injected TA were measured in situ using a floating microelectrode.11 Action potentials (APs) were measured at baseline, and following 5–7 minutes of 1 Hz isometric twitching to model low-intensity activity, known to induce KATP channel activation.11 We found that in control vivo-morpholino injected muscles (baseline: average for eight mice, 5 APs each; post-twitch: average for seven mice, 10 APs each) there was a significant hyperpolarization of resting membrane potential from baseline of 8 mV (−87.7 ± 0.9 versus −79.7 ± 0.9 mV, P < 0.05, Figure 2a,b), a reduction in action potential overshoot from baseline of ~14 mV (22.4 ± 4.4 versus 36.5 ± 2.4 mV, P < 0.05, Figure 2a,c) and a shortening of the duration at −40 mV of .06 msec (APD−40 mV; 0.52 ± 0.01 versus 0.58 ± 0.02 msec, P < 0.05, Figure 2a,d) in response to the twitching, consistent with the electrophysiologic influence of KATP channel activation.11 However, in the anti-Kir6.2 vivo-morpholino-treated muscles (baseline: average for seven mice, 5 APs each; post-twitch: average for seven mice, 10 APs each) no significant alterations were seen in resting membrane potential (−79.3 ± 1.0 versus −77.2 ± 0.7 mV, P = NS, Figure 2a,b), action potential overshoot (27.9 ± 3.7 versus 35.5 ± 2.8 mV, P = NS, Figure 2a,c), or APD−40 mV (0.60 ± 0.02 versus 0.62 ± 0.03 msec, P = NS, Figure 2a–d).
Figure 2.

Effect of Kir6.2 expression inhibition on muscle electrophysiology and force generation in situ. (a) Representative transmembrane action potentials (APs) obtained by floating microelectrode in situ from control (solid bars) versus anti-Kir6.2 (hatched bars) vivo-morpholino treated tibialis anterior (TA) muscles at baseline and following 5–7 minutes of isometric twitching at 1 Hz (twitch). (b) Summary statistics for baseline (control: average for eight mice, 5 APs each and anti-Kir6.2: average for seven mice, 5 APs each) and post-twitch (control: average for seven mice, 10 APs each and anti-Kir6.2: average for seven mice, 10 APs each) resting membrane potential, (c) action potential overshoot, and (d) AP duration at −40 mV (APD−40 mV). Representative tracings of in situ specific peak force over 10 minutes of 1 Hz twitching and single twitch waveforms (insets) from (e) control and (f) anti-Kir6.2 vivo-morpholino treated tibialis anterior. *P < 0.05 for post-twitch versus baseline action potential parameters.
To determine the effect of vivo-morpholino knock down of KATP channel expression on skeletal muscle mechanical function, we tested the in situ resting and specific peak force of anti-Kir6.2- versus control-treated TA muscle (Figure 2e,f). We found no differences in resting force between the two groups at baseline or after 7 minutes of 1 Hz isometric twitching (Table 1). However, the specific peak force, although similar at baseline, was significantly greater after 7 minutes of isometric twitching in the anti-Kir6.2 vivo-morpholino-treated group compared to controls (Table 2). Representative tracings of the time course indicate that the specific peak force initially increased in response to twitching in both cohorts. However, in control the specific peak force trended back toward baseline over time in contrast to the persistent elevation of specific peak force for anti-Kir6.2 (Figure 2e,f). As expected, there was no difference in the muscle length (L0) of the TA in the two groups (14.0 ± 0.3 versus 14.2 ± 0.1 mm for anti-Kir6.2 versus control vivo-morpholino treated mice, respectively, n = 10 each, P = NS). Together, these data indicate that local anti-Kir6.2 vivo-morpholino treatment induces knock-down of KATP channels sufficient to disrupt adjustment of sarcolemmal excitability in response to repeated muscle contractions.
Table 1. Muscle resting tension.

Table 2. Muscle peak twitch force.

Disruption of KATP channel–dependent expression by vivo-morpholino increases activity-related muscle thermogenesis and oxygen consumption
Contracting muscle uses energy for both force and heat generation. During isometric contraction, no external work is performed; thus, the muscle enthalpy is equal to the heat output.11,25 To measure heat output, we used implantable micro-thermocouples placed under the TA muscle during our in situ repetitive isometric twitch protocol. This data was then used to compare muscle energy consumption between control and anti-Kir6.2 vivo-morpholino treated mice. As expected, there was no difference in muscle mass between treatment groups (control 44.6 ± 1.33 mg, versus anti-Kir6.2 42.3 ± 0.4 mg, n = 4 each, P = NS). There was no difference in muscle temperature at rest (control 34.92 ± 0.09 versus anti-Kir6.2 34.88 ± 0.03 °C, n = 4 each, P = NS). However, we found that TA muscles with disrupted KATP channel function exhibited significantly greater activity-related increases in muscle temperature compared with corresponding controls (anti-Kir6.2 1.78 ± 0.14 versus control 1.21 ± 0.19 °C, n = 4 each, *P < 0.05, Figure 3c). Furthermore, when exposed to low-intensity treadmill exercise, previously demonstrated to be associated with KATP channel opening,9,11 mice treated with anti-Kir6.2 vivo-morpholino demonstrated significantly greater body surface temperature changes overlying the treated muscles compared to control vivo-morpholino treated mice (Figure 3a). Specifically, body surface temperature was assessed during exercise before any treatment, and after a series of injections of either anti-Kir6.2 or control vivo-morpholino into the bilateral gluteus superficialis, gluteus medius, quadriceps femoris, rectus femoris, biceps femoris, and semitendinous muscles (n = 4 mice each group with four treadmill runs/mouse before, and six treadmill runs/mouse after treatment in each group). As expected, there was no significant temperature difference between groups before treatment (30.15 ± 0.10 °C for the anti-Kir6.2 group versus 30.32 ± 0.07 °C for the control group, P = NS, Figure 3b, left panel). However, anti-Kir6.2 vivo-morpholino treatment resulted in a significant exercise-related increase in hind leg surface temperature (30.69 ± 0.09 °C, compared with before treatment 30.15 ± 0.08 °C, P < .05), approximately a half °C greater than the temperature observed in the control vivo-morpholino treatment mice (30.25 ± 0.08 °C, P < 0.05) which was unchanged from its baseline temperature (30.32 ± 0.07 °C, P = NS, Figure 3b, right panel). These results indicate that vivo-morpholino-induced reduction of Kir6.2 expression promotes muscle activity related thermogenesis.
Figure 3.

