

Abstract
Myasthenia gravis is an autoimmune disease characterized by muscle weakness due to neuromuscular junction (NMJ) damage by anti-acetylcholine receptor (AChR) auto-antibodies and complement. In experimental autoimmune myasthenia gravis (EAMG), which is induced by immunization with Torpedo AChR in CFA, anti-AChR IgG2b and IgG1 are the predominant isotypes in the circulation. Complement activation by isotypes such as IgG2b plays a crucial role in EAMG pathogenesis; this suggested the possibility that IgG1, which does not activate complement through the classical pathway, may suppress EAMG. In this study, we show that AChR-immunized BALB/c mice genetically deficient for IgG1 produce higher levels of complement-activating isotypes of anti-AChR, especially IgG3 and IgG2a, and develop increased IgG3/IgG2a deposits at the NMJ, as compared to wild type (WT) BALB/c mice. Consistent with this, AChR-immunized IgG1−/− BALB/c mice lose muscle strength and muscle AChR to a greater extent than AChR-immunized WT mice. These observations demonstrate that IgG1 deficiency leads to increased severity of EAMG associated with an increase in complement activating IgG isotypes. Further studies are needed to dissect the specific role or mechanism of IgG1 in limiting EAMG and that of EAMG exacerbating role of complement activating IgG3 and IgG2a in IgG1 deficiency.
Keywords: Myasthenia gravis, Experimental autoimmune myasthenia gravis, Autoimmunity, Acetylcholine receptor, IgG1, IgG3
1. Introduction
Muscle weakness and fatigue in autoimmune myasthenia gravis (MG) is caused by decreased activation of postsynaptic muscle AChR by ACh, due to antibody- and complement-mediated AChR destruction (Ha and Richman, 2015; Huda et al., 2014). A mouse model of experimental autoimmune MG (EAMG), which mimics MG, is induced by immunizing mice 2–3 times with Torpedo AChR in CFA (Christadoss et al., 2000). The predominant anti-AChR Ig isotype that develops after two immunizations of these mice is IgG2b, followed by IgG1 (Allman et al., 2011), which have features in common with human IgG1 and IgG4, respectively (Hussain and Kifayet, 1995; Nimmerjahn and Ravetch, 2010). By immuno-histochemical methods and radioimmunoassay, we and others have previously shown that IgG2b binding to AChR at the neuro-muscular junction (NMJ), triggers complement activation that leads to formation of the membrane attack complex (MAC), which depletes functional muscle AChRs at the NMJ (Nakano and Engel, 1993; Kusner and Kaminski, 2012; Tüzün and Christadoss, 2013). Complement activation by anti-AChR antibody is required for AChR depletion and EAMG development (Michaelsen et al, 2006). However, while mouse IgG2b (as well as mouse IgG2a and IgG3 and human IgG1 and IgG3) effectively activates the classical complement cascade, mouse IgG1 and human IgG2 and IgG4 have little or no ability to do this (Redpath et al, 1998). Consequently, while IgG2b anti-AChR is the critical isotype for EAMG pathogenesis (Tüzün et al, 2012), IgG1 anti- AChR antibody might suppress the development of this disorder by competing with complement-activating isotypes for AChR epitopes and/or inhibiting the production of complement-activating isotypes through negative feedback mechanisms. To test our hypothesis, we compared the responses of WT and IgG1-deficient BALB/c mice to Torpedo AChR/CFA immunization. BALB/c mice are relatively less susceptible to EAMG (Graus et al, 1993; Fuchs et al, 1976; Berman and Patrick, 1980). We find that IgG1 deficiency increases production of complement-activating isotypes of anti-AChR Ab, including IgG2a, IgG2b and IgG3, in Torpedo AChR/CFA-immunized BALB/c mice and increases their EAMG susceptibility.
