

Abstract
Isoflavonoids have been shown to inhibit tumor proliferation and metastasis by activating cell death pathways. As such, they have been widely studied as potential therapies for cancer prevention. The second generation synthetic isoflavan analogues ME-143 and ME-344 also exhibit anti-cancer effects, however their specific molecular targets have not been completely defined. To identify these targets, we examined the effects of ME-143 and ME-344 on cellular metabolism and found that they are potent inhibitors of mitochondrial oxidative phosphorylation (OXPHOS) complex I (NADH: ubiquinone oxidoreductase) activity. In isolated HEK293T mitochondria, ME-143 and ME-344 reduced complex I activity to 14.3% and 28.6% of control values respectively. In addition to the inhibition of complex I, ME-344 also significantly inhibited mitochondrial complex III (ubiquinol: ferricytochrome-c oxidoreductase) activity by 10.8%. This inhibition of complex I activity (and to a lesser extent complex III activity) was associated with a reduction in mitochondrial oxygen consumption. In permeabilized HEK293T cells, ME-143 and ME-344 significantly reduced the maximum ADP-stimulated respiration rate to 62.3% and 70.0% of control levels respectively in the presence of complex I-linked substrates. Conversely, complex II-linked respiration was unaffected by either drug. We also observed that the inhibition of complex I-linked respiration caused the dissipation of the mitochondrial membrane potential (ΔΨm). Blue native (BN-PAGE) analysis revealed that prolonged loss of ΔΨm results in the destabilization of the native OXPHOS complexes. In particular, treatment of 143B osteosarcoma, HeLa and HEK293T human embryonic kidney cells with ME-344 for 4 h resulted in reduced steady-state levels of mature complex I. Degradation of the complex I subunit NDUFA9, as well as the complex IV (ferrocytochrome c: oxygen oxidoreductase) subunit COXIV, was also evident. The identification of OXPHOS complex I as a target of ME-143 and ME-344 advances our understanding of how these drugs induce cell death by disrupting mitochondrial metabolism, and will direct future work to maximize the anti-cancer capacity of these and other isoflavone-based compounds.
Keywords: Isoflavonoids, anti-cancer, mitochondria, oxidative phosphorylation, complex I
Introduction
Isoflavones from the Leguminosae plant family have been widely studied as potential therapies for cancer prevention [1]. Phenoxodiol, one of the most well researched isoflavone derivatives, exhibits cytotoxic effects against various types of human cancers with high specificity [2-7]. Preclinical studies in chemoresistant ovarian cancer cells have shown that phenoxodiol exerts a pro-apoptotic effect by activating mitochondrial cell death signaling via caspase-2, Bid cleavage and proteasomal degradation of X-linked inhibitor of apoptosis protein (XIAP) [2,6,8]. Furthermore, phenoxodiol inhibits plasma membrane electron transport [9] and blocks the activity of the Ecto-NOX disulfide-thiol exchanger 2 (ENOX2 or tNOX) [10], a cancer-specific cell-surface protein with oxidative and protein disulfide-thiol interchange activity [11,12]. Importantly, phenoxodiol has little effect on the endogenous isoform, ENOX1 (cNOX), in either cancer or non-cancer cell lines [10].
ME-143 is a synthetic, second generation tNOX inhibitor based on the first generation compounds phenoxodiol and triphendiol. It exhibits greater anti-cancer potency than phenoxodiol in vitro, with IC50 values generally < 1 μm [13]. Dose escalation studies in patients with advanced solid tumors have shown that ME-143 is well tolerated when administered intravenously, and may be suitable for combination with cytotoxic chemotherapies [13]. Although ME-143, and its parent compound phenoxodiol, are known to inhibit plasma membrane electron transport, their other potential molecular targets remain to be defined.
A second isoflavone derivative, NV-128, has been shown to induce caspase-independent cell death in epithelial ovarian cancer stem cells via two independent mechanisms, both of which signal via the mitochondria. The first mechanism involves activation of the AMPKα1 pathway, leading to mTOR inhibition and mitochondrial depolarization. This causes the nuclear translocation of endonuclease G (EndoG), followed by cleavage of nuclear DNA and chromatin condensation [14,15]. The second mechanism involves induction of the mitochondrial MAP/ERK kinase/extracellular signal-regulated kinase pathway, resulting in the loss of mitochondrial membrane potential (ΔΨm) through Bax upregulation [15]. NV-128 also reduces the ATP/ADP ratio in vitro, which may contribute to mTOR inhibition [15]. Interestingly, NV-128 also significantly reduces the steady-state levels of the oxidative phosphorylation (OXPHOS) complex IV (ferrocytochrome c:oxygen oxidoreductase) subunits COXI and COXIV in a time-dependent manner [15]. This suggests that NV-128 may also inhibit mitochondrial OXPHOS to some degree, however this has not been determined.
ME-344, an active metabolite of NV-128, also induces cell death via activation of mitochondrial cell death signaling pathways. In a pre-clinical in vivo study in mice, ME-344 has been shown to decrease tumor burden of recurrent epithelial ovarian cancer (unpublished data). Phase I clinical studies of intravenously administered ME-344 in thirty patients with refractory solid tumors has revealed that the compound is well tolerated at weekly doses of 10 mg/kg [16]. Of these patients, ten have achieved stable disease or better, with an ongoing partial response in one patient with small cell lung cancer [16]. Phase 1b clinical trials of ME-344, in combination with the topoisomerase I inhibitor topotecan hydrochloride (Hycamtin®), have now begun in patients with solid small cell lung and ovarian cancers.
Although NV-128/ME-344 has been shown to inhibit mitochondrial ATP generation and reduce the steady-state levels of OXPHOS complex IV subunits, its specific mitochondrial target/s are unknown. Similarly, how ME-143 affects mitochondrial metabolism and/or signaling has not been determined.
To define the mechanism of action of ME-143 and ME-344, we treated different cell types in vitro with each drug and measured a range of metabolic parameters, including mitochondrial oxygen consumption and OXPHOS enzyme activities. Our results identified mitochondrial OXPHOS complex I (NADH: ubiquinone oxidoreductase) as a direct molecular target of both ME-143 and ME-344, with its inhibition associated with an instantaneous reduction of mitochondrial oxygen consumption and dissipation of the ΔΨm.