Suppression of skeletal muscle KATP channel expression increases thermogenesis and oxygen consumption. (a) Representative colorized infrared images of mice performing low-intensity (2 mW, ~7 m/minute at 15° incline for a 25 g mouse) exercise on the treadmill before and after control versus anti-Kir6.2 vivo-morpholino injection into bilateral gluteus superficialis, gluteus medius, quadriceps femoris, rectus femoris, biceps femoris and semitendinous muscles. The white dashed oval indicated by the arrow on the upper left mouse indicates the size and position of the limb area used for thermal analysis. (b) Summary data for hind limb temperature, as determined from thermal images, obtained after 6 minutes of 2 mW exercise. Data are displayed for control (white solid bars, n = 4 mice) and anti-Kir6.2 (hatched bars, n = 4 mice) vivo-morpholino treatment group mice before and after the series of injections (*P < 0.05 for anti-Kir6.2 versus control and Kir6.2 after versus before treatment). (c) In situ submuscular temperature from TA in response to repetitive low-frequency twitching for control (white solid bars, n = 4) and anti-Kir6.2 (hatched bars, n = 4) vivo-morpholino treated mice (*P < 0.05 for anti-Kir6.2 versus control). Representative plots of oxygen consumption (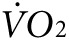 ) at rest and in response to initiation and maintenance of 2 mW exercise for control and anti-Kir6.2 vivo-morpholino treatment group mice (d) before the treatment has been delivered and (e) after the series of injections into bilateral hind limb muscles (same mice as in Figure 3a,b). The treadmill is stationary until minute 67 when it begins to move. (f) Summary statistics for the activity-related component of oxygen consumption (
) at rest and in response to initiation and maintenance of 2 mW exercise for control and anti-Kir6.2 vivo-morpholino treatment group mice (d) before the treatment has been delivered and (e) after the series of injections into bilateral hind limb muscles (same mice as in Figure 3a,b). The treadmill is stationary until minute 67 when it begins to move. (f) Summary statistics for the activity-related component of oxygen consumption (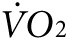 ) before and after delivery of control (solid bars, n = 3 mice) versus anti-Kir6.2 (hatched bars, n = 3 mice) vivo-morpholino injection into hind limb muscles (*P < 0.05 for anti-Kir6.2 versus control). (g) Summary statistics for the activity-related component of heat generation before and after delivery of control (solid bars, n = 3 mice) versus anti-Kir6.2 (hatched bars, n = 3 mice) vivo-morpholino injection into hind limb muscles (*P < 0.05 for anti-Kir6.2 versus control).
) before and after delivery of control (solid bars, n = 3 mice) versus anti-Kir6.2 (hatched bars, n = 3 mice) vivo-morpholino injection into hind limb muscles (*P < 0.05 for anti-Kir6.2 versus control). (g) Summary statistics for the activity-related component of heat generation before and after delivery of control (solid bars, n = 3 mice) versus anti-Kir6.2 (hatched bars, n = 3 mice) vivo-morpholino injection into hind limb muscles (*P < 0.05 for anti-Kir6.2 versus control).
We next assessed whether anti-Kir6.2 treatment of limb muscles results in a change in overall bodily oxygen consumption (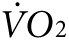 ) during low-intensity exercise. The same two groups of mice as used above for thermal imaging were tested on treadmills equipped with sensors for indirect calorimetry. All mice were tested for
) during low-intensity exercise. The same two groups of mice as used above for thermal imaging were tested on treadmills equipped with sensors for indirect calorimetry. All mice were tested for 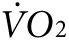 and CO2 production at rest and with exercise before any treatment, and then after treatment with anti-Kir6.2 or control vivo-morpholino (n = 3 each). Curves for
and CO2 production at rest and with exercise before any treatment, and then after treatment with anti-Kir6.2 or control vivo-morpholino (n = 3 each). Curves for 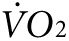 indicate fluctuating values as mice acclimate to the stationary treadmill, reaching a steady state at about 55–65 minutes (Figure 3d,e). At about 65 minutes, the treadmill begins to move causing an abrupt increase in
indicate fluctuating values as mice acclimate to the stationary treadmill, reaching a steady state at about 55–65 minutes (Figure 3d,e). At about 65 minutes, the treadmill begins to move causing an abrupt increase in 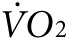 as mice respond to the new conditions. Between 60 and 80 minutes of exercise, the
as mice respond to the new conditions. Between 60 and 80 minutes of exercise, the 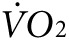 reaches a new steady state. The activity-specific component of
reaches a new steady state. The activity-specific component of 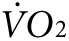 and heat were calculated as the difference between the steady-state level during exercise, taken at ~60 minutes after initiation of treadmill movement, and the steady-state resting level, taken after ~60 minutes of acclimation on the stationary treadmill. As expected, mice in the groups destined for either anti-Kir6.2 or control treatment, but having received no treatment yet, showed no difference in activity-related
and heat were calculated as the difference between the steady-state level during exercise, taken at ~60 minutes after initiation of treadmill movement, and the steady-state resting level, taken after ~60 minutes of acclimation on the stationary treadmill. As expected, mice in the groups destined for either anti-Kir6.2 or control treatment, but having received no treatment yet, showed no difference in activity-related 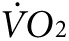 (2,172 ± 133 versus 2,301 ± 107 ml/kg/h, P = NS, Figure 3f, left panel) or heat (0.25 ± 0.01 versus 0.28 ± 0.01 kcal, P = NS, Figure 3g, left panel). However, 1 week after finishing the injection protocol, treadmill testing was repeated showing a 38% greater activity-related
(2,172 ± 133 versus 2,301 ± 107 ml/kg/h, P = NS, Figure 3f, left panel) or heat (0.25 ± 0.01 versus 0.28 ± 0.01 kcal, P = NS, Figure 3g, left panel). However, 1 week after finishing the injection protocol, treadmill testing was repeated showing a 38% greater activity-related 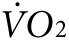 (2,585 ± 168 versus 1,868 ± 118 ml/kg/h, P < 0.05, Figure 3f, right panel) and a 24% greater activity-related heat production (0.31 ± 0.01 versus 0.25 ± 0.02 kcal, P < 0.05, Figure 3g, right panel) in the anti-Kir6.2 versus control vivo-morpholino groups. Therefore, KATP channel deficit in select skeletal muscles increased the energetic cost of low-intensity treadmill exercise.
(2,585 ± 168 versus 1,868 ± 118 ml/kg/h, P < 0.05, Figure 3f, right panel) and a 24% greater activity-related heat production (0.31 ± 0.01 versus 0.25 ± 0.02 kcal, P < 0.05, Figure 3g, right panel) in the anti-Kir6.2 versus control vivo-morpholino groups. Therefore, KATP channel deficit in select skeletal muscles increased the energetic cost of low-intensity treadmill exercise.