2. Materials and methods
2.1. Animals, EAMG model and clinical evaluation of EAMG
BALB/c IgG1-deficient (IgG1−/−) mice were generated as described (Jung et al., 1993). Age matched BALB/cJ mice (12 weeks, WT) were purchased from Jackson Laboratories (Bar Harbor, Maine). AChR from the electric organ of Torpedo californica was affinity purified with a neurotoxin affinity column (Wu et al., 2013). Mice were immunized and boosted at 4 and 8 weeks with 20 μg of affinity-purified Torpedo AChR emulsified in CFA (heat-killed Mycobacterium butyricum) (Wu et al., 2013). All IgG1−/− mice were maintained in a barrier facility according to the Animal Care and Use Committee guidelines of the University of Texas Medical Branch. Grip strength of mice was quantified with a Dynamometer (Chatillion Digital Force Gauge, Columbus Instruments, OH) twice a week for 5 weeks (Wu et al., 2013). Mice were bled 2 weeks after the second immunization and 4 weeks after the third immunization, prior to termination. Serum, triceps muscle, spleen, lymph nodes and carcasses were either used for in vitro culture or stored frozen at −80 °C.
2.2. ELISAs for anti-mouse AChR Ab
Levels of anti-mouse AChR Ab were determined by ELISA, using serum at a dilution of 1:2000 and mouse AChR (0.5 μg/mL) as a coating antigen. Horseradish peroxidase (HRP)-conjugated rat anti-mouse IgG1, IgG2a, IgG2b, or IgM and biotin-conjugated rat anti-mouse IgG3 mAb and streptavidin HRP (BD Bioscience) were used at a 1:1000 dilution. ABTS was used as substrate for an ELISA (Wu et al, 2013).
2.3. LN cytokines
At termination of mice, draining inguinal, popliteal, and axillary lymph node cells (LNCs) from mice were cultured in 48 well plates. Cells were stimulated with Torpedo AChR at a concentration of 2.5 μg/mL for 72 h and IFNγ, IL-6 and IL-4 concentrations in the culture supernatants were measured with ELISA kits from Invitrogen (Yang et al., 2005).
2.4. Radioimmunoassay for muscle AChR quantitation
Mouse muscle extract was prepared from frozen carcasses and AChR were quantified as described in our previous paper (Wu et al, 2013).
2.5. Immunofluorescence for IgG isotypes and C3 at NMJ
Frozen triceps muscle sections were mounted onto microscope slides, fixed in cold acetone, washed, blocked with 2% BSA and incubated at 4°C overnight with α-bungarotoxin (BTX) conjugated to Alexa 555, Alexa 350- and Alexa 488-conjugated anti-mouse IgG isotypes, anti-mouse IgM-FITC (Invitrogen) and anti-C3 (1:100) (Cedarlane, CA). Anti-C3 Ab-incubated sections were further probed with Alexa 350-conjugated secondary antibody (Invitrogen). After washing in PBS, the sections were air dried and photographed with an Olympus IX-70 microscope (10X, 20X) equipped with a DP-11 digital camera. Ig-isotypes and C3-binding to BTX sites in the muscle sections was identified and photographed (Wu et al, 2013; Huda et al, 2013). Fluorescence deposits colocalized at the NMJ (positive for BTX) from 100 microscopic fields of the frozen muscle sections from 5 samples were counted. Mean numbers were plotted for relative quantitation of Ig isotype-deposits and C3 at the NMJs.
2.6. Statistical analysis
Grip strength analysis and all other data were compared and evaluated by using Student’s t-test or the Mann–Whitney U test, where applicable. Calculated P values were considered significant at <0.05 (*), <0.01 (**), <0.001 (***) etc.
3. Results
3.1. Grip strength and clinical evaluation of CFA/AChR-immunized IgG1−/− and WT mice
Clinically relevant muscle weakness is generally induced in mice by two or three immunizations with Torpedo AChR in adjuvant. CFA/AChR immunization decreased the mean grip strength of IgG1−/− mice of both genders significantly more than that of gender-matched WT mice as early as 3 weeks after the initial immunization; this difference persisted for at least 5 weeks after a third immunization with CFA/AChR (Fig. 1, A, B). Following the third immunization with CFA/AChR, 80% of IgG1−/− - and 10% WT BALB/c mice exhibited muscle weakness. The grip strength of IgG1−/− mice that had been inoculated with CFA without AChR did not differ significantly from that of similarly treated BALB/c mice (Fig. 1, A). The mean body weights of IgG1−/− and WT immunized mice (with either CFA or CFA/AChR), both genders were comparable. (Fig. 1, C).
Fig. 1.