These findings provide new insights into how isoflavone-based compounds induce cell death by disrupting mitochondrial metabolism, in particular the identification of mitochondrial complex I as a target of ME-143 and ME-344.
Materials and methods
Reagents
ME-143 and ME-344 (MEI Pharma, San Diego, USA) were prepared as 10 mg/mL stock solutions in dimethylsulphoxide (DMSO, Sigma).
Cell culture
Cells were cultured at 37°C and 5% CO2 in Dulbecco’s Modified Eagle’s Medium (DMEM; Life Technologies) supplemented with 5% (v/v) fetal bovine serum (FBS, Life Technologies), Penicillin-Streptomycin (Life Technologies) and GlutaMAXTM (Life Technologies).
Measurement of oxygen consumption rate
High-resolution respirometry using intact cells was performed with an Oxygraph-2K oxygen electrode (Oroboros, Innsbruck, Austria). Approximately 7.5 × 107 cells were incubated in 2 mL Hank’s Balanced Salt Solution (HBSS, Life Technologies) at 37°C in each chamber. Basal respiration rates were recorded, followed by the addition of either DMSO [as a solute control, final concentration 0.1% (v/v)], ME-143 or ME-344 (at the final concentrations indicated) and treatment respiration rates recorded. Experiments were performed three times for each treatment. Mitochondrial respiration rates were calculated using DatLab software (version 4.3.4.51, Oroboros Instruments) and expressed as pmol O2/s/mg whole cell protein. For data analysis, treatment: basal respiration rate ratios were normalized to control (DMSO) rates. Significant differences between control and treated samples were determined using one-way ANOVA followed by Tukey’s multiple comparison test. For the dosage experiment, significant differences between control and treated samples were determined using two tailed t-tests.
Measurement of complex I or II-linked respiration
High-resolution respirometry using permeabilized cells was performed with an Oxygraph-2K oxygen electrode (Oroboros). Each 2 mL chamber was filled with intracellular media [10 mM NaCl, 135 mM KCl, 1 mM MgCl2, 5 mM KH2PO4, 20 mM HEPES, 5 mM ethylene glycol tetra-acetic acid (EGTA), 1.86 mM CaCl2, pH 7.1], followed by the injection of either DMSO [solute control, final concentration 0.1% (v/v)], ME-143 or ME-344 (at final concentrations of 10 μg/mL). Glutamate and malate (for complex I-linked respiration) or succinate (for complex II-linked respiration) were then added (final concentration of 5 mM), followed by the addition of 1 × 107 HEK293T cells. After 2.5 min, digitonin (Merck) was added (final concentration 50 μg/mL) to permeabilize the plasma membrane and allow the uptake of substrates. After a further 3 min, 125 or 200 nmol of ADP was added to measure complex I or II-linked state III respiration respectively. Once complete condensation of the added ADP to ATP was attained, oxygen flux was measured to determine state IV respiration rates. The experiments were performed three times for each treatment. Oxygen consumption rates were calculated using DatLab software and were expressed as pmol O2/s/million cells. Significant differences between control and treated samples were determined using two tailed t-tests.
Mitochondria isolation
Mitochondria were isolated according to McKenzie et al. [17]. Cell pellets were resuspended in isolation buffer [20 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES, pH 7.6), 220 mM mannitol, 70 mM sucrose, 1 mM ethylenediaminetetraacetic acid (EDTA), 0.5 mM Phenylmethylsulfonyl Fluoride (PMSF), 2 mg/mL bovine serum albumin (BSA)] and incubated on ice for 15 min. Cells were lysed using 20 strokes of a drill-fitted pestle in a glass homogenizer at 4°C. Cell debris and nuclei were pelleted by centrifugation at 800 g for 10 min at 4°C. Mitochondria were pelleted from the supernatant by centrifugation at 10,000 g for 20 min at 4°C and rinsed with isolation buffer without BSA at 10,000 g for 10 min at 4°C.
Spectrophotometric measurement of enzyme activity
The individual enzymatic activities of OXPHOS complexes I (NADH: ubiquinone oxidoreductase, EC 1.6.5.3), II (succinate: ubiquinone oxidoreductase, EC 1.3.5.1), II + III (succinate: cytochrome c oxidoreductase, EC 1.3.5.1 + EC 1.10.2.2), IV (ferrocytochrome c: oxygen oxidoreductase, EC 1.9.3.1) and citrate synthase (EC 4.1.3.7) were measured in isolated HEK293T mitochondria as described previously [18]. For all assays, enzymatic activity was measured in the presence of DMSO [solute control, final concentration 0.1% (v/v)], 10 μg/mL ME-143 or 10 μg/mL ME-344. Experiments were performed at least three times for each treatment. Significant differences between control and treated samples were determined using two tailed t-tests.
For measurement of complex I activity, isolated mitochondria were first disrupted by sonication with a Microson Ultrasonic homogenizer (Misonix, USA) fitted with a 3.2 mm microprobe. Three 0.7 s pulses were performed at a power setting of 1.5 (~4 W). 50 μg of sonicated mitochondria were subsequently used for each complex I assay. Complex I activity was also measured in the presence of the inhibitor rotenone (5 mg/mL).
Measurement of mitochondrial membrane potential (ΔΨm)
HEK293T cells were plated in 96-well plates at 3.5 × 104 cells per well and allowed to grow overnight. Cells were incubated in fluorescent dye media [DMEM supplemented with 10% (v/v) FBS, Penicillin-Streptomycin, GlutaMAX, 5 μg/mL Hoechst 33342 (Life Technologies), 20 nM tetramethylrhodamine, methyl ester (TMRM, Life Technologies) and 10 μM Verapamil (Sigma)] for 1 h at 37°C in a 5% CO2 incubator. Cells were imaged in 10 min intervals using an ArrayScanVti High Content Analysis Reader (Thermo Scientific, MA, US) with 365 nm (Hoechst) and 549 nm (TMRM) light beams. After the third set of images was acquired, fluorescent dye media (no treatment control), DMSO [solute control, final concentration 0.1% (v/v)], ME-143 or ME-344 (at final concentrations of 10 μg/mL) were added. Another three sets of images were taken before carbonyl cyanide 4-(trifluoromethoxy) phenylhydrazone (FCCP, Sigma) at a final concentration of 10 μM was added. A minimum of five replicates were performed.