Anti-Kir6.2 vivo-morpholino injection in selected limb muscles results in only minimal reduction in exercise tolerance
To determine whether anti-Kir6.2 vivo-morpholino inhibition of limb skeletal muscle KATP channel expression affects exercise tolerance, we challenged the same mice used for thermal imaging and calorimetry with a treadmill protocol of escalating speed and incline (Figure 4a) 1 week after their series of anti-Kir6.2 (n = 7 mice) or control (n = 3 mice) vivo-morpholino injections. We found that anti-Kir6.2 versus control morpholino injection resulted in a nonsignificant trend toward a small reduction in exertional tolerance as measured by total distance achieved (493 ± 23 versus 437 ± 38 meters, P < 0.4, Figure 4b), total duration of running (44.8 ± 1.2 versus 41.4 ± 2.1 minutes, P < 0.4, Figure 4c), and the tolerated workload (30.5 ± 1.8 versus 27.0 ± 2.4 J, P = 0.4, Figure 4d) which takes into consideration the increasing incline and velocity of the treadmill. These translate into a trend towards anti-Kir6.2 vivo-morpholino treatment-related reductions of 11.4, 7.6, and 11.5%, respectively. Thus, selective hind limb reduction of skeletal muscle KATP channel expression that results in increased oxygen consumption and heat generation could have a very modest negative effect on exercise tolerance that causes mice to drop out of exercise slightly earlier at the most vigorous extremes.
Figure 4.

Exercise tolerance is modestly reduced by anti-Kir6.2 vivo-morpholino injection into hind limb muscles. (a) Schematic of treadmill calculations for kinetic energy (Ek), potential energy (Ep), and workload (Ek + Ep). m = mass of the mouse, g = acceleration due to gravity, t = time on the treadmill, v = treadmill velocity, φ = angle of treadmill incline. Steps are proportional to the workload. Vertical lines indicate the mean time of exercise drop out for mice that have undergone control versus anti-Kir6.2 vivo-morpholino injection into bilateral gluteus superficialis, gluteus medius, quadriceps femoris, rectus femoris, biceps femoris, and semitendinous muscles as in Figure 3. Summary statistics for control (solid bars, n = 3 mice) versus anti-Kir6.2 (hatched bars, n = 7 mice) vivo-morpholino treatment group mice for exercise tolerance after treatment as represented by (b) distance completed, (c) duration of exercise, and (d) tolerated workload.
Discussion
This study demonstrates that skeletal muscle KATP channel expression and physiologic function can be locally manipulated through the use of anti-sense Kir6.2 oligomers delivered as vivo-morpholinos. We have also shown that such local treatment has a significant impact on skeletal muscle and overall bodily activity-related energy efficiency. Thus, these findings serve as a “proof-of-concept” for the strategy of targeting the KATP channels of specific muscles to modify energy efficiency.
We chose to target the Kir6.2 subunit of KATP channels based on previous studies in which expression or function of Kir6.2 was disrupted.9,10,11,12,16 In those studies, we found that potassium efflux via sarcolemmal KATP channels was activated by low-intensity muscle activity and opposed depolarizing currents thus shortening action potential duration and limiting calcium influx.9,10,11,12,16 The consequent decrease in calcium-dependent functions translates into less energy used for contractions, calcium re-sequestration, and heat production. Conversely, disruption of normal function of the Kir6.2 subunit augments ATP turnover and elicits an extra energetic cost of muscle performance.9,10,11,12,16 The findings in the current study further support this mechanism by confirming that local disruption of skeletal muscle KATP channels by anti-Kir6.2 vivo-morpholino attenuates activity-induced membrane hyperpolarization, overshoot reduction, and action potential duration shortening, resulting in increased muscle peak force development and thermogenesis.
Vivo-morpholinos were used in this study due to their attractive profile of stability, low toxicity, good cell penetration, and ease of use.26,27 These cell-penetrating morpholinos are oligomers covalently conjugated with a delivery moiety containing eight terminal guanidinium groups on a dendrimer scaffold.27 The guanidinium heads are predicted to interact with phosphates of phospholipids by electrostatic interaction and multiple hydrogen bonds, thereby permitting translocation across cell membranes.26,27 Within the cell, vivo-morpholinos cause steric blocking of the target RNA sequence. Thus, the anti-Kir6.2 RNA oligo used in the current study is presumed to exert its effects through binding mRNA to interfere with translation of the pore-forming Kir6.2 subunit of sarcolemmal KATP channels. The control vivo-morpholino utilizes an antisense sequence against RNA that is not present in mouse tissues.
Using the anti-Kir6.2 vivo-morpholino, we were able to obtain a significant reduction in KATP channel current (IKATP), comparable to the level found in our previous studies, in which we used transgenic expression of a dominant-negative mutant of the pore-forming subunit to manipulate KATP channel function.9,10,11,12 This is demonstrated both for the number of functional channels per cell-attached patch and for in situ KATP channel-dependent membrane and action potential characteristics.11
We found that disruption of KATP channel expression was induced only in the hind limb muscles that were injected with anti-Kir6.2 vivo-morpholino, while nearby untreated muscles and the heart were shown to be unaffected. This is of key importance to the therapeutic potential of this approach since extensive data documents the role of myocardial KATP channels in protection of the heart from injury due to ischemia, hypoxia, hypertension, as well as development of heart failure, and arrhythmias.13,16,28,29,30 Even acceleration of heart rate within the physiologic range, as might occur with exercise, is sufficient to activate cardiac KATP channels to optimize cardiac energetics.12 Thus, sparing of cardiac KATP channel function would be a central requirement of any therapy. As we did not see an effect of anti-Kir6.2 vivo-morpholino treatment in heart or nearby skeletal muscles, we assume intact KATP channel function in other non-treated Kir6.2-containing tissues as well. Indeed, this site-specific effect of intramuscular anti-Kir6.2 vivo-morpholino injections on KATP channel function indicates an important advantage over the use of known KATP channel blocking drugs. These KATP channel blockers work through binding the sulfonylurea receptor subunit of the channel and have high affinity to all three known isoforms (pancreatic SUR1, cardiac and skeletal muscle SUR2A, as well as smooth muscle SUR2B).21,22 Therefore, these drugs, even if delivered locally to avoid the systemic effects noted in the introduction, would affect both skeletal muscle (predominantly Kir6.2/SUR2A)31 and vascular smooth muscle (predominantly Kir6.1/SUR2B)32 KATP channels at the site of application. Blockade of vascular smooth muscle KATP channels would be expected to result in vasoconstriction and reduced blood flow.33,34 Furthermore, the half-life of sulfonylureas is between 4 and 10 hours such that even if these other limitations could be overcome, their use would entail frequent re-administration. In contrast, vivo-morpholinos are designed to be highly lipophilic, quickly and irreversibly entering cells on contact, and their effect on skeletal muscle protein expression can be substantial26,35,36,37,38,39,40 for 17 weeks or more post treatment.27,35,41
Energy efficiency is measured as the ratio of “useful work” to thermogenesis. When comparing animals for which “useful work” is matched by paired treadmill exercise, the difference in energy efficiency is proportional to the difference in heat generation or oxygen consumption (thermogenesis) between the mice.11 Here, we used low-intensity treadmill exercise (2 mW or ~7 m/min at 15° incline for a 25 g mouse) that could be considered comparable to the level of exertion that occurs during activities of daily living and that was previously shown to be sufficient to elicit KATP channel opening.9,11 As in previous studies with Kir6.2 knock-out or transgenic suppression of Kir6.2 function,9,11 reduction in skeletal muscle KATP channel expression by anti-Kir6.2 vivo-morpholino resulted in worsened energy efficiency as manifested by increased heat production and oxygen consumption determined by indirect calorimetry and infrared imaging of body surface temperature during treadmill exercise. It is particularly important that significant energetic effects of local skeletal muscle KATP channel knock-down were observed in response to this relatively gentle exercise challenge as it supports the concept that such an intervention could be used to enhance caloric consumption in obese individuals with limited exercise capacity who are able to perform activities of daily living but unlikely to tolerate a moderate- or high-intensity work-out. The effect of the anti-Kir6.2 vivo-morpholino treatment on muscle energy efficiency was confirmed by direct measurements of muscle temperature elevation in response to low-frequency repetitive twitches.