Grip strength in CFA- and CFA/AChR-immunized IgG1−/− vs. WT BALB/c mice. IgG1−/− and WT BALB/c mice were immunized twice with CFA/AChR. Both male and female CFA/AChR immunized IgG1−/− mice had significantly less grip strength than WT mice (p < 0.001 after second immunization) (A, B). Grip strengths of CFA immunized IgG1−/− and WT mice did not differ significantly (A, right). Differences in mean body weight of CFA- or CFA/AChR immunized IgG1−/− and WT mice were not significant (C). Fore limb grip strength from each mouse was the average value of 5 repeats, read from the digital dynamometer each time. Grip strength was determined twice a week for 5 weeks (n = 10 for IgG1−/− and WT mice, each gender, M — male; F — female).
3.2. Levels of complement-activating IgG isotypes are elevated in immunized IgG1−/− mice
IgG2a, IgG2b, and especially IgG3 levels of anti-AChR Ab were markedly elevated in IgG1−/− mice immunized with CFA/AChR as compared to immunized WT mice, while IgM levels were similar in both strains (Fig. 2). As expected, WT, but not IgG1−/− mice developed IgG1 specific anti-AChR Abs. Both wild type and IgG1−/− BALB/c mice immunized with CFA had only background levels of anti-AChR Abs and were not significantly different from one other (not shown).
Fig. 2.

Serum AChR antibody levels of IgG1−/− and WT mice. Serum level of anti-AChR auto-Ab was determined by ELISA using affinity purified mouse AChR as a coating antigen. A dramatic increase in anti-AChR IgG3 level was seen in all CFA/AChR immunized IgG1−/− mice (male and female), ***p < 0.001. The increase in serum IgG2a, IgG2b and IgG3, but not IgM anti-AChR Ab was significant in CFA/AChR immunized IgG1−/− mice compared to CFA/AChR immunized wild type mice, *p < 0.05, **p < 0.01. Results are representative of 3 independent experiments. Vertical bars represent standard error, n = 10 each for IgG1−/− and WT mice, M — male; F — female.
3.3. Cytokine secretion
We and others have previously shown that IL6 and Th1 cytokine, IFN-γ play an essential role in immunopathogenesis of EAMG (Deng et al., 2002; Feferman et al., 2005). Therefore, we tested if EAMG susceptibility of IgG1−/− BALB/c mice is similarly associated with an enhanced IL-6 and Th1 response. Indeed, LNCs from CFA/AChR immunized IgG1−/−, but not wild type mice, secreted elevated levels of IFN-γ and IL-6 (Fig. 3). Neither strain demonstrated increased IL-4 secretion (Fig. 3).
Fig. 3.
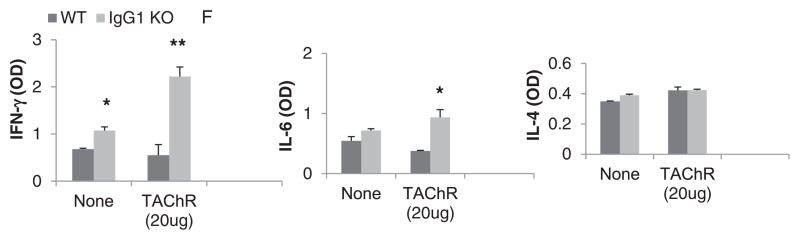
Cytokine secretion by Ag-activated LNCs. Cytokine levels in LN cell culture supernatant were measured by ELISA. IgG1−/− mice showed significantly higher levels of IFN-γ and IL-6 production upon AChR stimulation as compared to wild type mice, whereas IL-4 levels were comparable in both groups. Results are representative of 3 independent experiments. Vertical bars represent standard error, n= 10 each for IgG1−/− and WT mice. Only data obtained from male mice are shown.
3.4. Reduced quantity of functional AChR at NMJ in the muscles of IgG1−/− mice
To determine whether CFA/AChR immunization causes greater loss of AChR in IgG1−/− mice than in WT mice, as might be predicted by the greater loss of grip strength in the IgG1−/− mice, we assessed the levels of functional AChR in the membrane fraction in lysates prepared from the carcasses of CFA/AChR immunized IgG1−/− and WT mice. Results demonstrate a significant reduction of functional muscle AChR content in CFA/AChR immunized IgG1−/− group as compared to the CFA/AChR immunized WT group (*p < 0.05) (Fig. 4).
