

Abstract
Mollusks are the most morphologically disparate living animal phylum, they have diversified into all habitats, and have a deep fossil record. Monophyly and identity of their eight living classes is undisputed, but relationships between these groups and patterns of their early radiation have remained elusive. Arguments about traditional morphological phylogeny focus on a small number of topological concepts but often without regard to proximity of the individual classes. In contrast, molecular studies have proposed a number of radically different, inherently contradictory, and controversial sister relationships. Here, we assembled a data set of 42 unique published trees describing molluscan interrelationships. We used these data to ask several questions about the state of resolution of molluscan phylogeny compared with a null model of the variation possible in random trees constructed from a monophyletic assemblage of eight terminals. Although 27 different unique trees have been proposed from morphological inference, the majority of these are not statistically different from each other. Within the available molecular topologies, only four studies to date have included the deep sea class Monoplacophora; but 36.4% of all trees are not significantly different. We also present supertrees derived from two data partitions and three methods, including all available molecular molluscan phylogenies, which will form the basis for future hypothesis testing. The supertrees presented here were not constructed to provide yet another hypothesis of molluscan relationships, but rather to algorithmically evaluate the relationships present in the disparate published topologies. Based on the totality of available evidence, certain patterns of relatedness among constituent taxa become clear. The internodal distance is consistently short between a few taxon pairs, particularly supporting the relatedness of Monoplacophora and the chitons, Polyplacophora. Other taxon pairs are rarely or never found in close proximity, such as the vermiform Caudofoveata and Bivalvia. Our results have specific utility for guiding constructive research planning to better test relationships in Mollusca as well as other problematic groups. Taxa with consistently proximate relationships should be the focus of a combined approach in a concerted assessment of potential genetic and anatomical homology, whereas unequivocally distant taxa will make the most constructive choices for exemplar selection in higher level phylogenomic analyses.
Keywords: Aculifera, Conchifera, Mollusca, Serialia, supertree, Testaria
Yet the discoveries and techniques of molecular biology have now provided an appropriate source for recovering homology … Molecular phylogenies work not because DNA is ‘better,’ more real, or more basic than morphology, but simply because the items of a DNA program are sufficiently numerous and independent to ensure that degrees of simple matching accurately measure homology.
S.J. Gould 1986
Unfortunately, the early hope that molecular data would provide the final word in vetting phylogenetic relationships has not yet been realized for many diverse, conspicuous, and well-studied groups, including Mollusca. Within this phylum, the eight living classes are undisputedly monophyletic and well supported by morphological and molecular analyses. They span a startling range of body plans, from cephalopods to bivalves, but also show extensive convergence and body plan modification within most groups. This phylum presents a unique challenge in understanding the relatedness of several equally disparate clades and this topology therefore remains unresolved. This is partly due to the fact that in much of organismal biology, the focus of particular projects or even particular careers remains within a constrained subclade, and many trees therefore are constructed directly or indirectly with the intent to determine the placement of one taxon of interest, rather than the structure of the total group.
Morphological or historical topological hypotheses are conservative compared with most molecular trees (or compared with the vast diversity of living molluscan body plans), staying constrained to two competing ideas: Aculifera versus Testaria (Fig. 1). The former concept proposes a clade Aculifera combining the eight-plated chitons (class Polyplacophora) and the two vermiform or aplacophoran classes (Solenogastres, Caudofoveata), contrasted with a clade Conchifera containing the remaining five classes which are traditionally seen as having a “true” shell. This hypothesis is opposed by the Testaria concept, a clade of Conchifera and Polyplacophora, with the two aplacophoran classes as sister to all other molluscan groups. These two concepts have been used to frame arguments about the bauplan of the molluscan ancestor, but this is essentially a question of whether the tree is rooted between Aplacophora and all other classes (Testaria) or between “Aculifera” (Aplacophora + Polyplacophora), and all other classes. Interestingly, although these two rooting concepts have been extensively debated, relatively little attention has been paid to the specific interrelationships of the five classes within Conchifera. Several hypotheses have been proposed for sister relationships between pairs of clades (Fig. 1), but these again are discussed as isolated hypotheses without detailed evaluation of the total molluscan tree.
Figure 1.

Schematic topology of the major evolutionary hypotheses and sister relationships proposed for the eight living classes in Mollusca: Aculifera (Solenogastres + Caudofoveata + Polyplacophora), Aplacophora (Caudofoveata + Solenogastres), Conchifera (Monoplacophora + Bivalvia + Scaphopoda + Gastropoda + Cephalopoda), Cyrtosoma (Gastropoda + Cephalopoda; historically also including Monoplacophora), Diasoma (Bivalvia + Scaphopoda), Serialia (Polyplacophora + Monoplacophora), Testaria (Conchifera + Polyplacophora).
The traditional reductionist model of the “hypothetical ancestral mollusk” hampered the resolution of molluscan topology, and its influence may still be present (Lindberg and Ghiselin 2003). Most research, including recent molecular approaches, has focused on major topological hypotheses proposed to explain the polarity of characters based on the proximity to the last common ancestor of all living mollusks, rather than considering derived synapomorphies that could aid reconstruction of relationships between specific living classes (Sigwart and Sutton 2007; Kocot et al. 2011; Smith et al. 2011). This limited approach to considering total group topology of the eight clades has had a substantial impact on the interpretation of molecular phylogenies, and all molecular trees are interpreted based on whether they do (Kocot et al. 2011; Smith et al. 2011) or do not (Passamaneck et al. 2004; Giribet et al. 2006; Wilson et al. 2010) recover a tree compatible with one of these historical textbook drawings. Differences, at finer resolution than these major competing hypotheses, are considered to be disposable to the question of total group molluscan evolution, to the point where trees are often summarized as “generalised topologies” without reference to any specific research (Sigwart and Sutton 2007, figure 1) or reproduced inaccurately without comment (e.g., Lindberg et al. 2004, figure 16.5).
The lack of clear morphological synapomorphies for any subgroups among the classes of Mollusca creates a challenging situation where it may be possible to selectively evaluate data to support more or less any association between two classes chosen at random. Molecular data provide an independent source of information; however most gene-based phylogenies produced trees that are so radically dissimilar to traditional phylogeny (e.g., Giribet et al. 2006) that they are rejected by the community of experts (e.g., Wägele et al. 2009). Two recent phylogenomic studies have brought to bear many more gene fragments and these both recovered what were claimed to be “traditional” topologies (Kocot et al. 2011; Smith et al. 2011). Yet those studies support other controversial pairings among taxa and their results are two more unique new contributions to the pantheon of proposed molluscan trees; they do not exactly match any evolutionary hypothesis proposed with morphology. Given the deep divergence of mollusks in the early Cambrian (Lindberg and Ponder 1996; Lindberg et al. 2004) and the morphological disparity present in the limited number of living clades (at the class level), the positions of all eight branches are significant in understanding the morphological evolution of this phylum.
A broad variety of combinations linking molluscan taxa have been suggested but only a small fraction of the possible topologies are so far published; given eight taxa, there are 135,135 possible combinations of the internal relationships (for rooted, bifurcating trees). Our study was motivated in part by curiosity about which relationships have and have not been proposed in the literature. We conducted an extensive literature search to establish a complete set of all published topologies, which explicitly addressed molluscan interrelationships and included a majority of the subclades (i.e., at least five of eight). We then used this database of trees to quantitatively assess whether there is any consistent signal among all these many different topological hypotheses, or between molecular and morphological trees.