Since control animals were injected with the same volume and concentration of vivo-morpholino, we conclude that the rise in the temperature of anti-Kir6.2 vivo-morpholino treated mice is unlikely to be due to a local inflammatory reaction linked to the treatment such as injury from injection or toxicity of the vivo-morpholino. Rather, our results are consistent with a measurable increase in thermogenesis due to vivo-morpholino-related reduction of Kir6.2 expression. Although the 0.5 °C difference in activity-induced body surface warming observed between anti-Kir6.2 vivo-morpholino treated and control mice may appear small, the detection of any surface temperature change in an intact animal is notable given the redistribution of heat via blood flow and dissipation at the body surface. It would likely require a complex model to calculate the exact caloric cost of raising the surface or muscle temperature in the leg by 0.5 °C, but the indirect calorimetry indicating a 35% increase in 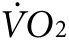 and a 24% increase in heat production supports the concept that the costs are substantial.
and a 24% increase in heat production supports the concept that the costs are substantial.
The observed changes in 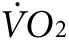 and heat production induced by anti-Kir6.2 vivo-morpholino treatment were smaller than that observed with transgenic and knock-out mouse models.9,11 This is expected since, in contrast to the studies in transgenic and KO models, not all skeletal muscles were affected in the present study. On the other hand, despite only treating a subset of hind limb muscles, the anti-Kir6.2 vivo-morpholino injection still resulted in a significant reduction in energy efficiency possibly because the acuity of the knock-down allows less opportunity for compensatory mechanisms to be established compared to knock-out and transgenic mouse models.
and heat production induced by anti-Kir6.2 vivo-morpholino treatment were smaller than that observed with transgenic and knock-out mouse models.9,11 This is expected since, in contrast to the studies in transgenic and KO models, not all skeletal muscles were affected in the present study. On the other hand, despite only treating a subset of hind limb muscles, the anti-Kir6.2 vivo-morpholino injection still resulted in a significant reduction in energy efficiency possibly because the acuity of the knock-down allows less opportunity for compensatory mechanisms to be established compared to knock-out and transgenic mouse models.
Regarding exercise tolerance, our findings here are also in line with what we would expect. Specifically, the myofiber calcium loading shown to be promoted by loss of KATP channel current initially promotes greater contractile force.9,11,13,16,17,18,19,20,42,43,44,45,46 At the same time, since it also promotes greater utilization of energetic resources, it negatively affects endurance. Indeed, reduced exercise endurance was shown in KO and transgenic mice with reduced or eliminated skeletal muscle KATP channels.9,11,16 In the present study, however, when we knock-down KATP channel expression in a limited number of muscles, we find that the treated mice are mostly able to keep up with their control-treated littermates, with a trend toward dropping out only a few minutes earlier after more than 40 minutes of escalating exercise effort. We hypothesize that there is no maximum degree of local skeletal muscle KATP channel knockdown that should be targeted by this approach to avoid muscle injury since complete elimination of skeletal muscle KATP channels in KO mice has been associated with fiber damage only after repeated extreme endurance exercise exposure.19,20 However, due to the possibility that fiber damage could occur in response to extreme exertion in anti-Kir6.2 vivo-morpholino treated muscles, limitation to moderate activities would be appropriate if such an approach were to be used therapeutically.
While our results indicate the potential for the use of vivo-morpholino targeting of Kir6.2 as a therapeutic strategy, the detailed pharmacokinetic and dynamic profile as well as the long-term clinical effect will need to be further defined. For instance, while the effect of other vivo-morpholino based therapies are long-lasting, the duration of anti-Kir6.2 vivo-morpholino treatment on skeletal muscle KATP channel-dependent functions is unknown, although we can conclude that the presented protocol has measurable effectiveness for at least a week based on our data that was acquired one week after the last anti-Kir6.2 vivo-morpholino injection. The optimal dose and delivery schedule also remains to be defined along with any long-term consequences of Kir6.2 expression suppression by vivo-morpholino. Finally, we selected a group of hind limb muscles due to their accessibility, size, and prominent use during treadmill exercise, but other or fewer treated muscles may also produce an acceptable energetic and exercise tolerance response such that the muscles selected could be tailored for particular subjects or exercise routines. Such elements as the optimal time-line, number and location of muscle injections and whether any long-term compensation exists to counteract the expected effects will be the subject of future studies.
In conclusion, this study serves as a paradigm for local manipulation of skeletal muscle KATP channel-related electrophysiology and energy consumption. If further validated, this strategy could potentially serve as a well-tolerated adjunct to low- or moderate-intensity exercise to increase its effectiveness for body weight control.
