

Dystroglycanopathies are characterized by deficient O-mannosyl glycosylation of α-dystroglycan (αDG) and represent an expanding genetically, biochemically, and clinically heterogeneous group of muscular dystrophies. Currently, there are 18 known genes leading to forms of α-dystroglycan–related dystrophy (αDG-RD), ranging in severity from a Walker-Warburg phenotype with severe brain malformations and hypotonia to milder childhood- or adult-onset limb-girdle muscular dystrophy (LGMD) phenotypes with or without intellectual disability.1,2
We report 3 siblings with mutations in a recently identified αDG gene, GDP-mannose pyrophosphorylase B (GMPPB),3 with variable degrees of weakness and a striking spectrum of early developmental cognitive involvement in all 3 patients, as well as epilepsy in one of the 3 siblings. Ocular abnormalities and elevated creatine kinase (CK) occurred in all. These patients highlight the interfamilial and intrafamilial phenotypic variability that can exist with GMPPB mutations.
Methods.
See appendix e-1 on the Neurology® Web site at Neurology.org for a description of the Methods.
Results.
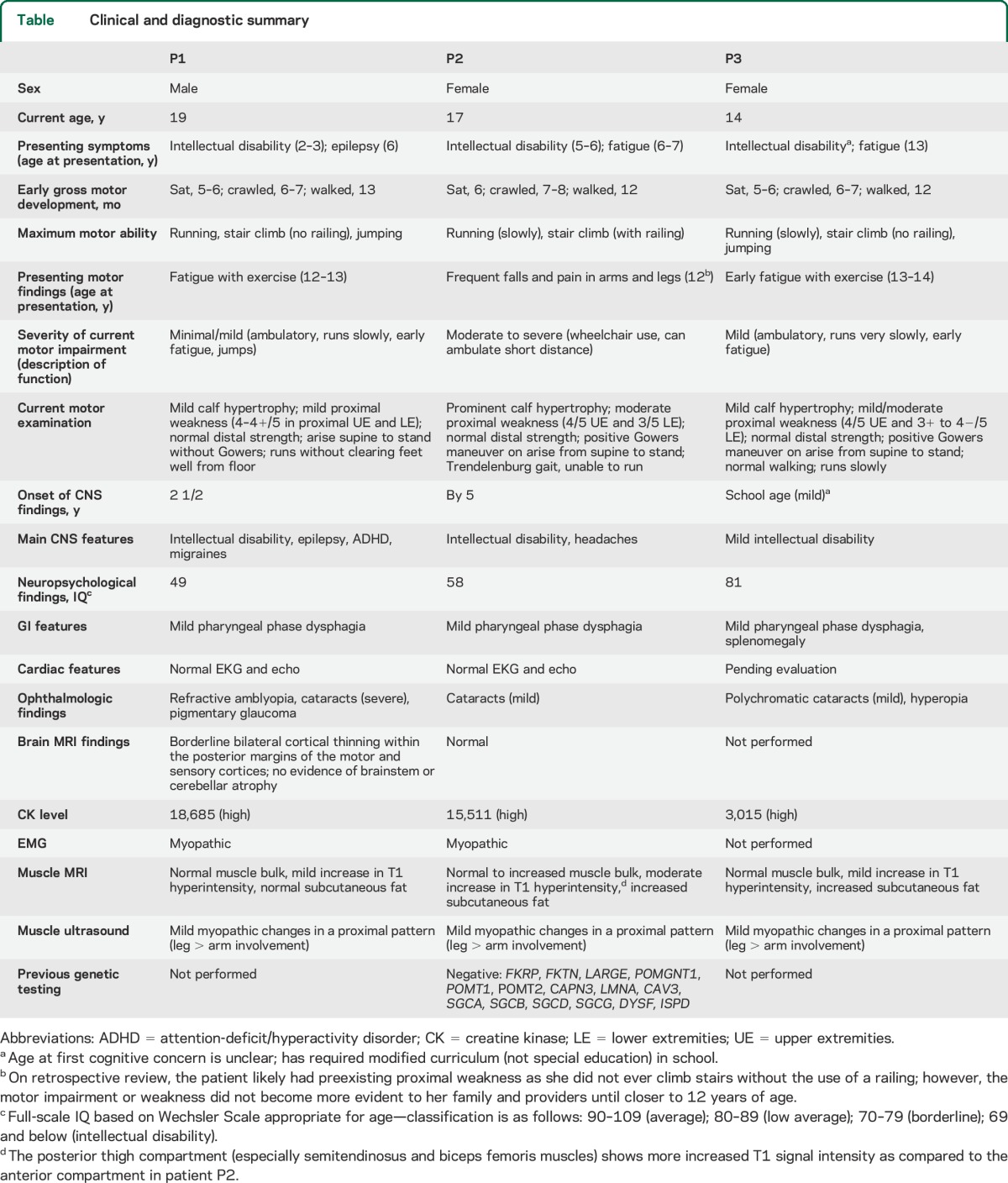
Patients P1, P2, and P3 are siblings ages 19, 17, and 14 years old, respectively, with an LGMD pattern but variable degrees of weakness and elevated CK levels (range 3,015–18,685) (table). Patient P1 had early epilepsy and intellectual disability, and later presented with mild fatigue with exercise (at 12–13 years old) and slow running, while patient P2 had unexplained fatigue and elevated liver function tests at age 6 years, and then more notable and progressive weakness since age 12 years, with near loss of ambulation since age 16 years. Additional findings include intractable epilepsy (1/3), variable degrees of intellectual disability (3/3), ocular findings (3/3), headaches (2/3), and unexplained splenomegaly (1/3). None of the patients has known cardiac involvement.
Table.
Clinical and diagnostic summary
Muscle biopsy in P2 (figure e-1) at age 11 years was dystrophic with reduced αDG glycodependent immunostaining in an irregular pattern, and with normal dystrophin, β-dystroglycan, and sarcoglycan staining. Laminin α2 (merosin) immunoreactivity was slightly decreased in a few fibers. Muscle imaging revealed variable degrees of myopathic changes in all 3 patients, and areas consistent with fatty infiltration in P2 and P3 (figure e-1).