Fig. 4.

Muscle AChR content in IgG1−/− immunized with CFA/AChR. BALB/c WT and IgG1−/− mice (8 weeks.) were immunized and boosted twice with CFA or CFA/AChR. Upon termination, muscle lysates were prepared from mice carcasses. Radioimmunoassay using 125I-α bungarotoxin reflected the functional muscle AChR in CFA/AChR immunized IgG1−/− mice versus WT as 4.7 ± 0.5 × 10−12 mol and 3.09 ± 0.14 × 10−12 α bungarotoxin binding sites respectively, *p < 0.05. CFA immunized IgG1−/− and WT mice had no significant difference in functional AChR levels in muscle. Vertical bars represent standard error, n=10 each for IgG1−/− and wild type mice. Result is representative of 3 independent experiments.
3.5. Antibody and complement binding to AChR in vivo
Antibody binding to AChR in muscle endplates was evaluated by simultaneously staining muscle sections for AChR with α-bungarotoxin (BTX) and Abs that are specific for each of the IgG isotypes and C3. Results (Fig. 5) showed that a considerably higher percentage of muscle endplates (BTX + ve) stained for IgG3 and IgG2a in AChR-immunized IgG1−/− than in WT mice; that IgG2b staining was similar (and less than staining in IgG1−/− mice for IgG3 and IgG2a) for both strains and that C3 staining was greater for the IgG1−/− than for WT mice.
Fig. 5.

Ig-isotype deposits inmuscle NMJs of CFA/AChR immunized IgG1−/− and WT mice. Representative cryostat sections of the right triceps muscles of mice were fixed and stained with alpha-BTX, and antibodies conjugated with Alexa dyes. The representative images of sections in the top and bottom panels showed: Ig-isotype deposits at the NMJ (BTX positive AChRs) (A, B). A semi-quantitative assessment of Ig-deposits at the NMJ indicates higher frequency of IgG3 and IgG2a deposits in CFA/AChR immunized IgG1−/− mice as compared to WT mice or CFA mice. Result is representative of 3 independent experiments. Vertical bars represent standard error, n = 7, each for IgG1−/− and WT mice.
4. Discussion
Because the destruction of AChRs in EAMG is complement-dependent, we hypothesized that IgG isotypes that fail to activate complement through the classical pathway, such as mouse IgG1 and human IgG2 and IgG4, might suppress the development and severity of EAMG by competing with complement-activating isotypes for binding to AChR isotypes and interfering with complement activation. We indeed found that IgG1 deficiency is associated with exacerbated EAMG in BALB/c background mice (used because this strain normally develops mild disease), as shown by decreased grip strength and decreased muscle AChR content in the IgG1−/− mice. However, our observations demonstrate a more complex connection between IgG1 deficiency and disease exacerbation than was hypothesized. Specifically, our data show that the absence of IgG1 results in the increased production of complement-activating IgG isotypes of anti-AChR Ab, especially IgG3. This may result from IgG1 suppression of B cell activation, possibly because this isotype, unlike others, binds more strongly to inhibitory than to stimulatory B cell FcγRs (McLay et al., 2002). That mechanism, however, would not explain the particularly large increase in IgG3 secretion in immunized IgG1−/− mice. This may result because the γ3 constant region gene is immediately upstream of the γ1 constant region gene; a loss of ability to isotype switch to IgG1 may result in default switching to IgG3.
A dominant IgG3 response would be expected to exacerbate EAMG. In addition to activating complement through the classical pathway, mouse IgG3, like human IgG3, has the ability to autoaggregate (Trendelenburg et al., 2005). This property should increase the binding of IgG3 to its ligand and, as a result of generating larger complexes, increase the ability to activate complement. Although this property is likely to prove beneficial to hosts infected with bacterial or viral pathogens by increasing pathogen killing, it also increases tissue damage in autoimmunity, as has been shown previously in a mouse model of systemic lupus erythematosus (Takahashi et al., 1991). In contrast, isotypes such as mouse IgG1 that have limited ability to activate effector mechanisms may confer a selective advantage by limiting inflammation and tissue damage in inflammatory and autoimmune disorders. Although we have not tested the ability of IgG1 antibody to limit or reverse the development of EAMG, these considerations raise the possibility that Abs of this isotype (or the analogous human isotype, IgG4) may be useful as a therapeutic in patients with a chronic, Ab-mediated disorder such as MG (Strait et al., 2014).