Materials and Methods
Mouse models. All animal protocols conform to the Guide for the Care and Use of Laboratory Animals generated by the Institute for Laboratory Animal Research, National Research Council of the National Academies. All protocols were approved by the University of Iowa Institutional Animal Care and Use Committee. Male C57BL/6 WT mice 6–12 weeks were used for all experiments. For harvesting of tissue samples and for all other nonsurvival experiments, mice were anesthetized (Avertin: 2.5% solution of tribromoethanol in 2-methyl-2-butanol; Sigma-Aldrich, St Louis, MO; 240 mg/kg i.p.). At the end of nonsurvival experiments, mice were euthanized by cardiectomy while under anesthesia. For survival experiments, including vivo-morpholino injections, mice were anesthetized with inhaled isoflurane (Piramal Healthcare, Andhra Pradesh, India) to maintain a respiratory rate of ~50–60 breaths per minute.
Vivo-morpholino. The vivo-morpholino (Gene Tools, LLC, Pacific Grove, CA) modified oligonucleotides include a standard negative control sequence (CCTCTTACCTCAGTTACAATTTATA, M.W. 10,238 Da) and the anti-Kir6.2 sequence (GGATAATGCCCTTTCGGGACAGCAT, M.W. 10,294 Da). Vivo-morpholinos were introduced at room temperature into anesthetized 6–8 wk-old C57/BL6 WT male mice as an intramuscular injection of 2 nmol/l in 50 µl normal saline (about 1 ng) into each targeted muscle using a 31 gauge needle (Previous studies indicate no histological evidence of tissue or organ toxicity or immune response at single IM doses up to 10 µg, single IV doses as high as .6 mg or cumulative IV doses as high as 3 mg for a 20 g mouse26). All protocols consist of vivo-morpholino injections into the selected muscles once weekly for 3 weeks. All tissue samples or physiologic data were collected 1 week after the final injection.
Experimental apparatus. Anesthetized mice were placed on a platform (809B; Aurora Scientific, Aurora, Ontario, Canada) heated by circulating water (6200 R20F; Thermo Fisher Scientific, Grand Island, NY). The TA muscle of one leg was exposed and the tendons attached to the force transducer (305C; Aurora Scientific). Tyrode's solution (in mmol/l: NaCl 36.5, KCl 5.4, CaCl2 2.5, MgCl2 0.53, glucose 5.5, and HEPES-NaOH 5.5, pH 7.4) warmed to 30.0 °C was dripped on the muscle at ~1 drop/second. The temperature of the platform on which the mouse was placed was maintained by circulating heated water at 34.8 °C. These two methods stabilized the temperature measured at the surface of the muscle at ~30 °C. A stimulator (Accupulser A310; World Precision Instruments, Sarasota, FL) routed through a stimulus isolation unit (A365 SIU; World Precision Instruments) was used to excite the sciatic nerve (variable amplitude, pulse width 0.5 ms using a custom electrode). Pacing triggers were generated and data collected at 25 kHz using pCLAMP software (Molecular Devices, Sunnyvale, CA) interfaced with a digital acquisition device (Digidata 1440A; Molecular Devices). The experimental apparatus, including force transduction platform, force transducer, microelectrode head stage (see below), and micromanipulators, was located on a vibration isolation table (VH3648; Newport, Irvine, CA) inside a Faraday cage (FC3648; Newport).
Microelectrode recordings. Pipettes were pulled on a horizontal puller (P-1000; Sutter Instrument, Novato, CA) using borosilicate glass (1B150F; World Precision Instruments). The tip of each pipette was filled with 3 M KCl. A 0.005-inch diameter silver wire coated with polytetrafluoroethylene (PTFE) was mechanically stripped at both ends and electrochemically plated with silver chloride on one end to produce an Ag/AgCl electrode. The wire was coiled around a cylinder (~1-cm diameter) by one turn to produce a helical spring-like shape in the middle. The micropipette was scored with a carbide utility blade ~1–2 mm above the shoulder and the shank gently separated from the tip. The silver chloride–coated end of the wire was inserted into the glass tip sufficiently to create enough friction to support the weight of the tip.11 The opposite end of the silver wire was attached to the amplifier head stage (CV203-BU; Molecular Devices). A silver chloride–coated reference wire was placed in contact with the lateral edge of the exposed muscle using a salt bridge. The tip resistance of the microelectrode was measured (7–20 MΩ), and zero potential was adjusted before each impalement with the floating microelectrode and reference in a bath of Tyrode's solution. Each impalement was performed with a new microelectrode. Action potentials were amplified (Axopatch 200B; Molecular Devices) and recorded at 25 kHz using pCLAMP software (Molecular Devices) interfaced with a digital acquisition device (Digidata 1440A; Molecular Devices). Action potentials were analyzed if all of the following criteria were met: (i) the measured potential showed a sharp drop upon microelectrode penetration, (ii) the resting membrane potential was more negative than −70 mV and was stable, (iii) the overshoot was >0 mV, and (iv) the stimulus artifact did not interfere with the upstroke of the action potential. The resting membrane potential was measured from the baseline before the action potential. Overshoot was defined as the difference in potential between the peak of the action potential and 0 mV. Action potential amplitude was defined as the difference between resting membrane potential and the peak potential. Action potential duration (APD) was defined as the duration at −40 mV11. Statistics were performed using the average parameter value for each mouse as the raw data such that n = number of mice, and with the same number of action potentials per mouse in each of the control and vivo-morpholino cohorts such that all action potentials are weighted equally. The first 5 (baseline) and 10 (post-twitch) action potentials meeting the above criteria were analyzed for each mouse.
Force measurements. The force transducer/controller (305C; Aurora Scientific) was set for isometric contraction. Muscle twitches were provoked by sciatic nerve stimulation. The resting tension on the muscle was set by incrementally adjusting the controller lever arm, and thus muscle length (L0) and resting tension, and then delivering a supramaximal stimulus to the sciatic nerve until a maximal twitch force was obtained. The resting tension at L0 was then recorded, typically ~75 mN. The stimulation amplitude was then set by plotting a twitch force versus stimulus amplitude curve and then setting the stimulator output at two times the threshold for a maximal twitch. Twitch force was normalized to the calculated muscle cross-sectional area (mN/cm2) using the muscle mass (in g) divided by the product of the presumed muscle density of 1.06 g/cm3 and the optimal fiber length (L0 × 0.6, in cm) and expressed as specific force.47,48 L0 was measured between suture knots on the muscle tendon and muscle mass was measured in muscles excised at the suture knots.11 To assess time-dependent changes, force was measured at 0 and 7 minutes of isometric twitching at 1 Hz.