Cell treatment with ME-143 and ME-344
Cells were cultured to 80% confluence and incubated in OptiMEM® (Life Technologies) supplemented with GlutaMAXTM for 4 h at 37°C. Cells were then treated with fresh media (OptiMEM® with GlutaMAXTM) containing 10 μg/mL ME-143 or 10 μg/mL ME-344 at 37°C for the time duration indicated. For controls, cells were incubated in fresh media with or without 0.1% (v/v) DMSO for 4 h. Following treatment, cells were harvested and washed in 1 × PBS before being pelleted by centrifugation at 2,300 g for 5 min for SDS-PAGE and BN-PAGE analyses.
Sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE)
Whole cell lysates (15 μg protein) were solubilized in loading dye [50 mM Tris-Cl pH 6.8, 0.1 M dithiothreitol (DTT), 2% (w/v) SDS, 10% (v/v) glycerol, and 0.05% (w/v) bromophenol blue] and boiled for 3 min prior to separation on a 12% SDS-polyacrylamide gel.
Blue Native PAGE (BN-PAGE)
BN-PAGE was performed as described with minor modifications [19,20]. Whole cell lysates (100 μg protein) were solubilized in 20 mM Bis-Tris pH 7.4, 50 mM NaCl, and 10% (v/v) glycerol containing 1% (v/v) Triton X-100 or 1% (w/v) digitonin for 30 min on ice. Insoluble material was pelleted by centrifugation at 21,000 g for 5 min at 4°C. Loading dye (final concentrations: 0.5% Coomassie Blue G, 50 mM ε-amino n-caproic acid, and 10 mM Bis-Tris pH 7.0) was added to the soluble fraction prior to separation for 15 h at 100 V/7 mA, 4°C on a 4-13% acrylamide-bisacrylamide gel in 70 mM ε-amino n-caproic acid and 50 mM Bis-Tris (pH 7.0). Blue cathode buffer [15 mM Bis-Tris pH 7.0, 50 mM tricine, 0.02% (w/v) Coomassie Blue G] was replaced by colorless cathode buffer once the dye front had migrated through approximately one-third of the gel. Anode buffer contained 50 mM Bis-Tris pH 7.0.
Western blotting and immunodetection
Western blotting was performed on PVDF membranes (Millipore) using a semi-dry transfer method [21] as described previously [22]. Membranes were blocked with 10% (w/v) skim milk powder in 1 × PBS containing 0.05% (v/v) Tween-20 for 2 h. Membranes were probed overnight at 4°C with primary antibodies against NDUFA9 [23], NDUFS5 (kind gift from M. Ryan, Monash University, Melbourne, Australia), SDHA (Abcam, ab14715), UQCRC1 (Abcam, ab110252), COXI (Abcam, ab14705) and COXIV (Cell Signaling, 4844S), then for 2 h at room temperature with anti-mouse or anti-rabbit HRP-conjugated secondary antibodies (Sigma). Proteins were detected using ECL or ECL Prime reagents (GE Healthcare) with a MicroChemi chemiluminescence detection system (DNR Bio-Imaging Systems).
Statistical analysis
All measurements were collected from at least three independent experiments. Statistical analysis was performed using GraphPad Prism 6.01 and Microsoft Excel software. Significant differences between control and treated samples were determined using two tailed t-tests. For oxygen consumption rates of intact cells, the significant differences between treatments were determined using one-way ANOVA followed by Tukey’s multiple comparison test.
Results
ME-344, but not ME-143, disrupts the stability of the OXPHOS complexes
Previous studies have shown that the isoflavone NV-128 decreases the levels of the mitochondrial OXPHOS complex IV subunits COXI and COXIV in epithelial ovarian cancer cells [15]. We therefore examined whether ME-143 and ME-344 affect the steady-state levels of other OXPHOS complex structural subunits in 143B osteosarcoma and HeLa cancer cells, HEK293T cells and non-cancerous primary human skin fibroblasts.
In HEK293T cells, a protein of slightly lower molecular weight than COXIV was detected by SDS-PAGE and western blotting following 1 h of ME-344 treatment (Figure 1A, marked with an ‘*’). This smaller protein was consistently observed in HEK293T cells following ME-344 treatment and is most likely a specific COXIV degradation product. It was not observed in any other cell type examined.
Figure 1.

ME-344, but not ME-143, disrupts OXPHOS complex stability. SDS-PAGE and BN-PAGE analyses of mitochondrial proteins from (A) HEK293T cells, (B) 143B osteosarcoma cells (C) HeLa cells or (D) primary human fibroblasts following ME-143 or ME-344 treatment for the time duration indicated. Subunits of complex I (NDUFA9), complex II (SDHA), complex III (UQCRC1) and complex IV (COXIV) were detected by SDS-PAGE and western blotting. Mature complex I (CI), complex II (CII), the complex III homodimer (CIII2), complex IV (CIV) and the complex I/complex III2 supercomplex (CI/CIII2) were detected by BN-PAGE and western blotting. ME-344 disrupts the stability of native complex I in HEK293T, 143B and HeLa cells but not primary human fibroblasts following 4 h of treatment.
In 143B osteosarcoma cells, the levels of COXIV and the complex I subunit NDUFA9 were reduced following 2 h of ME-344 treatment (Figure 1B). No other OXPHOS subunits were affected by ME-344 in any other cell lines studied.
ME-143 had no effect on the steady-state levels of the individual OXPHOS complex subunits except for NDUFA9, which was slightly reduced in HeLa cells after 2 h of treatment (Figure 1C).
We next investigated whether ME-143 and ME-344 have any effect on the OXPHOS complexes by examining their native, mature forms by blue native (BN)-PAGE and western blotting. Whole cells were solubilized in 1% Triton X-100 to resolve the OXPHOS complexes in their monomeric form. Due to the protein/detergent ratio used, some complex I/complex III2supercomplex (CI/CIII2) was also resolved (Figure 1).
Following 4 h of ME-344 treatment, native complex I steady-state levels were reduced in HEK293T cells (Figure 1A), 143B osteosarcoma cells (Figure 1B) and HeLa cells (Figure 1C). In contrast, native complex I remained intact in primary human fibroblasts (Figure 1D). Native complexes II, III and IV were not affected by ME-344 in any of the cell lines investigated.