Acknowledgments
This study was partly supported by R01 AI072040 (FDF, RS and PC), the Myasthenia Gravis Foundation of IL, the Association Francaise contre les Myopathies (14473) and the Muscular Dystrophy Association (217696) and a Merit Award from the Department of Veterans Affairs (FDF).
References
- Allman W, Saini SS, Tuzun E, Christadoss P. Characterization of peripheral blood acetylcholine receptor-binding B cells in experimental myasthenia gravis. Cell Immunol. 2011;271(2):292–298. doi: 10.1016/j.cellimm.2011.07.007. [DOI] [PubMed] [Google Scholar]
- Berman PW, Patrick J. Linkage between the frequency of muscular weakness and loci that regulate immune responsiveness inmurine experimental-myasthenia gravis. J Exp Med. 1980;152(3):507–520. doi: 10.1084/jem.152.3.507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christadoss P, Poussin M, Deng C. Animal models of myasthenia gravis. Clin Immunol. 2000;94:75–87. doi: 10.1006/clim.1999.4807. [DOI] [PubMed] [Google Scholar]
- Deng C, Goluszko E, Tüzün E, Yang H, Christadoss P. Resistance to experimental autoimmune myasthenia gravis in IL-6-deficient mice is associated with reduced germinal center formation and C3 production. J Immunol. 2002;169(2):1077–1083. doi: 10.4049/jimmunol.169.2.1077. [DOI] [PubMed] [Google Scholar]
- Feferman T, Maiti PK, Berrih-Aknin S, Bismuth J, Bidault J, Fuchs S, Souroujon MC. Overexpression of IFN-induced protein 10 and its receptor CXCR3 in myasthenia gravis. J Immunol. 2005;174(9):5324–5331. doi: 10.4049/jimmunol.174.9.5324. [DOI] [PubMed] [Google Scholar]
- Fuchs S, Nevo D, Tarrab-Hazdai R, Yaar I. Strain differences in the autoimmune response of mice to acetylcholine receptors. Nature. 1976;263:329–330. doi: 10.1038/263329a0. [DOI] [PubMed] [Google Scholar]
- Graus YM, van Breda Vriesman PJ, de Baets MH. Characterization of anti-acetylcholine receptor (AChR) antibodies from mice differing in susceptibility for experimental autoimmune myasthenia gravis (EAMG) Clin Exp Immunol. 1993;92:506–513. doi: 10.1111/j.1365-2249.1993.tb03429.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ha JC, Richman DP. Myasthenia gravis and related disorders: pathology and molecular pathogenesis. Biochim Biophys Acta. 2015;1852 (4):651–657. doi: 10.1016/j.bbadis.2014.11.022. [DOI] [PubMed] [Google Scholar]
- Huda R, Tüzün E, Christadoss P. Complement C2 siRNA mediated therapy of myasthenia gravis in mice. J Autoimmun. 2013;42:94–104. doi: 10.1016/j.jaut.2013.01.003. [DOI] [PubMed] [Google Scholar]
- Huda R, Tüzün E, Christadoss P. Targeting complement system to treat myasthenia gravis. Rev Neurosci. 2014;25(4):575–583. doi: 10.1515/revneuro-2014-0021. [DOI] [PubMed] [Google Scholar]
- Hussain R, Kifayet A. Immunoglobulin G1 (IgG1) and IgG3 antibodies are markers of progressive disease in leprosy. Infect Immun. 1995;63(2):410–415. doi: 10.1128/iai.63.2.410-415.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung S, Rajewsky K, Radbruch A. Shutdown of class switch recombination by deletion of a switch region control element. Science. 1993;259 (5097):984–987. doi: 10.1126/science.8438159. [DOI] [PubMed] [Google Scholar]