Muscle temperature. Anesthetized mice were placed on the experimental apparatus as described above. The TA tendons of one leg were exposed and attached to the force transducer (305C; Aurora Scientific). A flexible implantable thermocouple microprobe IT-23 (Physitemp Instruments, Clifton, NJ) was inserted under the TA muscle fascia adjacent to the tibia without disruption of muscle integrity. The skin was closed with 6.0 Ethilon suture to prevent heat loss. The TA was subjected to repetitive isometric contractions via sciatic nerve stimulation as described above. Differential thermal analysis of TA muscle (heated platform with constant temperature was used as a reference) was conducted using a BAT-10 Multipurpose Thermometer (Physitemp Instruments, Inc.) that allows measuring of differential temperature in the physiologica range with 0.01 °C accuracy.
Body surface temperature. The body surface temperature of the mice was imaged using a high-resolution infrared camera (A655sc Thermal Imager; FLIR Systems, Wilsonville, OR). Quantitative analysis of infrared images was performed using FLIR ResearchIR software version 3.4.13039.1003. Anti-Kir6.2 versus control vivo-morpholino group mice were simultaneously imaged in adjacent lanes on a multilane treadmill after 6 minutes of treadmill exercise at 7–9 m/minute and 15° incline (~2 mW for a 25 g mouse).9,11,16 Images were obtained every 1 second. The side by side imaging of mice in the same camera frame allows simultaneous assessment and direct comparison of the body surface heating in the different mouse models providing an advantage over other methods for assessing body temperature that would be limited by the accuracy and concordance of two separate temperature probes.
Single channel recordings. Single FDB muscle fibers were enzymatically isolated,9,49 and single channel currents were recorded in the cell-attached mode using an Axopatch 200B amplifier (Molecular Devices) integrated with a TE2000-U microscope (Nikon, Tokyo, Japan). The holding potential was −60 mV. Pipettes were filled with internal pipette solution (in mmol/l: KCl 140, CaCl2 1, MgCl2 1, HEPES-KOH 5, pH 7.3), resistance of 5–7 MΩ. Cells were bathed in internal solution (in mmol/l: KCl 140, MgCl2 1, EGTA 5, HEPES-KOH 5, glucose 5, pH 7.3). Experiments were performed at 30 °C using a temperature controller TC2r (Cell MicroControls, Norfolk, VA). KATP channel opening was induced by superfusion with pinacidil and DNP. Data were recorded at 25 kHz using pCLAMP software (Molecular Devices) interfaced with a digital acquisition device (Digidata 1440A; Molecular Devices).
Indirect calorimetry and treadmill exercise. Oxygen consumption (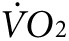 ) and CO2 generation (
) and CO2 generation (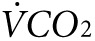 ) during low-intensity physical activity (2 mW) were measured using a modular treadmill connected to the Oxymax indirect calorimetry system (Columbus Instruments, Columbus, OH) as described.9,11 Heat was calculated using the manufacturer's software (Heat = CV ×
) during low-intensity physical activity (2 mW) were measured using a modular treadmill connected to the Oxymax indirect calorimetry system (Columbus Instruments, Columbus, OH) as described.9,11 Heat was calculated using the manufacturer's software (Heat = CV × 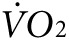 , and CV = 3.815 + 1.232 × RER, where CV is the calorific value and RER is the respiratory exchange ratio). Five days before exercise tests, mice were acclimated daily for 45 minutes on a nonmoving treadmill followed by 15 minutes at a velocity of 3.5 m/minute. For indirect calorimetry and thermal imaging testing, the treadmill velocity, set for a constant workload rate of 2 mW (~7 m/minute at 15° incline for a 25 g mouse) was adjusted to body weight for each animal as W/(m⋅g)/sin(φ), where m is body weight; g, acceleration due to gravity (9.8 m/s2); φ, treadmill inclination.9,11,16 To test exercise tolerance, the exercise protocol consisted of stepwise increases in either incline or velocity at 3-minute intervals until mice were no longer able to match the treadmill speed.9,11,16
, and CV = 3.815 + 1.232 × RER, where CV is the calorific value and RER is the respiratory exchange ratio). Five days before exercise tests, mice were acclimated daily for 45 minutes on a nonmoving treadmill followed by 15 minutes at a velocity of 3.5 m/minute. For indirect calorimetry and thermal imaging testing, the treadmill velocity, set for a constant workload rate of 2 mW (~7 m/minute at 15° incline for a 25 g mouse) was adjusted to body weight for each animal as W/(m⋅g)/sin(φ), where m is body weight; g, acceleration due to gravity (9.8 m/s2); φ, treadmill inclination.9,11,16 To test exercise tolerance, the exercise protocol consisted of stepwise increases in either incline or velocity at 3-minute intervals until mice were no longer able to match the treadmill speed.9,11,16
Western blotting. Protein extracts were prepared by homogenizing tissue in NaCl 50 mmol/l, Tris-base 50 mmol/l, EDTA 5 mmol/l, NaF 50 mmol/l, pH 7.4 supplemented with 1.5% Triton X-100 and protease inhibitors. Electrophoresis was performed on 3–8% gradient Nu-Page Tris-Acetate gel and transferred to PVDF (polyvinylidene difluoride) membranes (Invitrogen, Thermo Fisher Scientific). Rabbit polyclonal antibodies against Kir6.2, and secondary anti-rabbit HRP-conjugated antibodies were used (Santa Cruz Biotechnology, Dallas, TX). Densitometric analysis of western blots was performed using Adobe Photoshop (Adobe Systems, San Jose, CA).
Statistics. Results are expressed as mean ± standard error of the mean. Comparisons between two groups were made using the two-sided Student's t-test and between more than two groups using analysis of variance. A P value < 0.05 was considered statistically significant. Sigma Plot 11 was used for all statistical analyses.
Acknowledgments
This work was supported by the National Institutes of Health HL113089 to Denice Hodgson-Zingman (University of Iowa), DK092412 to L.Z. (University of Iowa), and the VA Merit Review Program 1I0BX000718 to L.Z. (University of Iowa). There are no conflicts of interest.