ME-143 did not reduce the steady-state levels of the native OXPHOS complexes in any of the cell lines. Instead, levels of several complexes were elevated in some cell types. Complex I levels were increased in 143B osteosarcoma cells, HeLa cells, and primary human fibroblasts following ME-143 treatment (Figure 1B-D). ME-143 also increased complex III levels in HeLa cells and primary human fibroblasts (Figure 1C and 1D) and complex IV levels in HeLa cells (Figure 1C).
In summary, ME-344 reduced the steady-state levels of native complex I following 4 h of treatment. This was evident in HEK293T cells, 143B osteosarcoma cells and HeLa cells, but not in primary human fibroblasts. In contrast, ME-143 did not reduce the amount of native OXPHOS complexes in the mitochondria, with levels actually increased in some cases.
ME-143 and ME-344 inhibit mitochondrial oxygen consumption
We observed that ME-143 and ME-344 can alter the steady-state levels of the native OXPHOS complexes; in particular, ME-344 reduces the levels of mature complex I (Figure 1). In addition, NV-128 (the parent compound of ME-344) has been shown to reduce the ΔΨm and cellular ATP/ADP ratios in vitro [14,15]. To determine if these effects of ME-143 and ME-344 are associated with the inhibition of OXPHOS activity, we assessed the oxygen consumption rates in the presence of either ME-143 or ME-344 in intact cultured cells using glucose as a substrate.
ME-143 (at a final concentration of 20 µg/mL) significantly reduced mitochondrial respiration in 143B osteosarcoma and HeLa cancer cells, HEK293T cells and primary human skin fibroblasts (Figure 2). The greatest inhibition was observed in HEK293T cells, with residual respiration 76.7 ± 1.4% of control values (Figure 2). ME-344 (20 µg/mL) also significantly reduced mitochondrial respiration in all of the cell types tested (Figure 2). In addition, ME-344 inhibition of respiration was significantly greater than ME-143 in HEK293T cells and HeLa cells, with residual rates of 66.7 ± 5.0% and 73.6 ± 2.1% of control values respectively (Figure 2).
Figure 2.
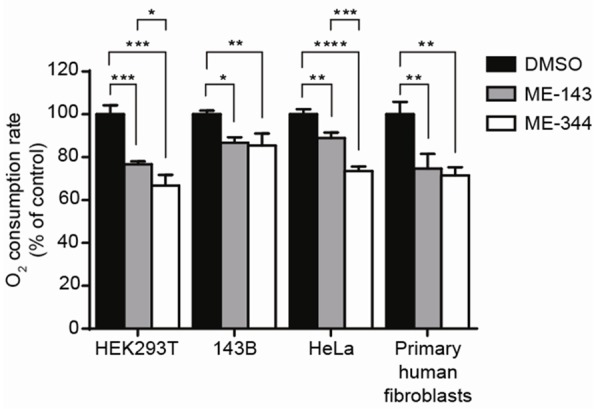
Both ME-143 and ME-344 inhibit respiration of intact cultured cells. Oxygen consumption of HEK293T cells, 143B osteosarcoma cells, HeLa cells and primary human fibroblasts was measured in the presence of 20 μg/mL ME-143 (■) or 20 μg/mL ME-344 (□). Both ME-143 and ME-344 significantly inhibit respiration in each cell type. DMSO (■) was used as a negative control. Data is expressed as mean ± s.d., n = 3. *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001, ****p ≤ 0.0001.
We also performed a titration with both ME-143 and ME-344 to determine the minimum concentrations required to inhibit respiration. Significant inhibition of oxygen consumption in HEK293T cells was achieved with 7.5 µg/mL ME-143 (Figure 3A). ME-344 was more potent at inhibiting mitochondrial respiration, requiring only 5 µg/mL to significantly decrease oxygen flux (Figure 3B).
Figure 3.
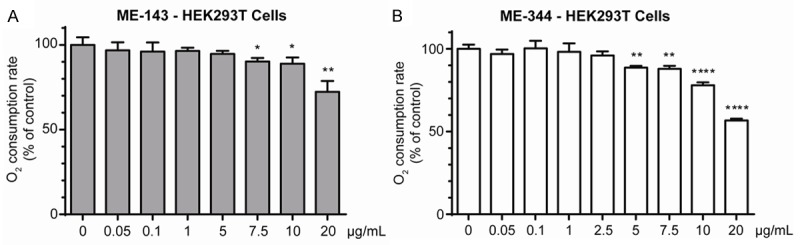
ME-344 is a more potent inhibitor of mitochondrial respiration than ME-143. Oxygen consumption was measured in intact HEK293T cells in the presence of varying concentrations of ME-143 (A) or ME-344 (B). Respiration was significantly inhibited in the presence of 7.5 μg/mL ME-143 or 5 μg/mL ME-344. DMSO was used as a negative control. Data is expressed as mean ± s.d., n = 3. *p ≤ 0.05, **p ≤ 0.01, ****p ≤ 0.0005.
These results indicate that ME-143 and ME-344 can both significantly inhibit mitochondrial respiration by approximately 25 to 35%. To determine if this inhibition is at OXPHOS complex I or complex II, we measured respiration rates in permeabilized cells using various substrates that specifically stimulate complex I or II-linked respiration. In addition, we measured the effects of ME-143 and ME-344 on both ADP-stimulated (state III) and ADP-limited (state IV) respiration.
In the presence of the complex I-linked substrates glutamate and malate, both ME-143 and ME-344 significantly reduced the maximum ADP-stimulated respiration rate (state III) to 62.3% and 70.0% of control levels respectively (Table 1A). Conversely, complex II-linked state III respiration rates (in the presence of succinate) were not significantly affected by either compound (Table 1B). This suggests that both ME-143 and ME-344 inhibit respiration by directly acting at complex I, as electron transfer through complex II was unaffected.
Table 1.