Methods
We identified a set of 42 trees that have appeared in the literature and online since 1926; we ended our sampling in summer 2013 (Table 1). Some trees have been redrawn and republished many times in the literature. Multiple authors have derived others. Each unique topology is only counted once. We have attempted to record the earliest instance for each tree but not the complete publication history of a given topological construct. All trees have been included in the electronic Supplementary Material available on Dryad at http://dx.doi.org/10.5061/dryad.b4m2c. (Supplementary Table S1). The source trees were categorized as either morphological or molecular, and our further analyses were conducted on two partitions separately: Morphological trees () and molecular trees ().
Table 1.
Sources for all unique hypotheses of molluscan phylogeny (including at least five of eight classes), representing the first instance a given topology appeared in the literature. (1) This figure included a polytomy (Kano 2012 figure 4); the authors published a second, explicitly less-preferred tree with the polytomy resolved (Kano 2012 figure 5), which we added to tree dissimilarity analysis but otherwise excluded; (2) http://palaeos.com/metazoa/mollusca/mollusca.htm (content by M. Alan Kazlev 2007; archived at http://wayback.archive.org/web/*/http://www.palaeos.com; (3) http://www.pearl-guide.com/pearl-producing-mollusks.shtml (content by Jeremy Sheperd ca. 2007); (4) http://www.ucmp.berkeley.edu/taxa/inverts/mollusca/mollusca.php
| Source | Data type | Generalized topology | |
|---|---|---|---|
| Brusca and Brusca 2002, fig 20.55 | morphology | Testaria | (C) |
| Dogelya 1940, schema 7 | morphology | Testaria | * |
| Dogelya 1940, schema 1 | morphology | Aculifera | |
| Dogelya 1940, schema 2 | morphology | Aculifera | |
| Dogelya 1940, schema 5 | morphology | Testaria | * |
| Dunn et al. 2008, figure 1 | molecular | other | |
| Giribet et al. 2006, figure 2 | molecular | other (Serialia) | (S) |
| Götting 1980, p. 25 | morphology | Testaria | (C) |
| Gubanov 1998, figure 1 | morphology | other | |
| Haszprunar 2000, figure 2 | morphology | Testaria | |
| Iijima et al. 2006, figure 2 (Hox5a clade) | molecular | other | * |
| Kano 2012, figure 4 [1] | molecular | other (Serialia) | |
| Kocot et al. 2011, figure 2 | molecular | Aculifera | (C) |
| Lieb and Todt 2008, figure 2 | molecular | other (∼Aculifera) | |
| Naef 1926, p. 99 | morphology | Aculifera | * |
| Palaeos.com [online only 2] | morphology | Testaria | (C) |
| Passamaneck et al. 2004, figure 2 (minimum evolution) | molecular | other | |
| Passamaneck et al. 2004, figure 6A (maximum parsimony) | molecular | other | * |
| Passamaneck et al. 2004, figure 6B (maximum likelihood) | molecular | other | (S) |
| Pearl-Guide.com [online only 3] | morphology | other | * |
| Peel 1991, figure 46 | morphology | Testaria | |
| Runnegar 1996, figure 6.5 | morphology | Testaria | (C) |
| Runnegar and Pojeta 1974, figure 1 | morphology | other | |
| Runnegar and Pojeta 1974, figure 4 | morphology | other | |
| “Runnegar and Pojeta 1974” as reproduced in Runnegar 1996, figure 6.1B | morphology | other | * |
| Salvini-Plawen and Steiner 1996 figure 2.4 | morphology | Testaria | (C) |
| Salvini-Plawen 1985 figure 42 | morphology | Testaria | (C) |
| Salvini-Plawen 2006, figure 14 after Haszprunar and Wanninger 2000 | morphology | Testaria | * |
| Scheltema 1993, figure 12 | morphology | Aculifera | * |
| Sigwart and Sutton 2007, figure 1b | morphology | Testaria | * |
| Smith et al. 2011, figure 2 | molecular | Aculifera | (C) |
| Steiner and Reynolds 2003, figure 3A | morphology | other | |
| Stoeger et al., in press | molecular | other (Serialia) | (S) |
| UCMP molluscan phylogeny [online only 4], after Sigwart and Sutton 2007 | morphology | Aculifera | * |
| Vinther et al. 2012, figure 3 | molecular | other (∼Aculifera) | (C) |
| Wägele et al. 2009, figure 5 | molecular | other | |
| Waller 1998, figure 1 | morphology | Testaria | |
| “Waller 1998” as reproduced in as reproduced in Lindberg et al. 2004, figure 16.5 | morphology | other | |
| Wilson et al. 2010, figure 1 | molecular | other (Serialia) | (S) |
| Winnepenninckx et al. 1996, figure 4a (excluding heterodont bivalves) | molecular | other | (S) |
| Yochelson 1978, figure 1 | morphology | other (∼Serialia) | * |
| Yu 1990, text-figure 6 | morphology | Testaria | * |
Notes: *Trees including polytomies that were excluded from tree dissimilarity analyses; mutually significantly similar trees in two clusters are noted (C) Conchifera, or (S) Serialia.
Tree Dissimilarity
We calculated distance metrics between pairwise combinations of trees using the software TOPD (TOPological Distance), a Perl script to calculate the difference between two trees by various methods (Puigbò et al. 2007). We applied the “nodal” methods, which calculated root-mean-square distance between matrices of intertaxon separation for the two trees. The TOPD “random method” compares this nodal distance between two test trees to the distances from randomly generated trees, and thereby statistically tests the null hypothesis that separation between the two given trees is not different to random. TOPD calculates the nodal distance () separating the two test trees, and a 1 standard deviation (SD) confidence interval around the mean nodal distance (nodal distance random) between the two test trees and 100 random trees. Importantly, this method allows comparison of trees that have unequal taxonomic representation, using the “unpruned” results returned. If is within the span of the confidence interval, any difference between the two trees is equivalent to random noise. If the distance is greater than the maximum confidence limit, then the two trees are statistically different within 1 SD (i.e., the difference is better than random). In cases where is less than the minimum confidence limit, the two test trees are similar or equivalent topologies.
Calculation of distances within TOPD does not allow the inclusion of polytomies (i.e., three or more branches from a node). Therefore, such trees were excluded a priori from this aspect of analysis, leaving a set of 13 molecular and 16 morphological source trees that represent the most “structured” trees (Table 1); a further subset of 19 pairs were discarded because they had less than 50% overlap in included taxa, generating 387 pairwise comparisons.
Proximity of Clades
Inspecting the patterns of internodal distances in more detail, for each source tree we recorded the topological separation between individual taxa (classes) by counting the nodes separating any pair. In this approach, sister taxa or members of a polytomy have a separation of one node, etc., but the root is not counted as a node (e.g., in Fig. 1 the separation between any two clades would be 1, as they are all sisters in the illustrated polytomy, regardless of the arbitrary root between Bivalvia and Monoplacophora). Median internodal distances were calculated for every taxon pair among the data partitions (molecular, morphology, and total set). These median distances were then compared using a multi-dimensional scaling plot implemented in R (cmdscale command, R Core Development Team 2013). Most historical literature did not differentiate the two currently recognized aplacophoran classes: therefore, in the morphology tree set these two were condensed where present as “Aplacophora.”
Molluscan Supertrees
The supertrees presented here were not constructed to provide yet another hypothesis of molluscan relationships, but rather to algorithmically evaluate the relationships present in the disparate topologies proposed over the last 86 years (Table 1) (McArthur and Harasewych 2003). Similar to the TOPD analysis discussed above, the supertree approach provided a method that allowed the summarization of tree topologies with unequal taxonomic representation, which precluded tree comparisons using more traditional consensus methods where all taxa are shared amongst the trees. However, supertree reconstruction assumptions, methods, and performance are not uncontroversial (Gatesy and Springer 2004, Williams 2004, Ren et al. 2009, Kupczok 2011), and we therefore used several different methods in an attempt to avoid possible artifacts.