- Kusner LL, Kaminski HJ. The role of complement in experimental autoimmune myasthenia gravis. Ann N Y Acad Sci. 2012;1274:127–132. doi: 10.1111/j.1749-6632.2012.06783.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLay J, Leonard E, Peterson S, Shapiro D, Greenspan NS, Schreiber JR. γ3 gene-disrupted mice selectively deficient in the dominant IgG subclass made to bacterial polysaccharides. II. Increased susceptibility to fatal pneumococcal sepsis due to absence of anti-polysaccharide IgG3 is corrected by induction of anti-polysaccharide IgG1. J Immunol. 2002;168:3437–3443. doi: 10.4049/jimmunol.168.7.3437. [DOI] [PubMed] [Google Scholar]
- Michaelsen TE, Thommesen JE, Ihle O, Gregers TF, Sandin RH, Brekke OH, Sandlie I. A mutant human IgG molecule with only one C1q binding site can activate complement and induce lysis of target cells. Eur J Immunol. 2006;36(1):129–138. doi: 10.1002/eji.200535178. [DOI] [PubMed] [Google Scholar]
- Nakano S, Engel AG. Myasthenia gravis: quantitative immunocytochemical analysis of inflammatory cells and detection of complement membrane attack complex at the end-plate in 30 patients. Neurology. 1993;43:1167–1172. doi: 10.1212/wnl.43.6.1167. [DOI] [PubMed] [Google Scholar]
- Nimmerjahn F, Ravetch JV. Antibody-mediated modulation of immune responses. Immunol Rev. 2010;236:265–275. doi: 10.1111/j.1600-065X.2010.00910.x. [DOI] [PubMed] [Google Scholar]
- Redpath S, Michaelsen T, Sandlie I, Clark MR. Activation of complement by human IgG1 and human IgG3 antibodies against the human leucocyte antigen CD52. Immunology. 1998;93 (4):595–600. doi: 10.1046/j.1365-2567.1998.00472.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strait RT, Posgai MT, Mahler A, Barasa N, Jacob CO, Köhl J, Ehlers M, Stringer K, Shanmukappa SK, Witte D, Hossain MM, Khodoun M, Herr AB, Finkelman FD. IgG1 protects against renal disease in a mouse model of cryoglobulinemia, 2015. Nature. 2014;517 (7535):501–504. doi: 10.1038/nature13868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi S, Nose M, Sasaki J, Yamamoto T, Kyogoku M. IgG3 production in MRL/lpr mice is responsible for development of lupus nephritis. J Immunol. 1991;147:515–519. [PubMed] [Google Scholar]
- Trendelenburg M, Fossati-Jimack L, Cortes-Hernandez J, Turnberg D, Lewis M, Izui S, Cook HT, Botto M. The role of complement in cryoglobulin-induced immune complex glomerulonephritis. J Immunol. 2005;175:6909–6914. doi: 10.4049/jimmunol.175.10.6909. [DOI] [PubMed] [Google Scholar]
- Tüzün E, Christadoss P. Complement associated pathogenic mechanisms in myasthenia gravis. Autoimmun Rev. 2013;12(9):904–911. doi: 10.1016/j.autrev.2013.03.003. [DOI] [PubMed] [Google Scholar]
- Tüzün E, Allman W, Ulusoy C, Yang H, Christadoss P. Novel animal models of acetylcholine receptor antibody-related myasthenia gravis. Ann N Y Acad Sci. 2012;1274:133–139. doi: 10.1111/j.1749-6632.2012.06773.x. [DOI] [PubMed] [Google Scholar]
- Wu B, Goluszko E, Huda R, Tüzün E, Christadoss P. Experimental autoimmune myasthenia gravis in the mouse. Curr Protoc Immunol. 2013;100:15.8.1–15.8.26. doi: 10.1002/0471142735.im1508s100. [DOI] [PubMed] [Google Scholar]
- Yang H, Tüzün E, Alagappan D, Yu X, Scott BG, Ischenko A, Christadoss P. IL-1 receptor antagonist-mediated therapeutic effect inmurine myasthenia gravis is associated with suppressed serum proinflammatory cytokines, C3, and anti-acetylcholine receptor IgG1. J Immunol. 2005;175:2018–2025. doi: 10.4049/jimmunol.175.3.2018. [DOI] [PubMed] [Google Scholar]