Mitochondrial respiration in permeabilized HEK293T cells
| A. Complex I-linked substrates | |||
|
| |||
|---|---|---|---|
| Respiration rate pmol O2/s/million cells | State III/State IV | ||
|
| |||
| State III | State IV | ||
| DMSO | 24.7 ± 1.1 | 9.8 ± 1.8 | 2.6 ± 0.4 |
| ME-143 | 15.4 ± 0.9** | 7.4 ± 0.4 | 2.1 ± 0.2 |
| ME-344 | 17.3 ± 1.5* | 7.4 ± 1.8 | 2.4 ± 0.5 |
|
| |||
| B. Complex II-linked substrates | |||
|
| |||
| Respiration rate pmol O2/s/million cells | State III/State IV | ||
|
| |||
| State III | State IV | ||
|
| |||
| DMSO | 46.1 ± 3.9 | 20.1 ± 2.3 | 2.3 ± 0.1 |
| ME-143 | 41.2 ± 4.8 | 18.7 ± 3.5 | 2.2 ± 0.3 |
| ME-344 | 41.0 ± 1.9 | 19.8 ± 0.9 | 2.1 ± 0.1 |
DMSO was used as a solute control. Data is expressed as mean ± s.d., n = 3.
p ≤ 0.005;
p ≤ 0.0005.
The respiratory control ratio (state III/state IV), which is a useful marker of mitochondrial substrate oxidization capacity and proton leak across the mitochondrial inner membrane, was slightly reduced by ME-143 and ME-344 in the presence of complex I-linked substrates (Table 1A). This indicates a trend towards reduced ATP synthesis capacity, however this reduction was not statistically significant.
In summary, both ME-143 and ME-344 reduce mitochondrial oxygen consumption. As respiration via complex II is unaffected, this reduction is most likely due to the inhibition of electron flux through complex I.
ME-143 and ME-344 directly inhibit complex I enzymatic activity
Our respiration experiments revealed that ME-143 and ME-344 inhibit mitochondrial oxygen consumption, particularly in the presence of complex I-linked substrates. We next assessed OXPHOS complex enzymatic activities spectrophotometrically in isolated HEK293T cell mitochondria in the presence or absence of ME-143 or ME-344 to confirm that complex I is the primary site of inhibition.
Complex I activity was significantly reduced by both ME-143 and ME-344 to 14.3% and 28.6% of control activity respectively (Table 2). This inhibition was comparable to rotenone, a known complex I inhibitor (37.1% of control activity). Activities of complex II, complex IV and the mitochondrial matrix enzyme citrate synthase were not affected by either ME-143 or ME-344 (Table 2).
Table 2.
Specific OXPHOS enzyme activities in isolated HEK293T mitochondria
| Activity nmol/min/mg | ||||
|---|---|---|---|---|
|
| ||||
| DMSO | ME-143 | ME-344 | Rotenone | |
| Complex I | 10.5 ± 3.4 | 1.5 ± 0.2*** | 3.0 ± 2.9** | 3.9 ± 2.5* |
| Complex II | 27.8 ± 1.2 | 26.5 ± 1.0 | 24.5 ± 4.4 | - |
| Complex II + III | 54.5 ± 1.5 | 54.2 ± 2.3 | 48.6 ± 2.1* | - |
| Complex IV | 11.5 ± 5.0 | 11.3 ± 1.4 | 12.2 ± 0.8 | - |
| Citrate synthase | 157.4 ± 12.9 | 168.8 ± 4.9 | 153.2 ± 2.6 | - |
DMSO was used as a solute control. Data is expressed as mean ± s.d., n ≥ 3.
p ≤ 0.05;
p ≤ 0.01;
p ≤ 0.0005.
Interestingly, complex II + III activity was also significantly reduced to 89.2% of control activity by ME-344 (Table 2). As complex II is the rate limiting step of this coupled assay [18] (and complex II activity is unaffected by ME-344), this result suggests that ME-344 also inhibits electron flux through complex III.
In summary, these results confirm that complex I is the principal OXPHOS target for both ME-143 and ME-344. In addition, ME-344 also has an inhibitory effect on complex III activity.
ME-143 and ME-344 induce dissipation of the mitochondrial membrane potential (ΔΨm)
The energy derived from electron flux through OXPHOS complexes I, II, III and IV is used to pump protons out of the mitochondrial matrix and across the mitochondrial inner membrane to generate the mitochondrial membrane potential (ΔΨm). The ΔΨm is used to drive the mitochondrial F1Fo-ATP synthase to condense ADP and inorganic phosphate to generate ATP [24].
Reduction of the ΔΨm has been observed previously following treatment for 1 h of epithelial ovarian cancer cells with NV-128 (the parent compound of ME-344) [14]. To determine if ME-143 and ME-344 also alter the ΔΨm, we incubated HEK293T cells with the potentiometric dye TMRM to measure the ΔΨm in real time (Figure 4).
Figure 4.
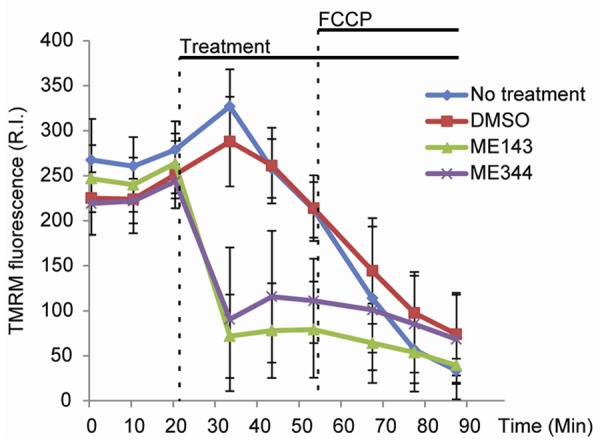
Both ME-143 and ME-344 dissipate the mitochondrial membrane potential (ΔΨm). The ΔΨm was measured in real time in HEK293T cells following the addition of 10 μg/mL ME-143 or 10 μg/mL ME-344. Cells with no treatment or with DMSO added were used as negative controls. Both ME-143 and ME-344 quickly reduced mitochondrial TMRM fluorescence, indicating the dissipation of the ΔΨm. Data is expressed as mean ± s.d.; R.I., relative intensity. Measurements were collected from at least 5 replicate wells for each treatment condition.