Four programs were used to construct supertrees:;1) Clann 3.0 (Creevey and McInerney 2005), (2) Rainbow 0.3 (Chen et al. 2004), (3) RF-Supertrees 2.0 (Bansal et al. 2010), and (4) PhySIC_IST (Scornavacca et al. 2008). These programs provided different algorithms and approaches for the construction of supertrees, including: (1) matrix representation using parsimony analysis (mrp), (2) matrix representation using flipping (mrf), (3) most similar supertree method (dfit), (4) maximum split fit (sfit), (5) average consensus (avcon), (6) maximum quartet fit (qfit), (7) Robinson–Foulds distance (RF), and (8) phylogenetic signal with induction and noncontradiction (PHYsic_IST). With these programs we ultimately constructed eight molluscan supertrees from two partitions (morphological and molecular) using seven analytical methods (Table 2). Additional program parameters and settings for each analysis are included in Table 2.
Table 2.
Parameters and results of supertree analyses
| Partition (# of trees) | Analysis | Reps | # of trees | Program score | Recovered tree (as labeled in Fig. 3) | Software |
|---|---|---|---|---|---|---|
| Molecular (15 trees) | DFIT | All trees | 1 | 1439 | d | Clann 3.0 |
| SFIT | All trees | 1 | 1439 | d | Clann 3.0 | |
| AVCON | n/a | 1 | n/a | a | Clann 3.0 | |
| QFIT | All trees | 1 | 5.54 | d | Clann 3.0 | |
| RF | 50 | 38 | 79 | b (strict), d (majority rule) | RF-Supertrees 2.0 | |
| MRF | 90 | 1 | 47 flips | c | Rainbow 0.3 | |
| MRP | 90 | 2 | 140 steps | e, f | Rainbow 0.3 | |
| Morphology (27 trees) | DFIT | All trees | 1 | 21.52 | g | Clann 3.0 |
| SFIT | All trees | 1 | 20.25 | g | Clann 3.0 | |
| AVCON | n/a | 1 | n/a | h | Clann 3.0 | |
| QFIT | All trees | 1 | 4.91 | g | Clann 3.0 | |
| RF | 50 | 51 | 103 | g (strict and majority rule) | RF-Supertrees 2.0 | |
| MRF | 90 | 1 | 41 flips | g | Rainbow 0.3 | |
| MRP | 90 | 1 | 207 steps | g | Rainbow 0.3 |
Notes: “Reps” refers to the number of random addition sequences performed by Rainbow 0.3 and the number of ratchet search iterations performed by RF-Supertrees; program scores are those reported by each of the respective software programs.
DFIT = most similar supertrees; SFIT = maximum split fit; AVCON = average consensus; QFIT = maximum quartet fit; RF = Robinson–Foulds distance; MRF = matrix representation with flipping; MRF = matrix representation with parsimony.
Our attempt to construct supertrees using the conservative consensus approach of PHYsic_IST only produced a four-taxon tree from the molecular partition source trees ([Cephalopoda, Caudofoveata), (Gastropoda, Bivalvia]) The inability of the PHYsic_IST analysis to place the remaining four groups was likely due to the absence of these taxa from many of the source trees and the conservative nature of this method (McMorris and Wilkinson 2011). In contrast, analysis of the morphological partition produced a larger seven taxon tree: ((Polyplacophora, (Monoplacophora, Cephalopoda, (Scaphopoda, Bivalvia))), (Caudofoveata, Solenogastres))). However, this tree was lacking Gastropoda, which was likely excluded because of its highly variable placement in the source trees. These trees were not further considered because of the lack of taxa compared with the complete supertrees generated by the more liberal optimization methods. However, the PHYsic_IST morphological tree did recover Scaphopoda + Bivalvia and Caudofoveata + Solenogastres (Aplacophora) clades, which were also recovered by the majority of the more liberal optimization analyses of the morphological trees (see below).
Symmetric distance differences between supertrees were calculated using PAUP 4b10 (Swofford 2002). The TreeSetVis Package (Amenta and Klinger 2002) for the Mesquite system for phylogenetic computing (Maddison and Maddison 2011) was used to visualize the distributions of the source trees and supertrees in a tree space defined by the distribution of 5000 random eight-taxon molluscan trees (Hillis et al. 2005). The visualization process uses multidimensional scaling (MDS) of the Robinson–Foulds tree-to-tree distances amongst the trees to position them in tree space (Amenta and Klinger 2002).
Results
Less than half () of the published trees include all eight taxa. The only clade present in all trees is Bivalvia. All of the other classes are absent from at least three of the trees (e.g., Scaphopoda, Cephalopoda) or up to eight (Solenogastres) and the monoplacophorans are absent from six (22%) of the morphological trees but 71% of the molecular trees. Not surprisingly, all published molecular trees include Gastropoda and Bivalvia, and all but one (Kano 2012) have included Caudofoveata. A strict consensus tree of the published topologies for total group Mollusca produced a completely unstructured polytomy with no sister relationships among any of the eight taxa. Although new trees have been continuously proposed from 1926 until the present day, the advent of molecular phylogenetics has not impinged on new variants of morphological trees appearing in published literature (Fig. 2).
Figure 2.
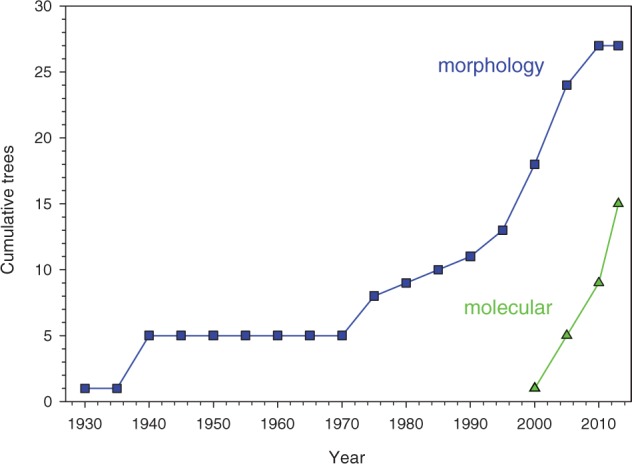
Cumulative number of new unique topologies published for molluscan evolution, from 1926–2012, from morphological (squares, ) and molecular (triangles, ) studies.
Tree Dissimilarity
Among the morphological trees examined, all trees are either similar (57.8% of comparisons), or the split distance between them is within the confidence intervals generated by comparison to random trees. Comparison within the molecular tree set finds only 45.3% are similar; 9.3% of pairs are different within the molecular data partition. Distances separating molecular trees and morphological trees show them to be similar in 27.6% of pairs, and different in 20.4% of cases. The remainder of comparisons are within the scope of random variation (1 SD of comparison with random trees) or are similar in their split distances to the mean of random trees.
Proximity of Clades
Among morphological trees, the median intermodal distances separating the taxa show strong similarity between Bivalvia + Scaphopoda, and between Cephalopoda + Gastropoda. The latter is closer to Monoplacophora (Fig. 3a). The proximity of all these conchiferan taxa collectively, reflects testarian and aculiferan topologies that are the predominant structure in morphological topologies. Molecular trees have the largest median internodal distances and classes do not form clusters but are mutually separated (Fig. 3b). The total group data set recovers clusters uniting Cephalopoda, Gastropoda, and Scaphopoda, separate from a close proximity between Monoplacophora and Bivalvia. Monoplacophora and Bivalvia are within two nodes of each other in 50% of molecular and 50% of morphological trees; Polyplacophora is closest to this pair. Aplacophora (Solenogastres + Caudofoveata) are most distant from any other taxa (Fig. 3c). In the total data set, several taxa are consistently distant; monoplacophoran are always more than two nodes separated from either aplacophoran group, and Caudofoveata in particular is always more than two nodes distant from Bivalvia (Supplementary Fig. S1 available on Dryad at http://dx.doi.org/10.5061/dryad.b4m2c).