Sequencing of GMPPB revealed compound heterozygous mutations: in exon 1 c.79G>C (p.Asp27His), previously reported,3 and a maternally inherited heterozygous novel sequence variant c.790C>T (p.Gln264*) in all 3 affected siblings. Paternal segregation testing was not available, but the c.79G>C mutation was not present in the mother.
Discussion.
Many of the αDG-RD genes encode glycosyltransferases that add O-linked mannose to αDG or contribute to the synthesis of the final LARGE-dependent glycan that binds to extracellular matrix ligands or are kinases or proteins of unknown function acting along the same pathway towards this final glycoepitope.3–5 In contrast, GMPPB function is important for the production of GDP mannose, the major mannosyl donor necessary for 4 mannosylation dependent pathways of which αDG mannosylation is one. While there could thus be impairment of multiple GDP-mannose dependent pathways, GMPPB patients do not seem to show the extent of multisystem organ involvement often seen in patients with typical congenital disorders of glycosylation (CDG syndromes).1,3,6
The 3 siblings reported here are notable for their variable severity of weakness, intellectual disability, and ophthalmologic findings. All but 1 patient thus far described in the literature with GMPPB mutations has had intellectual disability, while intractable epilepsy was seen in 3/10 previously reported patients.3,6 However, there has been no report of affected patients with a mild later onset motor phenotype and severe CNS findings. Carss et al.3 reported patients with severe intellectual disability who had early-onset CMD phenotype with severe motor weakness. Muscle imaging, not previously reported in this disorder, reveals increased T1 hyperintensity and seems to correlate with the degree of weakness without a selective pattern (figure e-1).
There are currently no clear genotype/phenotype correlations. The degree of intrafamilial variability in our patients is striking. The p.Asp27His identified in our family was previously reported in a boy with LGMD who had normal cognitive function and absent eye findings. The phenotype in our patients with significant brain and lens involvement is likely due to compound heterozygosity with the loss of function mutation p.Gln264*. Another patient heterozygous for a null mutation (p.Arg74*) in GMPPB (associated with the missense p.Asp334Asn mutation) presented with a muscle-eye-brain phenotype, while the phenotype of homozygous null or 2 different heterozygous null GMPPB mutations is unknown.