We found that the addition of either 10 μg/mL ME-143 or 10 μg/mL ME-344 (Figure 4, ‘Treatment’) induced a reduction in mitochondrial TMRM fluorescence, indicating a loss of the ΔΨm. This dissipation of ΔΨm occurred quickly following the addition of the drugs, and was comparable in magnitude to treatment with the protonophore FCCP, a compound that is commonly used to dissipate ΔΨm (Figure 4).
These results indicate that the inhibition of complex I-linked respiration by ME-143 or ME-344 inhibits respiratory chain electron flux to a sufficient degree that the generation of the ΔΨm by the OXPHOS complexes is significantly impaired.
Discussion
Changes in cellular bioenergetics are one of the key hallmarks of cancer. In 1956, Warburg made the important observation that cancer cells utilize glycolysis for energy production even in the presence of oxygen [25]. This reliance on ‘aerobic glycolysis’ implicated altered mitochondrial metabolism in tumor biogenesis, in particular the potential disruption of OXPHOS. However, it is now apparent that many tumor types retain a functional OXPHOS system that is essential for survival. This allows for the possibility to design novel mitochondrial anti-cancer drugs that target the OXPHOS enzyme complexes.
Complex I is the first protein complex of the mitochondrial respiratory chain, a series of enzymes involved in energy generation via OXPHOS. Complex I oxidizes NADH derived from the tricarboxylic acid (TCA) cycle, using the donated electrons to reduce ubiquinone. This redox reaction is associated with the translocation of protons across the mitochondrial inner membrane, generating a membrane potential (ΔΨm). The ΔΨm is then utilized by the F1Fo-ATP synthase to condense ADP and inorganic phosphate (Pi) to ATP.
A wide range of compounds, including pesticides and antibiotics, have been shown to target complex I and inhibit its enzymatic activity. Acetogenins, such as rolliniastatin and bullatacin, are potent complex I inhibitors that are cytotoxic in mammary adenocarcinoma and ovarian and hepatocellular carcinoma [26,27]. Other complex I inhibitors, including rhein and diphenyliodonium, have been shown to induce apoptosis in multiple cancer cell lines by causing the overproduction of reactive oxygen species (ROS) [28].
One of the most well-studied specific complex I inhibitors, rotenone, is a member of the rotenoid family of naturally occurring isoflavonoids produced by Leguminosae plants. We found that the synthetic isoflavan analogues ME-143 and ME-344 are also potent inhibitors of complex I. This inhibition causes an immediate reduction of mitochondrial respiration and loss of the ΔΨm, with the subsequent destabilization of the OXPHOS complexes.
Both ME-143 and ME-344 significantly inhibit the ability of complex I to oxidize NADH, impeding electron flux through the OXPHOS complexes. In addition, ME-344 inhibits complex III enzymatic activity, resulting in a further reduction in electron flux. In intact cells, inhibition of complex I caused a rapid reduction of mitochondrial oxygen consumption, with ME-344 exhibiting greater toxicity due to the additional inhibition of complex III (Figure 2).
Following ME-344 treatment of HEK293T, HeLa and 143B cells, the prolonged dissipation of the ΔΨm for up to 4 h destabilized the mature OXPHOS complexes, in particular complex I. Degradation of the complex I subunit NDUFA9 and the complex IV subunit COXIV was also observed. This corresponds to the previously described loss of COXIV following NV-128 treatment [15]. However, the degradation of the OXPHOS complexes is not likely a primary signal for cell death induction as proposed previously, but is instead a result of prolonged ΔΨm dissipation due to mitochondrial respiratory inhibition.
Loss of OXPHOS complex stability was not observed following ME-143 treatment. This may be due in part to its lower capacity to inhibit mitochondrial respiration (we found that ME-143 inhibits complex I only whereas ME-344 inhibits both complex I and III). In fact, ME-143 treatment appears to increase the steady-state levels of complex I in some cell types, possibly as a compensatory mechanism in response to respiratory inhibition.
Interestingly, OXPHOS complex stability was not affected in primary fibroblasts by ME-143 or ME-344 (Figure 1D), whereas reduced steady-state levels of complex I were observed in 143B and HeLa cancer cells and transformed HEK293T cells following ME-344 treatment (Figure 1A-C). These results may help to explain the tumor specificity of ME-344, however, the mechanism which determines OXPHOS complex destabilization only in tumor-derived cells remains to be defined.
The identification of complex I as a target of ME-143 and ME-344 aids our understanding of the previously described effects of isoflavone derivatives on mitochondrial function and their link to mitochondrial cell death induction. Treatment of epithelial ovarian cancer cells with the ME-344 precursor, NV-128, was shown to decrease ATP levels, induce mitochondrial ROS generation, dissipate the ΔΨm and decrease steady-state levels of the complex IV subunits COXI and COXIV [14,15]. These metabolic changes were associated with mTOR signaling inhibition and Bax translocation to the outer mitochondrial membrane. In addition, markers of autophagic induction, including elevated LC3-II levels and beclin-1 translocation to the mitochondria, were observed [14].
Based on our new findings, in combination with previous reports of the parent compound NV-128 [14,15], we propose a model for the mechanism by which ME-344 induces cell death in cancer cells (Figure 5).
Figure 5.
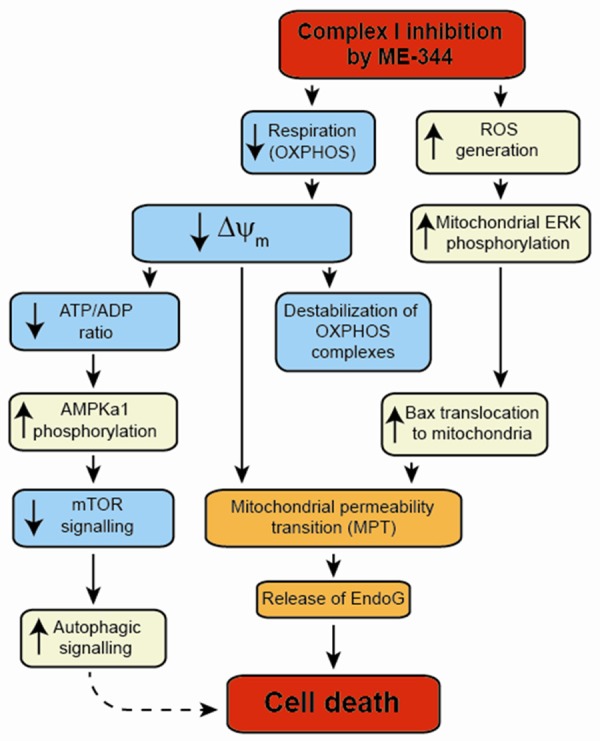
ME-344 induces changes in mitochondrial function that lead to cell death. ME-344 inhibition of complex I reduces mitochondrial respiration, resulting in the dissipation of the ΔΨm. This leads to the destabilization of the mature OXPHOS complexes, a reduced ATP/ADP ratio, and the induction of mitochondrial permeability transition (MPT). The reduced ATP/ADP ratio increases AMPKα1 levels, subsequently reducing mTOR signaling via phosphorylated S6 kinase to activate autophagic signaling. Inhibition of complex I also causes the generation of ROS, which induces the phosphorylation of mitochondrial ERK, leading to Bax translocation to the mitochondrial outer membrane. In conjunction with the prolonged dissipation of the ΔΨm, Bax translocation induces MPT, releasing EndoG to the nucleus to initiate cell death.