Figure 3.

Multidimensional scaling plot of median internodal distances separating the eight molluscan classes (e.g., sister taxa are separated by one node). The space across the plot, noted by the two axes, can be read as the number of nodes separating any pair of taxa across a hypothetical tree for total group Mollusca.
Molluscan Supertrees
A total of eight molluscan supertrees were constructed (a–h, Table 2). For the molecular data partition, the DFIT, SFIT, QFIT, and the RF majority rule consensus trees all produced a single tree (Fig. 4d). The strict consensus of the RF supertrees (Fig. 4b), the AVCON analysis (Fig. 4a), and MRF (Fig. 4c) analyses produced another set of unique trees, whereas the MRP analysis produce two equally parsimonious unique trees (Fig. 3e, f). All of the molecular supertrees with the exception of the AVCON analysis reconstructed a sister relationship of Monoplacophora + Polyplacophora (Serialia) and a clade composed of Scaphopoda, Cephalopoda, Caudofoveata, and Solenogastres. A Bivalvia + Gastropoda clade was present in one of the MRP trees (Fig. 4e), the MRF tree (Fig. 4c), and in the DFIT, SFIT, QFIT, and RF (majority rule) molecular supertrees. The AVCON tree (Fig. 4a) was the most dissimilar to the other molecular supertrees. The only set of sister relationships shared with the morphological supertree was Caudofoveata + Solenogasteres (Aplacophora) which was present in MRP supertrees (Figs. 4e and 3f).
Figure 4.

Multidimensional scaling plot of 5000 randomly generated trees including eight molluscan classes (open circles) and the eight supertrees derived from 42 unique, published source trees topologies (Table 1; Supplementary Figure S2 availabe on Dryad at http://dx.doi.org/10.5061/dryad.b4m2c). Supertrees a–f (solid circles) are derived from molecular trees (), g and h (open squares) are derived from the morphological data partition (). Supertree reconstruction methods included: (a) AVCON (Clann) Supertree; (b) RF (RFS) strict consensus Supertree; (c) MRF (Rainbow) Supertree; (d) DFIT, SFIT, QFIT (Clann) and RF (RFS) majority rule consensus Supertree; (e and f) MRP (Rainbow) SupertreesAll of the above programs and options produce the same Supertree topology; and (g) from morphological source trees, with the exception of AVCON (Clann) Supertree (h).
In the morphological data partition, six of the seven supertree analyses produced identical trees (Fig. 4d); only the AVCON analysis produced a different tree (Fig. 4h). Tree scores, tree notation, permutation tail probability tests, source tree fit, and related data for each of the molecular and morphological supertree analyses reported here are presented in Supplementary Table S2 and Supplementary Table S3, respectively, available on Dryad at http://dx.doi.org/10.5061/dryad.b4m2c. Pairwise comparisons of symmetric distances between the supertrees ranged between 2 and 13 (Supplementary Table S4 available on Dryad at http://dx.doi.org/10.5061/dryad.b4m2c). The highest symmetric distance values were associated with the AVCON supertrees [Fig. 3a (molecular) and 3g (morphological)] (mean symmetric distance = 12.14 and 11.57, respectively) and are substantially higher than symmetric distance values for the remaining trees. Average symmetric distance distance among the remaining molecular trees was only 7.0 indicating greater similarity, and the morphological mean symmetric distance is similar at 7.29. Surprisingly, the AVCON morphology supertree (Fig. 4h) did not substantially differ (mean symmetric distance = 5.4) from the molecular supertrees with the exception of the AVCON molecular supetree (Fig. 4a).
The source trees were widely distributed over tree space (Supplementary Fig. S2 available on Dryad at http://dx.doi.org/10.5061/dryad.b4m2c). The majority of the morphological source trees were grouped into two clusters, one at the centre of tree space and the other near the edge. These two groupings accounted for 77% of the morphological source trees, whereas a single grouping of primarily incomplete trees account for 60% of the molecular source trees on the edge of the tree space. The remaining source trees were scattered throughout tree space. Some supertrees also showed a somewhat clustered distribution in tree space. With the exception of both the molecular (Fig. 4a) and morphological (Fig. 4h) AVCON supertrees, and the strict consensus tree of the molecular RF analysis (Fig. 4b), both the morphological supertree (Fig. 4g) and the remaining molecular supertrees (Fig. 3c–f), formed an arc along one edge of tree space (Fig. 4).
Discussion
We do not seek to endorse or to refute any particular hypothesis that has been proposed in the literature but to assess the topological disparity of published molluscan phylogenies. We have deliberately avoided commentary on the position or contribution of any specific published tree within our analyses. Indeed, in the course of this analysis we have done what we criticize in some of our colleagues, by adding yet more topologies to the published literature. But the topologies proposed to date are substantially more constrained than random arrangements of taxa (Figs. 3 and 4), and the “one true tree” for living Mollusca undoubtedly resides somewhere within the tree space presented here in Fig. 4.
All published trees are hypotheses subject to further scrutiny and additional data, but the reuse of molecular sequences for rare taxa (e.g., Caudofoveata sequence(s) used in most molecular studies) may bias analyses by the inclusion of contaminated sequences (e.g., Norén and Jondelius 1997; Bourlat et al. 2003). Contamination has been demonstrated for some aplacophoran (Meyer et al. 2010; Kocot 2013) and monoplacophoran sequences (Wilson et al. 2010). Although phylogenomic data may provide great advances, the current volume of missing data used in these analyses is problematic (Roure et al. 2013).
Morphological trees have a long history and many are topologically similar (Table 1, Supplementary Fig. S2 available on Dryad at http://dx.doi.org/10.5061/dryad.b4m2c), yet our MDS results of clade proximity show they are not as constrained as expected (Fig. 3a). Molecular analyses contribute greater topological dissimilarity and are significantly different from each other and from most morphological topologies. The disparity of topologies, and particularly the distance between morphology and molecular data partitions, is captured by the supertree analysis, by the limited number of morphological supertrees (Fig. 3g, h) and their differences with the molecular supertrees (Fig. 3a–f). Hence although there is some consistency, within each of the separate data partitions (Supplementary Table S4 available on Dryad at http://dx.doi.org/10.5061/dryad.b4m2c), there are superficially unresolvable differences between the two, which is at the root of what we aim to overcome with this study.
Competing Concepts
Three controversial competing hypotheses are typically invoked in discussions of molluscan phylogeny: Aculifera (a clade Polyplacophora + aplacophorans sister to remaining Mollusca); Testaria (aplacophorans (Polyplacophora (all remaining Mollusca))); and Serialia (a clade Polyplacophora + Monoplacophora). Critically, these topological concepts do not ascribe positional relationships to any other classes (Fig. 1). Phylogenetic results therefore are evaluated essentially with respect to the position of Polyplacophora.
The relative lack of critical discussion over other ingroup relationships has founded a very weak framework for discussion of the overall molluscan tree. Although we have not attempted to document the history of all published trees, many trees have been republished repeatedly, as topological concepts have been developed (and subsequently supported) by sequential analyses within research groups. There are few examples of the derivation of the same tree by independent analyses. This underlies some of the frustration in matching molecular and morphological topologies.
Hypothetically, the total set of unique topologies is now increased from 42 instances in the literature to 48 with the inclusion of our new supertrees and others published since our sampling closed in summer 2013. Only two of the supertrees overlap with published phylogenies or topological hypotheses. One molecular supertree (Fig. 4b) is effectively identical to one of the early trees to recover “Serialia” (Wilson et al. 2010), although that study recovered Solenogastres outside of Mollusca (an artifact attributable to contamination, and that branch was excluded from our metaanalysis, Supplemenatry Table S1 available on Dryad at http://dx.doi.org/10.5061/dryad.b4m2c). The morphological supertree (Fig. 4g) is, comfortingly, identical or nearly identical to several textbook illustrations (e.g., Brusca and Brusca 2002). That study likely inadvertently published a unique new topology (Table 1), but they succeeded in genuinely reflecting a total consensus based on available morphological data, and the dominance of the Testaria hypothesis.