Our findings implicate that intellectual disability or epilepsy may be the predominant early manifestations of disease in patients with GMPPB mutations or other genotypic forms of α-dystroglycan-related dystrophies. Our observation suggests the usefulness of early CK determination in all patients with intellectual disability, developmental delay, or otherwise unexplained epilepsy.
Supplementary Material
Footnotes
Supplemental data at Neurology.org
Author contributions: Diana X. Bharucha-Goebel, MD: drafting/revising the manuscript for content, including medical writing for content; acquisition of data; study concept or design. Erin Neil, DO: drafting/revising the manuscript for content, including medical writing for content; acquisition of data. Sandra Donkervoort, MS, CGC: drafting/revising the manuscript for content, including medical writing for content; acquisition of data; study concept or design. Jahannaz Dastgir, DO: acquisition of data. Edythe A. Wiggs, PhD: drafting/revising the manuscript for content, including medical writing for content; acquisition of data. Thomas L. Winder, PhD: acquisition of data. Steven A. Moore, MD, PhD: drafting/revising the manuscript for content, including medical writing for content; acquisition of data. Susan Iannaccone, MD: drafting/revising the manuscript for content, including medical writing for content; acquisition of data. Carsten G. Bönnemann, MD: acquisition of data; study supervision or coordination; study concept or design; drafting/revising the manuscript for content, including medical writing for content.
Study funding: Supported by NIH grant U54, NS053672 for the Iowa Wellstone Muscular Dystrophy Cooperative Research Center (S.A.M.). C.G.B. is supported by intramural funds of the National Institute of Neurological Disorders and Stroke/NIH. D.X.B.-G. is supported by grant NIH-T32-AR056993.
Disclosure: D. Bharucha-Goebel receives support from grant NIH-T32-AR056993. E. Neil, S. Donkervoort, J. Dastgir, and E. Wiggs report no disclosures relevant to the manuscript. T. Winder was employed by Prevention Genetics, LLC, and now works at Invitae Corp. S. Moore is partially funded by NIH grant U54, NS053672. He is performing fee-for-service muscle biopsy assays for Sarepta Therapeutics. S. Iannaccone consults for the American Academy of Neurology, National Institute of Health and Welfare, Santhera Pharmaceutics, and Sarepta Therapeutics; serves as an editorial member of the American Academy of Neurology; has received reimbursements from Lilly USA LLC; has received investment interests from Sarepta Therapeutics; and has received revenue from GlaxoSmithKline, ISIS Pharmaceuticals, Lilly USA LLC, Muscular Dystrophy Association, PTC Therapeutics, and the Spinal Muscular Atrophy Foundation. C. Bönnemann is supported by intramural funds of the National Institute of Neurological Disorders and Stroke/NIH. Go to Neurology.org for full disclosures.
References
- 1.Godfrey C, Foley AR, Clement E, Muntoni F. Dystroglycanopathies: coming into focus. Curr Opin Genet Dev 2011;21:278–285. [DOI] [PubMed] [Google Scholar]
- 2.Mercuri E, Messina S, Bruno C, et al. Congenital muscular dystrophies with defective glycosylation of dystroglycan. Neurology 2009;72:1802–1809. [DOI] [PubMed] [Google Scholar]
- 3.Carss KJ, Stevens E, Foley AR, et al. Mutations in GDP-mannose pyrophosphorylase B cause congenital and limb-girdle muscular dystrophies associated with hypoglycosylation of α-dystroglycan. Am J Hum Genet 2013;93:29–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yoshida-Moriguchi T, Willer T, Anderson ME, et al. SGK196 is a glycosylation-specific O-mannose kinase required for dystroglycan function. Science 2013;341:896–899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Live D, Wells L, Boons GJ. Dissecting the molecular basis of the role of the O-mannosylation pathway in disease: α-dystroglycan and forms of muscular dystrophy. Chem Bio Chem 2013;14:2392–2402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Raphael AR, Couthouis J, Sakamuri S, et al. Congenital muscular dystrophy and generalized epilepsy caused by GMPPB mutations. Brain Res 2014;1575:66–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.