ME-344 directly inhibits complex I, causing an immediate reduction of mitochondrial oxygen consumption that results in the dissipation of the ΔΨm. The prolonged reduction of the ΔΨm, over approximately 2 to 4 h, subsequently leads to the destabilization of the mature OXPHOS complexes (see Figure 1). As these complexes are disassembled, their individual structural subunits are degraded and lost (see Figure 1).
The reduction of mitochondrial respiration and loss of the ΔΨm also result in a decrease of the ATP/ADP ratio [15]. This signals an increase in AMPKα1 levels and a reduction in mTOR signaling via phosphorylated S6 kinase, resulting in activation of the autophagic signaling pathway (although this does not appear to be the primary mechanism of cell death induction [14]).
ME-344 inhibition of complex I (and to a lesser extent complex III) also causes the generation of ROS [15]. This ROS induces the phosphorylation of mitochondrial ERK, leading to the translocation of Bax to the mitochondrial outer membrane. In conjunction with the prolonged dissipation of the ΔΨm, Bax translocation induces mitochondrial permeability transition (MPT), resulting in the release of pro-apoptotic molecules, including EndoG [14]. Thus, the inhibition of mitochondrial complex I-linked respiration by ME-344 results in the activation of multiple signaling pathways that lead to cell death induction associated with mitochondrial permeability transition.
In conclusion, ME-143 and ME-344 inhibit mitochondrial respiration by acting directly at OXPHOS complex I. This finding provides new insights into how synthetic isoflavone-based compounds induce cell death and will direct future work to maximize their anti-cancer cytotoxicity.
Acknowledgements
We thank Ian Trounce and Nicole Van Bergen for assistance with the complex I activity assay. This work was supported by the Victorian Government’s Operational Infrastructure Support Program. MMcK is supported by an Australian Research Council Future Fellowship, MIMR-PHI Institute of Medical Research, Monash University, the Australian Mitochondrial Disease Foundation, the William Buckland Foundation and MEI Pharma.
Disclosure of conflict of interest
This project was funded by MEI Pharma, San Diego, USA, who supplied the compounds ME-143 and ME-344 used in this study.
References
- 1.Brown DM, Kelly GE, Husband AJ. Flavonoid compounds in maintenance of prostate health and prevention and treatment of cancer. Mol Biotechnol. 2005;30:253–270. doi: 10.1385/MB:30:3:253. [DOI] [PubMed] [Google Scholar]
- 2.Kamsteeg M, Rutherford T, Sapi E, Hanczaruk B, Shahabi S, Flick M, Brown D, Mor G. Phenoxodiol--an isoflavone analog--induces apoptosis in chemoresistant ovarian cancer cells. Oncogene. 2003;22:2611–2620. doi: 10.1038/sj.onc.1206422. [DOI] [PubMed] [Google Scholar]
- 3.Aguero MF, Facchinetti MM, Sheleg Z, Senderowicz AM. Phenoxodiol, a novel isoflavone, induces G1 arrest by specific loss in cyclin-dependent kinase 2 activity by p53-independent induction of p21WAF1/CIP1. Cancer Res. 2005;65:3364–3373. doi: 10.1158/0008-5472.CAN-04-2429. [DOI] [PubMed] [Google Scholar]
- 4.Axanova L, Morre DJ, Morre DM. Growth of LNCaP cells in monoculture and coculture with osteoblasts and response to tNOX inhibitors. Cancer Lett. 2005;225:35–40. doi: 10.1016/j.canlet.2004.11.017. [DOI] [PubMed] [Google Scholar]
- 5.Brown JM, Attardi LD. The role of apoptosis in cancer development and treatment response. Nat Rev Cancer. 2005;5:231–237. doi: 10.1038/nrc1560. [DOI] [PubMed] [Google Scholar]
- 6.Sapi E, Alvero AB, Chen W, O’Malley D, Hao XY, Dwipoyono B, Garg M, Kamsteeg M, Rutherford T, Mor G. Resistance of ovarian carcinoma cells to docetaxel is XIAP dependent and reversible by phenoxodiol. Oncol Res. 2004;14:567–578. doi: 10.3727/0965040042707943. [DOI] [PubMed] [Google Scholar]
- 7.Silasi DA, Alvero AB, Rutherford TJ, Brown D, Mor G. Phenoxodiol: pharmacology and clinical experience in cancer monotherapy and in combination with chemotherapeutic drugs. Expert Opin Pharmacother. 2009;10:1059–1067. doi: 10.1517/14656560902837980. [DOI] [PubMed] [Google Scholar]
- 8.Alvero AB, O’Malley D, Brown D, Kelly G, Garg M, Chen W, Rutherford T, Mor G. Molecular mechanism of phenoxodiol-induced apoptosis in ovarian carcinoma cells. Cancer. 2005;106:599–608. doi: 10.1002/cncr.21633. [DOI] [PubMed] [Google Scholar]
- 9.Herst PM, Petersen T, Jerram P, Baty J, Berridge MV. The antiproliferative effects of phenoxodiol are associated with inhibition of plasma membrane electron transport in tumour cell lines and primary immune cells. Biochem Pharmacol. 2007;74:1587–1595. doi: 10.1016/j.bcp.2007.08.019. [DOI] [PubMed] [Google Scholar]
- 10.Morré DJ, Chueh PJ, Yagiz K, Balicki A, Kim C, Morre DM. ECTO-NOX target for the anticancer isoflavene phenoxodiol. Oncol Res. 2007;16:299–312. doi: 10.3727/000000006783980973. [DOI] [PubMed] [Google Scholar]