Of the source trees, 9 include Aculifera (from both molecular and morphology-based studies) and 15 Testaria (all morphology); the remaining trees have other combinations of taxa including Serialia (), but some trees represent combinations of taxa that do not test these clades (e.g., by excluding aplacophorans). The traditional testarian framework suggests a progressive mode of evolution from a primitive shell-less (spicule-bearing) condition through a multi-shelled state (like that seen in chitons) toward the production of a single or bivalved shell as seen in Conchifera. This does not inherently require the ancestral mollusk to necessarily be worm-like, as such a scenario is equally possible with an early-deriving pedomorphic aplacophoran lineage (Lindberg and Ponder 1996). Interestingly, no molecular study has ever recovered Testaria. But the dominance of this concept in the molluscan phylogenetic literature, among morphological studies, is visibly the driving force in establishing the topologies of our supertree (Fig. 4g), and accounts for the distances separating taxa among morphological topologies (Fig. 3a). The fact that over half of all unique topologies we have identified in the morphological literature are variants on the Testaria clade demonstrates that it is arguably well supported by morphological inference. However, the fact that there are so many different trees primarily means that within this simple framework the placement of the more recently derived conchiferan branches has often been done without significant evolutionary hypotheses being attached to their placements. It is the branching order, and the ancestral relationships among the supposedly “higher” classes that still hold some of the most important questions in the evolution of molluscan body plan variability.
Completeness
Including or excluding exemplars of various classes controls the number of potential relationships tested among the clades in any given study. As more taxa are included in a phylogenetic data set the possible topological arrangements expand rapidly and so does the computational challenge, yet increased taxon sampling is well known to improve accuracy of phylogenetic reconstruction (e.g., Lecointre et al. 1993; Hillis 1998; Poe 1998; Hedtke et al. 2006). Much early simulation work on this question was done in a context of parsimony analysis but the same principle applies irrespective of method (Heath et al. 2008). Missing taxa may (controversially) have a more detrimental effect on the accuracy of phylogenetic reconstruction than including taxa with large volumes of missing or ambiguous data (Wiens and Morrill 2011). This point is particularly relevant in a context of a new era of character dense but (so far) taxon-poor phylogenomic data sets. Among mollusks particularly, where there is extensive morphological and molecular diversity within as well as between clades, dense taxon sampling may prove to be essential to accurate phylogenetic reconstruction.
No class is universally present in all 42 available topologies. Some are more obviously absent; for example monoplacophoran tissues suitable for DNA analyses have only been available since 2006 (Giribet et al. 2006). In some molecular studies, tissues were limited or mollusks were included in context of larger scale metazoan phylogeny (Winnepenninckx et al. 1996; Dunn et al. 2008). However, where others are missing from morphological studies it alludes to the study motivation being driving by understanding the position of a particular taxon (Runnegar and Pojeta 1974). Paleontological analyses are limited by the applicability of morphological characters to shells and soft-bodied taxa (Sigwart and Sutton 2007). Morphological trees are not less guilty of limited taxon sampling than molecular trees.
The majority of published phylogenies for Mollusca, both morphological and molecular, do not include all eight constituent classes. Although there was historical debate about whether the two vermiform aplacophoran groups represented two distinct classes, both groups have been recognized as mollusks since the 1870s. Before 1952 monoplacophorans were known only from fossils (Lindberg 2009), thus some early trees are as complete as would be expected given the state of knowledge at the time (e.g. Dogelya 1940, schema 5, 7). However, many trees simply excluded classes that were not of immediate interest to the specific study (e.g. Dogelya 1940, schema 1; Gubanov 1998, figure 1).
The first material of a monoplacophoran suitable for DNA sequencing provided the first molecular phylogeny with all eight classes and highly controversial results (Giribet et al. 2006); the proposed sister relationship of Monoplacophora + Polyplacophora was never suggested from morphology (but see Wingstrand 1985, figure 25), but has been repeatedly recovered in subsequent molecular studies (Wilson et al. 2010; Kano 2012; Stöger et al. 2013). A similarly controversial result arose in the sole phylogenomic study able to include a monoplacophoran (Smith et al. 2011) which recovered a clade Cephalopoda + Monoplacophora; this had been proposed previously based on conjecture about fossil relationships (Gubanov 1998; Kröger et al. 2011), but there are critical gaps in the connection between morphological argument and the topology recovered by molecular data (see below).
Toward Total Evidence
Historically entrenched ideas inform interpretations of novel hypotheses, and hence a summary of past ideas may not directly produce the most accurate evolutionary scenario. This resonates with similar debates over resolving molecular and morphological topologies in other major metazoan (indeed eukaryotic) clades, most comparably with arthropods (Rota-Sabelli et al. 2011), acoelomorph flatworms (Philippe et al. 2011), and echinoderms (Janies et al. 2011). Yet work on molluscan relationships has focused on the validity or not of restricted large-scale topological concepts, which are in fact driven by a very small number of taxa. Putting these much discussed hypotheses aside, we can make some robust inferences about the general interrelatedness of the classes: For example, the close association of cephalopods and gastropods (i.e., Cyrtostoma) is not supported by molecular data. The strong spreading of the classes away from each other in terms of median distance of molecular analyses (Fig. 3b) reflects the high degree of conflict among published studies.
Is the state of knowledge about molluscan phylogeny converging on a genuine robust hypothesis? One aspect to answer this conundrum is assessing the disparity between phylogenetic signals from different, more or less independent, data partitions. Clearly molecular and morphological trees for mollusks produce very different arrangements of taxa (Fig. 3). Constructing supertrees provides one approach to putting together trees with variable taxon sampling within the group (Bininda-Emonds 2004; Page 2004). Using several different analytical approaches, the morphological consensus consistently favors a testarian topology, and most analyses of molecular-derived source trees recover a Serialia clade (Fig. 3b–f). Yet, recent phylogenomic data, and some anatomical evidence, supports a dichotomy of Aculifera and Conchifera. For example, the recent phylogenomic analyses of Kocot et al. (2011) and Smith et al. (2011) support the “conchiferan” hypothesis. In fact the topology of Kocot et al. (2011) matches exactly with the morphological tree of Dogelya (1940), schema 1). The clade Bivalvia + Gastropoda referred to as Pleistomollusca (Kocot et al. 2011) has been recovered in several other molecular studies (Dunn et al. 2008; Lieb and Todt 2008; Wägele et al. 2009; Vinther et al. 2012); the only time this sister relationship has been suggested through morphological evidence was that single early tree of Dogelya (1940). Although both Kocot et al. (2011) and Smith et al. (2011) recovered a clade comprising Bivalvia + Gastropoda + Scaphopoda, the arrangement is not necessarily well supported. A clade comprising these three taxa was first proposed by Naef (1926)) and appeared in one of Dogelya's (1940, schema 5) testarian hypotheses as well. However, only one other morphological study supported a relationship uniting those three classes (Runnegar 1996). Thus, although these molecular studies agree in their support of a deep dichotomy in the phylum (Kocot 2013), it is imprecise to say that they provide consensus with morphologically derived hypotheses.