References
- Blair SN, Nichaman MZ. The public health problem of increasing prevalence rates of obesity and what should be done about it. Mayo Clin Proc. 2002;77:109–113. doi: 10.4065/77.2.109. [DOI] [PubMed] [Google Scholar]
- Flegal KM, Carroll MD, Kit BK, Ogden CL. Prevalence of obesity and trends in the distribution of body mass index among US adults, 1999-2010. JAMA. 2012;307:491–497. doi: 10.1001/jama.2012.39. [DOI] [PubMed] [Google Scholar]
- Yanovski SZ, Yanovski JA. Obesity. N Engl J Med. 2002;346:591–602. doi: 10.1056/NEJMra012586. [DOI] [PubMed] [Google Scholar]
- Tseng YH, Cypess AM, Kahn CR. Cellular bioenergetics as a target for obesity therapy. Nat Rev Drug Discov. 2010;9:465–482. doi: 10.1038/nrd3138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wadden TA, Berkowitz RI, Womble LG, Sarwer DB, Phelan S, Cato RK, et al. Randomized trial of lifestyle modification and pharmacotherapy for obesity. N Engl J Med. 2005;353:2111–2120. doi: 10.1056/NEJMoa050156. [DOI] [PubMed] [Google Scholar]
- Barsh GS, Schwartz MW. Genetic approaches to studying energy balance: perception and integration. Nat Rev Genet. 2002;3:589–600. doi: 10.1038/nrg862. [DOI] [PubMed] [Google Scholar]
- Schwartz MW, Woods SC, Seeley RJ, Barsh GS, Baskin DG, Leibel RL. Is the energy homeostasis system inherently biased toward weight gain. Diabetes. 2003;52:232–238. doi: 10.2337/diabetes.52.2.232. [DOI] [PubMed] [Google Scholar]
- Spriet LL, Hargreaves M.Overview of exercise metabolism. Hargreaves M, Spriet LL.eds.) Exercise Metabolism Human Kinetics; Champaign, IL; 2006 [Google Scholar]
- Alekseev AE, Reyes S, Yamada S, Hodgson-Zingman DM, Sattiraju S, Zhu Z, et al. Sarcolemmal ATP-sensitive K(+) channels control energy expenditure determining body weight. Cell Metab. 2010;11:58–69. doi: 10.1016/j.cmet.2009.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Z, Burnett CM, Maksymov G, Stepniak E, Sierra A, Subbotina E, et al. Reduction in number of sarcolemmal KATP channels slows cardiac action potential duration shortening under hypoxia. Biochem Biophys Res Commun. 2011;415:637–641. doi: 10.1016/j.bbrc.2011.10.125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Z, Sierra A, Burnett CM, Chen B, Subbotina E, Koganti SR, et al. Sarcolemmal ATP-sensitive potassium channels modulate skeletal muscle function under low-intensity workloads. J Gen Physiol. 2014;143:119–134. doi: 10.1085/jgp.201311063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zingman LV, Zhu Z, Sierra A, Stepniak E, Burnett CM, Maksymov G, et al. Exercise-induced expression of cardiac ATP-sensitive potassium channels promotes action potential shortening and energy conservation. J Mol Cell Cardiol. 2011;51:72–81. doi: 10.1016/j.yjmcc.2011.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flagg TP, Enkvetchakul D, Koster JC, Nichols CG. Muscle KATP channels: recent insights to energy sensing and myoprotection. Physiol Rev. 2010;90:799–829. doi: 10.1152/physrev.00027.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nichols CG, Lederer WJ. Adenosine triphosphate-sensitive potassium channels in the cardiovascular system. Am J Physiol. 1991;261 6 Pt 2:H1675–H1686. doi: 10.1152/ajpheart.1991.261.6.H1675. [DOI] [PubMed] [Google Scholar]
- Gong B, Miki T, Seino S, Renaud JM. A K(ATP) channel deficiency affects resting tension, not contractile force, during fatigue in skeletal muscle. Am J Physiol Cell Physiol. 2000;279:C1351–C1358. doi: 10.1152/ajpcell.2000.279.5.C1351. [DOI] [PubMed] [Google Scholar]
- Zingman LV, Hodgson DM, Bast PH, Kane GC, Perez-Terzic C, Gumina RJ, et al. Kir6.2 is required for adaptation to stress. Proc Natl Acad Sci USA. 2002;99:13278–13283. doi: 10.1073/pnas.212315199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thabet M, Miki T, Seino S, Renaud JM. Treadmill running causes significant fiber damage in skeletal muscle of KATP channel-deficient mice. Physiol Genomics. 2005;22:204–212. doi: 10.1152/physiolgenomics.00064.2005. [DOI] [PubMed] [Google Scholar]
- MacIntosh BR, Holash RJ, Renaud JM. Skeletal muscle fatigue–regulation of excitation-contraction coupling to avoid metabolic catastrophe. J Cell Sci. 2012;125 Pt 9:2105–2114. doi: 10.1242/jcs.093674. [DOI] [PubMed] [Google Scholar]
- Cifelli C, Bourassa F, Gariépy L, Banas K, Benkhalti M, Renaud JM. KATP channel deficiency in mouse flexor digitorum brevis causes fibre damage and impairs Ca2+ release and force development during fatigue in vitro. J Physiol. 2007;582 Pt 2:843–857. doi: 10.1113/jphysiol.2007.130955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cifelli C, Boudreault L, Gong B, Bercier JP, Renaud JM. Contractile dysfunctions in ATP-dependent K+ channel-deficient mouse muscle during fatigue involve excessive depolarization and Ca2+ influx through L-type Ca2+ channels. Exp Physiol. 2008;93:1126–1138. doi: 10.1113/expphysiol.2008.042572. [DOI] [PubMed] [Google Scholar]
- Gribble FM, Reimann F. Sulphonylurea action revisited: the post-cloning era. Diabetologia. 2003;46:875–891. doi: 10.1007/s00125-003-1143-3. [DOI] [PubMed] [Google Scholar]
- Selvin E, Bolen S, Yeh HC, Wiley C, Wilson LM, Marinopoulos SS, et al. Cardiovascular outcomes in trials of oral diabetes medications: a systematic review. Arch Intern Med. 2008;168:2070–2080. doi: 10.1001/archinte.168.19.2070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aguilar-Bryan L, Bryan J. Molecular biology of adenosine triphosphate-sensitive potassium channels. Endocrine Rev. 1999;20:101. doi: 10.1210/edrv.20.2.0361. [DOI] [PubMed] [Google Scholar]
- Clement JP, 4th, Kunjilwar K, Gonzalez G, Schwanstecher M, Panten U, Aguilar-Bryan L, et al. Association and stoichiometry of K(ATP) channel subunits. Neuron. 1997;18:827–838. doi: 10.1016/s0896-6273(00)80321-9. [DOI] [PubMed] [Google Scholar]