The only phylogenomic study to date that included all eight classes (Smith et al. 2011) was compared with ideas proposed in paleontological literature, and this deserves careful consideration. The hypothesis of Gubanov (1998) was really concerned with the clade Cyrtostoma (Monoplacophora + Gastropoda + Cephalopoda) and not a relationship linking monoplacophorans and cephalopods per se. Within Cyrtostoma, diverse molluscan forms could be envisioned as progressing from a (monoplacophoran-like) helcionellid ancestor (Gubanov 1998), with no specific fossil evidence to support a transitional form or sister relationship linking cephalopods to any other particular group; they remain an oddity which is assumed have arisen from a monoplacophoran-like ur-mollusk. But this is a significant historical funnel that tilts any comparison between monoplacophorans and other mollusks. If we accept a priori that monoplacophorans are anatomically similar to a hypothetical common ancestor of the conchiferan classes, and those other derived classes are adaptive forms, then it is straightforward to construct an argument that cephalopods, or members of any class, are derived from a monoplacophoran-like ancestor. Thus such arguments presented as “consensus” are misleading (e.g., Kröger et al. 2011); any proposed sister relationship between two clades should share identifiable synapomorphies.
Conclusions
Although we cannot claim to yet have a robust topology for the molluscan portion of the Tree of Life there are specific informative points that have been brought to light; for example, the support for Cyrtostoma or Diasoma (Scaphopoda + Bivalvia) is relatively weak (Fig. 3). The Serialia hypothesis has been considered contentious, but in fact is equally plausible as myriad others and worthy of due consideration. Interpretation of relationships between monoplacophorans and other taxa must urgently be considered in terms of synapomorphies uniting living clades, rather than the monoplacophoran as a putative ancestral form. This perspective is fundamental to the Testaria concept, which has dominated morphological interpretation of molluscan relationships although it is robustly evident that there is no consensus support for this topology. There are positive and intriguing results here too: The relationships among cephalopods, aplacophorans, and scaphopods deserve more attention (Fig. 4; Stöger et al. 2013). And these analyses can be applied to deeper questions outside molluscan phylogeny. There is strong evidence that Caudofoveata is very distantly related to Bivalvia (Fig. 5; Supplementary Fig. S1 available on Dryad at http://dx.doi.org/10.5061/dryad.b4m2c). Exemplars from these two taxa (or alternatively Polyplacophora and Cephalopods; Fig. S1) could therefore make an appropriate choice to represent pan-molluscan diversity within deeper metazoan phylogenetic analyses.
To date there has been no explicit combined analysis that simultaneously analyzed molecular and morphological data for a broad sampling of total group Mollusca, although this has been repeatedly identified as a priority area for resolving questions of molluscan phylogenetics. There have been studies in which representatives of several classes have served as outgroups for more comprehensive analyses of specific classes—Bivalvia (Giribet and Wheeler 2002), Cephalopoda (Lindgren et al. 2004), and Gastropoda (Aktipis et al. 2008)—but these studies had limited sampling outside of the focus clade and crucially did not include non-mollusks and hence have no way to root an ingroup molluscan topology (These limitations are explicitly, or implicitly in the case of some morphological studies, included in all the trees we considered herein). There are several limiting paradigms within the scope of molluscan evolution, which may be correct, but equally may inadvertently confound the interpretation of phylogenetic data when they are not expressed as testable hypotheses. Clearly, from the points raised here, any future study that claims to investigate pan-molluscan relationships must include exemplars—and multiple species—from all eight classes.
Supplementary Material
Data available from the Dryad Digital Repository: http://dx.doi.org/10.5061/dryad.b4m2c.
Acknowledgments
Many colleagues have contributed to discussions that inspired and improved this work; in particular, we thank John Huelsenbeck and Nick Matzke for advice on data analyses. The manuscript also benefited from the comments of an anonymous reviewer, Frank Anderson, Benoit Dayrat, and Gonzalo Giribet. JDS wishes to thank Humboldt State University, Arcata, CA for travel funds that supported research in California. Partial funding for Open Access publication was provided by the Berkeley Research Impact Initiative (BRII), University of California, Berkeley.
References
- Aktipis S.W., Giribet G., Lindberg D.R., Ponder W.F. Gastropoda: an overview and analysis. In: Ponder W.F., Lindberg D.R., editors. Phylogeny and evolution of the Mollusca. Berkeley: University of California Press; 2008. pp. 201–237. [Google Scholar]
- Amenta N., Klingner J. 2002. Case study: Visualizing sets of evolutionary trees. 8th IEEE Symposium on Information Visualization 2002:71-74.
- Bansal M.S., Burleigh J.G., Eulenstein O., Fernez-Baca D. Robinson–Foulds Supertrees. Algorithms for Molecular Biology 2010. 2010;5:18. doi: 10.1186/1748-7188-5-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baum B.R., Regan M.A. The MRP method. In: Bininda-Emonds O.R.P., editor. Phylogenetic supertrees: combining information to reveal the tree of life. Dordrecht: Springer Science; 2004. pp. 17–34. [Google Scholar]
- Bininda-Emonds O.R.P. New uses for old phylogenies. In: Bininda-Emonds O.R.P., editor. Phylogenetic supertrees: combining information to reveal the tree of life. Dordrecht: Springer Science; 2004. pp. 3–14. [Google Scholar]
- Bourlat S. J., Nielsen C., Lockyer A.E., Timothy D., Littlewood J., Telford M.J. Xenoturbella is a deuterostome that eats molluscs. Nature. 2003;424:925–928. doi: 10.1038/nature01851. [DOI] [PubMed] [Google Scholar]
- Brusca R.C., Brusca G.J. Invertebrates. 2nd. Sunderland (MA): Sinauer Associates Inc.; 2002. [Google Scholar]
- Burleigh J.G., Eulenstein O., Fernández-Baca D., Sanderson M.J. MRF Supertrees. In: Bininda-Emonds O.R.P., editor. Phylogenetic supertrees: combining information to reveal the tree of life. Dordrecht: Springer Science; 2004. pp. 65–85. [Google Scholar]
- Chen D., Eulenstein O., Fernández-Baca D. Rainbow: a toolbox for phylogenetic supertree construction and analysis. Bioinformatics. 2004;20:2872–2873. doi: 10.1093/bioinformatics/bth313. [DOI] [PubMed] [Google Scholar]
- Creevey C.J., Fitzpatrick D.A., Philip G.A., Kinsella R.J., O'Connell M.J., Travers S.A, Wilkinson M., McInerney J.O. Does a tree-like phylogeny only exist at the tips in the prokaryotes? Proc. Roy Soc. Lond, B ser Biol. Sci. 2004;271:2551–2558. doi: 10.1098/rspb.2004.2864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Creevey C.J., McInerney J.O. Clann: investigating phylogenetic information through supertree analyses. Bioinformatics. 2005;21:390–392. doi: 10.1093/bioinformatics/bti020. [DOI] [PubMed] [Google Scholar]
- Dogelya V.A. Molluscan Phylogeny. In: Dogelya V.A., Zenkevicha L.A., editors. Manual of zoology. Vol. II. Moscow: Academy of Science of the USSR; 1940. pp. 610–617. [in Russian] [Google Scholar]
- Dunn C.W., Hejnol A., Matus D.Q., Pang K., Browne W.E., Smith S.A., Seaver E., Rouse G.W., Obst M., Edgecombe G.D., Sørenson M.V., Haddock S.H.D., Schmidt-Rhaesa A., Okusu A., Kristensen R.M., Wheeler W.C., Martindale M.Q., Giribet S. Broad phylogenomic sampling improves resolution of the animal tree of life. Nature. 2008;452:745–749. doi: 10.1038/nature06614. [DOI] [PubMed] [Google Scholar]
- Gatesy J., Springer M.S. A critique of matrix representation with parsimony supertrees. In: Bininda-Emonds O.R.P., editor. Phylogenetic supertrees: combining information to reveal the tree of life. Dordrecht: Springer Science; 2004. pp. 369–388. [Google Scholar]
- Giribet G., Okusu A., Lindgren A.R., Huff S. W., Schrödl M., Nishiguchi M.K. Evidence for a clade composed of molluscs with serially repeated structures: monoplacophorans are related to chitons. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:7723–7728. doi: 10.1073/pnas.0602578103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giribet G., Wheeler WC. On bivalve phylogeny: a high-level analysis of the Bivalvia (Mollusca) based on combined morphology and DNA sequence data. Invertebr. Biol. 2002;121:271–324. [Google Scholar]
- Götting K.J. Argumente für die Deszendenz der Mollusken von metameren Antezendenten. Zool[Arguments for the descent of molluscs from metameric ancestors.]. Jb. Anat. 1980;103:211–218. [Google Scholar]
- Gould S.J. Evolution and the triumph of homology, or why history matters. Am. Sci. 1986;74:60–69. [Google Scholar]
- Gubanov A.P. The early Cambrian molluscan evolution and its palaeogeographic implications. Acta Univ. Carolinae. Geol. 1998;42:419–422. [Google Scholar]
- Haszprunar G., Wanninger A. Molluscan muscle systems in development and evolution. J. Zoolog. Syst. Evol. Res. 2000;38:157–163. [Google Scholar]
- Haszprunar G. Is the Aplacophora monophyletic? A cladistic point of view. Am. Malacol. Bull. 2000;15:115–130. [Google Scholar]
- Heath T.A., Hedtke S.M., Hillis D.M. Taxon sampling and the accuracy of phylogenetic analyses. J. Syst. Evol. 2008;46:239–257. [Google Scholar]
- Hedtke S.M., Townsend T.M., Hillis D.M. Resolution of phylogenetic conflict in large data sets by increased taxon sampling. Syst. Biol. 2006;55:522–529. doi: 10.1080/10635150600697358. [DOI] [PubMed] [Google Scholar]
- Hillis D.M. Taxonomic sampling, phylogenetic accuracy, and investigator bias. Syst. Biol. 1998;47:3–8. doi: 10.1080/106351598260987. [DOI] [PubMed] [Google Scholar]
- Hillis D.M., Heath T.A., St John K. Analysis and visualization of tree space. Syst. Biol. 2005;54:471–482. doi: 10.1080/10635150590946961. [DOI] [PubMed] [Google Scholar]
- Iijima M., Akiba N., Sarashina I., Kuratani S., Endo K. Evolution of Hox genes in molluscs: a comparison among seven morphologically diverse classes. J. Molluscan Stud. 2006;72:259–266. [Google Scholar]
- Janies D.A., Voight J.R., Daly M. Echinoderm phylogeny including Xyloplax a progenetic asteroid. Syst. Biol. 2011;60:420–438. doi: 10.1093/sysbio/syr044. [DOI] [PubMed] [Google Scholar]
- Kano Y., Kimura S., Kimura T., Warén A. Living Monoplacophora: morphological conservatism or recent diversification? Zool. Script. 2012;41:471–488. [Google Scholar]
- Kocot K.M., Cannon J.T., Todt C., Citarella M.R., Kohn A.B., Meyer A., Santos S.R., Schander C., Moroz L.L., Lieb B., Halanych K.M. Phylogenomics reveals deep molluscan relationships. Nature. 2011;477:452–456. doi: 10.1038/nature10382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kocot K.M. Recent advances and unanswered questions in deep molluscan phylogenetics. American Malacological Bulletin. 2013;31:195–208. [Google Scholar]
- Kröger B., Vinther J., Fuchs D. Cephalopod origin and evolution: a congruent picture emerging from fossils, development and molecules. Bioessays. 2011;33:602–613. doi: 10.1002/bies.201100001. [DOI] [PubMed] [Google Scholar]
- Kupczok A. Consequences of different null models on the tree shape bias of supertree methods. Syst Biol. 2011;60:218–225. doi: 10.1093/sysbio/syq086. [DOI] [PubMed] [Google Scholar]
- Lecointre G., Philippe H., Van Le H.L., Le Guyader H. Species sampling has a major impact on phylogenetic inference. Mol. Phylogenet. Evol. 1993;2:205–224. doi: 10.1006/mpev.1993.1021. [DOI] [PubMed] [Google Scholar]
- Lieb B., Todt C. Hemocyanin in mollusks? A molecular survey and new data on hemocyanin genes in Solenogastres and Caudofoveata. Mol. Phylogenet. Evol. 2008;49:382–385. doi: 10.1016/j.ympev.2008.06.005. [DOI] [PubMed] [Google Scholar]
- Lindberg D.R. Monoplacophorans and the origin and relationships of mollusks. Evo. Edu. Outreach. 2009;2:191–203. [Google Scholar]
- Lindberg D.R., Ghiselin M.T. 2003. Fact, theory, and tradition in the study of molluscan origins. Proc. Calif. Acad. Sci. 54:663-686.
- Lindberg D.R., Ponder W.F. An evolutionary tree for the Mollusca: branches or roots? In: Taylor J., editor. Origin and evolutionary radiation of the Mollusca. Oxford: Oxford University Press; 1996. pp. 67–75. [Google Scholar]
- Lindberg D.R., Ponder W.F., Haszprunar G. The Mollusca: relationships and patterns from their first half-billion years. In: Cracraft J., Donoghue M.J., editors. Assembling the tree of life. New York: Oxford University Press; 2004. pp. 252–278. [Google Scholar]
- Lindgren A.R., Giribet G., Nishiguchi M.K. A combined approach to the phylogeny of Cephalopoda (Mollusca) Cladistics. 2004;20:454–486. doi: 10.1111/j.1096-0031.2004.00032.x. [DOI] [PubMed] [Google Scholar]
- Maddison W.P., Maddison D.R. 2011. Mesquite: a modular system for evolutionary analysis. Version 2.75. Available from: URL http://mesquiteproject.org (last accessed March 6, 2015).