- Barclay CJ, Woledge RC, Curtin NA. Is the efficiency of mammalian (mouse) skeletal muscle temperature dependent. J Physiol. 2010;588 Pt 19:3819–3831. doi: 10.1113/jphysiol.2010.192799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu B, Li Y, Morcos PA, Doran TJ, Lu P, Lu QL. Octa-guanidine morpholino restores dystrophin expression in cardiac and skeletal muscles and ameliorates pathology in dystrophic mdx mice. Mol Ther. 2009;17:864–871. doi: 10.1038/mt.2009.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morcos PA, Li Y, Jiang S. Vivo-Morpholinos: a non-peptide transporter delivers Morpholinos into a wide array of mouse tissues. Biotechniques. 2008;45:613–4, 616, 618 passim. doi: 10.2144/000113005. [DOI] [PubMed] [Google Scholar]
- Hodgson DM, Zingman LV, Kane GC, Perez-Terzic C, Bienengraeber M, Ozcan C, et al. Cellular remodeling in heart failure disrupts K(ATP) channel-dependent stress tolerance. EMBO J. 2003;22:1732–1742. doi: 10.1093/emboj/cdg192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seino S, Miki T. Physiological and pathophysiological roles of ATP-sensitive K+ channels. Prog Biophys Mol Biol. 2003;81:133–176. doi: 10.1016/s0079-6107(02)00053-6. [DOI] [PubMed] [Google Scholar]
- Kane GC, Behfar A, Dyer RB, O'Cochlain DF, Liu XK, Hodgson DM, et al. KCNJ11 gene knockout of the Kir6.2 KATP channel causes maladaptive remodeling and heart failure in hypertension. Hum Mol Genet. 2006;15:2285–2297. doi: 10.1093/hmg/ddl154. [DOI] [PubMed] [Google Scholar]
- Davis-Taber R, Choi W, Feng J, Hoogenboom L, McNally T, Kroeger P, et al. Molecular characterization of human SUR2-containing K(ATP) channels. Gene. 2000;256:261–270. doi: 10.1016/s0378-1119(00)00338-3. [DOI] [PubMed] [Google Scholar]
- Yamada M, Isomoto S, Matsumoto S, Kondo C, Shindo T, Horio Y, et al. Sulphonylurea receptor 2B and Kir6.1 form a sulphonylurea-sensitive but ATP-insensitive K+ channel. J Physiol. 1997;499 (Pt 3):715–720. doi: 10.1113/jphysiol.1997.sp021963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chutkow WA, Pu J, Wheeler MT, Wada T, Makielski JC, Burant CF, et al. Episodic coronary artery vasospasm and hypertension develop in the absence of Sur2 K(ATP) channels. J Clin Invest. 2002;110:203–208. doi: 10.1172/JCI15672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miki T, Suzuki M, Shibasaki T, Uemura H, Sato T, Yamaguchi K, et al. Mouse model of Prinzmetal angina by disruption of the inward rectifier Kir6.1. Nat Med. 2002;8:466–472. doi: 10.1038/nm0502-466. [DOI] [PubMed] [Google Scholar]
- Jearawiriyapaisarn N, Moulton HM, Sazani P, Kole R, Willis MS. Long-term improvement in mdx cardiomyopathy after therapy with peptide-conjugated morpholino oligomers. Cardiovasc Res. 2010;85:444–453. doi: 10.1093/cvr/cvp335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goyenvalle A, Babbs A, Powell D, Kole R, Fletcher S, Wilton SD, et al. Prevention of dystrophic pathology in severely affected dystrophin/utrophin-deficient mice by morpholino-oligomer-mediated exon-skipping. Mol Ther. 2010;18:198–205. doi: 10.1038/mt.2009.248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crisp A, Yin H, Goyenvalle A, Betts C, Moulton HM, Seow Y, et al. Diaphragm rescue alone prevents heart dysfunction in dystrophic mice. Hum Mol Genet. 2011;20:413–421. doi: 10.1093/hmg/ddq477. [DOI] [PubMed] [Google Scholar]
- Widrick JJ, Jiang S, Choi SJ, Knuth ST, Morcos PA. An octaguanidine-morpholino oligo conjugate improves muscle function of mdx mice. Muscle Nerve. 2011;44:563–570. doi: 10.1002/mus.22126. [DOI] [PubMed] [Google Scholar]
- Wu B, Xiao B, Cloer C, Shaban M, Sali A, Lu P, et al. One-year treatment of morpholino antisense oligomer improves skeletal and cardiac muscle functions in dystrophic mdx mice. Mol Ther. 2011;19:576–583. doi: 10.1038/mt.2010.288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taniguchi-Ikeda M, Kobayashi K, Kanagawa M, Yu CC, Mori K, Oda T, et al. Pathogenic exon-trapping by SVA retrotransposon and rescue in Fukuyama muscular dystrophy. Nature. 2011;478:127–131. doi: 10.1038/nature10456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moulton JD, Jiang S. Gene knockdowns in adult animals: PPMOs and vivo-morpholinos. Molecules. 2009;14:1304–1323. doi: 10.3390/molecules14031304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Light PE, Comtois AS, Renaud JM. The effect of glibenclamide on frog skeletal muscle: evidence for K+ATP channel activation during fatigue. J Physiol. 1994;475:495–507. doi: 10.1113/jphysiol.1994.sp020088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matar W, Nosek TM, Wong D, Renaud J. Pinacidil suppresses contractility and preserves energy but glibenclamide has no effect during muscle fatigue. Am J Physiol Cell Physiol. 2000;278:C404–C416. doi: 10.1152/ajpcell.2000.278.2.C404. [DOI] [PubMed] [Google Scholar]
- Matar W, Lunde JA, Jasmin BJ, Renaud JM. Denervation enhances the physiological effects of the K(ATP) channel during fatigue in EDL and soleus muscle. Am J Physiol Regul Integr Comp Physiol. 2001;281:R56–R65. doi: 10.1152/ajpregu.2001.281.1.R56. [DOI] [PubMed] [Google Scholar]
- Renaud JM. Modulation of force development by Na+, K+, Na+ K+ pump and KATP channel during muscular activity. Can J Appl Physiol. 2002;27:296–315. doi: 10.1139/h02-017. [DOI] [PubMed] [Google Scholar]
- Gong B, Legault D, Miki T, Seino S, Renaud JM. KATP channels depress force by reducing action potential amplitude in mouse EDL and soleus muscle. Am J Physiol Cell Physiol. 2003;285:C1464–C1474. doi: 10.1152/ajpcell.00278.2003. [DOI] [PubMed] [Google Scholar]
- Brooks SV, Faulkner JA. Contractile properties of skeletal muscles from young, adult and aged mice. J Physiol. 1988;404:71–82. doi: 10.1113/jphysiol.1988.sp017279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hakim CH, Li D, Duan D. Monitoring murine skeletal muscle function for muscle gene therapy. Methods Mol Biol. 2011;709:75–89. doi: 10.1007/978-1-61737-982-6_5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lueck JD, Rossi AE, Thornton CA, Campbell KP, Dirksen RT. Sarcolemmal-restricted localization of functional ClC-1 channels in mouse skeletal muscle. J Gen Physiol. 2010;136:597–613. doi: 10.1085/jgp.201010526. [DOI] [PMC free article] [PubMed] [Google Scholar]