- Meyer A., Todt C., Mikkelsen N.T., Lieb B. Fast evolving 18S rRNA sequences from Solenogastres (Mollusca) resist standard PCR amplification and give new insights into mollusk substitution rate heterogeneity. BMC Evolutionary Biology. 2010;10:70. doi: 10.1186/1471-2148-10-70. doi:10.1186/1471-2148-10-70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McArthur A.G., Harasewych M.G. Molecular systematics of the major lineages of the Gastropoda. In: Lydeard C., Lindberg D.R., editors. Molecular systematics and phylogeography of mollusks. Washington: Smithsonian Books; 2003. pp. 140–160. [Google Scholar]
- McMorris F. R., Wilkinson M. Conservative supertrees. Syst. Biol. 2011;60:232–238. doi: 10.1093/sysbio/syq091. [DOI] [PubMed] [Google Scholar]
- Naef A. Studien zur generellen Morphologie der Mollusken 3, Teil : Die typischen Beziehungen der Weichtiere untereinander und das Verhältnis ihrer Urformen zu anderen Cölomaten. Erg. Fort. Zool. 1926;6:27–124. [Google Scholar]
- Norén M., Jondelius U. Xenoturbella's molluscan relativest. Nature. 1997;390:31–32. [Google Scholar]
- Page R.D.M. Taxonomy, supertrees, and the tree of life. In: Bininda-Emonds O.R.P., editor. Phylogenetic supertrees: combining information to reveal the tree of life. Dordrecht: Springer Science; 2004. pp. 247–265. [Google Scholar]
- Passamaneck Y.J., Schander C., Halanych K.M. Investigation of molluscan phylogeny using large-subunit and small-subunit nuclear rRNA sequences. Mol. Phylogenet. Evol. 2004;32:25–38. doi: 10.1016/j.ympev.2003.12.016. [DOI] [PubMed] [Google Scholar]
- Peel J.S. Functional morphology of the class Helcionelloida nov., and the early evolution of Mollusca. In: Simonetta A.M., Conway Morris S., editors. The early evolution of Metazoa and the significance of problematic taxa. Cambridge: Cambridge University Press; 1991. pp. 157–177. [Google Scholar]
- Philippe H., Brinkmann H., Copley R.R., Moroz L.L, Nakano H., Poustka A.J., Wallberg A., Peterson K.J., Telford M.J. 2011. Acoelomorph flatworms are deuterostomes related to Xenoturbella Nature 470:255–258. [DOI] [PMC free article] [PubMed]
- Piaggio-Talice R., Burleigh J.G., Eulenstein O. Quartet supertrees. In: Bininda-Emonds O.R.P., editor. Phylogenetic supertrees: combining information to reveal the tree of life. Dordrecht: Springer Science; 2004. pp. 173–191. [Google Scholar]
- Poe S. Sensitivity of phylogeny estimation to taxonomic sampling. Syst. Biol. 1998;47:18–31. doi: 10.1080/106351598261003. [DOI] [PubMed] [Google Scholar]
- Puigbò P., Garcia-Vallvé S., McInerney J.O. TOPD/FMTS: a new software to compare phylogenetic trees. Bioinformatics. 2007;23:1556–1558. doi: 10.1093/bioinformatics/btm135. [DOI] [PubMed] [Google Scholar]
- Ren F., Tanaka H., Yang Z. A likelihood look at the supermatrix-supertree controversy. Gene. 2009;441:119–125. doi: 10.1016/j.gene.2008.04.002. [DOI] [PubMed] [Google Scholar]
- Rota-Stabelli O., Campbell L., Brinkmann H., Edgecombe G.D., Longhorn S.J., Peterson K.J., Pisani D., Philippe H., Telford M.J. 2011. A congruent solution to arthropod phylogeny: phylogenomics, microRNAs and morphology support monophyletic Mandibulata. Proc Roy Soc Lond., B Biol Sci 278:298–306. [DOI] [PMC free article] [PubMed]
- Roure B., Baurain D., Philippe H. Impact of missing data on phylogenies inferred from empirical phylogenomic datasets. Mol. Phylogenet. Evol. 2013;30:197–214. doi: 10.1093/molbev/mss208. [DOI] [PubMed] [Google Scholar]
- Runnegar B. Early evolution of the Mollusca: the fossil record. In: Taylor J., editor. Origin and evolutionary radiation of the Mollusca. Oxford: Oxford University Press; 1996. pp. 77–87. [Google Scholar]
- Runnegar B., Pojeta J., Jr Molluscan phylogeny: the paleontological viewpoint. Science. 1974;186:311–317. doi: 10.1126/science.186.4161.311. [DOI] [PubMed] [Google Scholar]
- Scheltema A.H. Aplacophora as progenetic aculiferans and the coelomate origin of mollusks as the sister taxon of Sipuncula. Biol. Bull. 1993;184:57–78. doi: 10.2307/1542380. [DOI] [PubMed] [Google Scholar]
- Scornavacca C., Berry V., Lefort V., Douzery E. J., Ranwez V. PhySIC_IST: cleaning source trees to infer more informative supertrees. BMC Bioinformatics. 2008;9:413. doi: 10.1186/1471-2105-9-413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sigwart J.D., Sutton M.D. 2007. Deep molluscan phylogeny: synthesis of palaeontological and neontological data. Proc. Roy. Soc. Lond. B. Biol. Sci. 274:2413-2419. [DOI] [PMC free article] [PubMed]
- Smith S.A., Wilson N.G., Goetz F.E., Feehery C., Andrade S.C.S., Rouse G.W., Giribet G., Dunn C.W. Resolving the evolutionary relationships of molluscs with phylogenomic tools. Nature. 2011;480:364–367. doi: 10.1038/nature10526. [DOI] [PubMed] [Google Scholar]
- Steiner G., Reynolds P.D. Molecular systematics of the Scaphopoda. In: Lydeard C., Lindberg D.R., editors. Molecular systematics and phylogeography of mollusks. Washington: Smithsonian Books; 2003. pp. 123–139. [Google Scholar]
- Stöger O., Sigwart J.D., Kano Y., Knebelsberger T., Marshall B.A., Schwabe E., Schrödl M. The Continuing Debate on Deep Molluscan Phylogeny: evidence for Serialia (Mollusca, Monoplacophora + Polyplacophora) BioMed Res. Int. 2013 doi: 10.1155/2013/407072. Article ID 407072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swofford D.L. 2002. PAUP software Version. 4.0 b10: phylogenetic analysis using parsimony, Sinauer Associates, Sunderland, Mass. USA.
- Vinther J., Sperling E.A., Briggs D. E., Peterson K.J. 2012. A molecular palaeobiological hypothesis for the origin of aplacophoran molluscs and their derivation from chiton-like ancestors. Proc. Roy. Soc. Lond. B. Biol. Sci. 279:1259-1268. [DOI] [PMC free article] [PubMed]
- von Salvini-Plawen L. Early evolution and the primitive groups. In: Trueman E.R., Clarke M.R., editors. The Mollusca vol. 10: Evolution. Waltham (MA): Academic Press; 1985. pp. 59–150. [Google Scholar]
- von Salvini-Plawen L. The significance of the Placophora for molluscan phylogeny. Venus. 2006;65:1–17. [Google Scholar]
- von Salvini-Plawen L., Steiner G. Synapomorphies and plesiomorphies in higher classification of Mollusca. In: Taylor J., editor. Origin and evolutionary radiation of the Mollusca. Oxford: Oxford University Press; 1996. pp. 29–51. [Google Scholar]
- Wägele J.W., Letsch H., Klussmann-Kolb A., Mayer C., Misof B., Wägele H. Phylogenetic support values are not necessarily informative: the case of the Serialia hypothesis (a mollusk phylogeny) Front. Zool. 2009;6:12. doi: 10.1186/1742-9994-6-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waller T.R. Origin of the molluscan class Bivalvia and a phylogeny of major groups. In: Johnston P., editor. Bivalves: an eon of evolution. Paleobiological studies honoring Norman D. Newell. Calgary: University of Calgary Press; 1998. pp. 1–45. [Google Scholar]
- Wiens J.J., Morrill M.C. Missing data in phylogenetic analysis: reconciling results from simulations and empirical data. Syst. Biol. 2011;60:719–731. doi: 10.1093/sysbio/syr025. [DOI] [PubMed] [Google Scholar]
- Williams D.M. Supertrees, components and three-item data. In: Bininda-Emonds O.R.P., editor. Phylogenetic supertrees: combining information to reveal the tree of life. Dordrecht: Springer Science; 2004. pp. 389–408. [Google Scholar]
- Wilson N.G., Rouse G.W., Giribet G. Assessing the molluscan hypothesis Serialia (Monoplacophora + Polyplacophora) using novel molecular data. Mol. Phylogenet. Evol. 2010;54:187–193. doi: 10.1016/j.ympev.2009.07.028. [DOI] [PubMed] [Google Scholar]
- Wingstrand K.G. On the anatomy and relationships of Recent Monoplacophora. Galathea Repts. 1985;16:7–94. [Google Scholar]
- Winnepenninckx B., Backeljau T., De Wachter R. Investigation of molluscan phylogeny on the basis of 18S rRNA sequences. Mol. Phylogenet. Evol. 1996;13:1306–1317. doi: 10.1093/oxfordjournals.molbev.a025577. [DOI] [PubMed] [Google Scholar]
- Yochelson E.L. Alternative approach to interpretation of phylogeny of ancient mollusks. Malacologia. 1978;17:165–191. [Google Scholar]
- Yu W. The first radiation of shelled molluscs. Palaeontol. Cathayana. 1990;5:139–170. [Google Scholar]