- 11.Morré DJ, Chueh PJ, Lawler J, Morré D. The Sulfonylurea-Inhibited NADH Oxidase Activity of HeLa Cell Plasma Membranes has Properties of a Protein Disulfide-Thiol Oxidoreductase with Protein Disulfide-Thiol Interchange Activity. J Bioenerg Biomembr. 1998;30:477–487. doi: 10.1023/a:1020594214379. [DOI] [PubMed] [Google Scholar]
- 12.Morre DJ, Morre DM. Cell surface NADH oxidases (ECTO-NOX proteins) with roles in cancer, cellular time-keeping, growth, aging and neurodegenerative diseases. Free Radic Res. 2003;37:795–808. doi: 10.1080/1071576031000083107. [DOI] [PubMed] [Google Scholar]
- 13.Pant S, Burris HA 3rd, Moore K, Bendell JC, Kurkjian C, Jones SF, Moreno O, Kuhn JG, McMeekin S, Infante JR. A first-in-human dose-escalation study of ME-143, a second generation NADH oxidase inhibitor, in patients with advanced solid tumors. Invest New Drugs. 2014;32:87–93. doi: 10.1007/s10637-013-9949-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Alvero AB, Montagna MK, Chen R, Kim KH, Kyungjin K, Visintin I, Fu HH, Brown D, Mor G. NV-128, a novel isoflavone derivative, induces caspase-independent cell death through the Akt/mammalian target of rapamycin pathway. Cancer. 2009;115:3204–3216. doi: 10.1002/cncr.24397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Alvero AB, Montagna MK, Holmberg JC, Craveiro V, Brown D, Mor G. Targeting the Mitochondria Activates Two Independent Cell Death Pathways in Ovarian Cancer Stem Cells. Mol Cancer Ther. 2011;10:1385–1393. doi: 10.1158/1535-7163.MCT-11-0023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bendell JC, Patel MR, Infante JR, Kurkjian CD, Jones SF, Pant S, Burris HA 3rd, Moreno O, Esquibel V, Levin W, Moore KN. Phase 1, open-label, dose escalation, safety, and pharmacokinetics study of ME-344 as a single agent in patients with refractory solid tumors. Cancer. 2014 doi: 10.1002/cncr.29155. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.McKenzie M, Lazarou M, Ryan MT. Chapter 18 Analysis of respiratory chain complex assembly with radiolabeled nuclear- and mitochondrial-encoded subunits. Methods Enzymol. 2009;456:321–339. doi: 10.1016/S0076-6879(08)04418-2. [DOI] [PubMed] [Google Scholar]
- 18.Trounce IA, Kim YL, Jun AS, Wallace DC. Assessment of mitochondrial oxidative phosphorylation in patient muscle biopsies, lymphoblasts, and transmitochondrial cell lines. Methods Enzymol. 1996;264:484–509. doi: 10.1016/s0076-6879(96)64044-0. [DOI] [PubMed] [Google Scholar]
- 19.Schagger H, von Jagow G. Blue native electrophoresis for isolation of membrane protein complexes in enzymatically active form. Anal Biochem. 1991;199:223–231. doi: 10.1016/0003-2697(91)90094-a. [DOI] [PubMed] [Google Scholar]
- 20.Trounce IA, Crouch PJ, Carey KT, McKenzie M. Modulation of ceramide-induced cell death and superoxide production by mitochondrial DNA-encoded respiratory chain defects in Rattus xenocybrid mouse cells. Biochim Biophys Acta. 2013;1827:817–825. doi: 10.1016/j.bbabio.2013.03.012. [DOI] [PubMed] [Google Scholar]
- 21.Harlow E, Lane D. Using Antibodies: A Laboratory Manual. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 1999. [Google Scholar]
- 22.McKenzie M, Lazarou M, Thorburn DR, Ryan MT. Analysis of mitochondrial subunit assembly into respiratory chain complexes using Blue Native polyacrylamide gel electrophoresis. Anal Biochem. 2007;364:128–137. doi: 10.1016/j.ab.2007.02.022. [DOI] [PubMed] [Google Scholar]
- 23.McKenzie M, Lazarou M, Thorburn DR, Ryan MT. Mitochondrial respiratory chain supercomplexes are destabilized in Barth Syndrome patients. J Mol Biol. 2006;361:462–469. doi: 10.1016/j.jmb.2006.06.057. [DOI] [PubMed] [Google Scholar]
- 24.McKenzie M. Mitochondrial DNA Mutations and Their Effects on Complex I Biogenesis: Implications for Metabolic Disease. In: St John JC, editor. Mitochondrial DNA, Mitochondria, Disease and Stem Cells. New York: Springer; 2013. [Google Scholar]
- 25.Warburg O. On the origin of cancer cells. Science. 1956;123:309–314. doi: 10.1126/science.123.3191.309. [DOI] [PubMed] [Google Scholar]
- 26.Chih HW, Chiu HF, Tang KS, Chang FR, Wu YC. Bullatacin, a potent antitumor annonaceous acetogenin, inhibits proliferation of human hepatocarcinoma cell line 2.2. 15 by apoptosis induction. Life Sci. 2001;69:1321–1331. doi: 10.1016/s0024-3205(01)01209-7. [DOI] [PubMed] [Google Scholar]
- 27.Oberlies NH, Croy VL, Harrison ML, McLaughlin JL. The Annonaceous acetogenin bullatacin is cytotoxic against multidrug-resistant human mammary adenocarcinoma cells. Cancer Lett. 1997;115:73–79. doi: 10.1016/s0304-3835(97)04716-2. [DOI] [PubMed] [Google Scholar]
- 28.Pathania D, Millard M, Neamati N. Opportunities in discovery and delivery of anticancer drugs targeting mitochondria and cancer cell metabolism. Adv Drug Deliv Rev. 2009;61:1250–1275. doi: 10.1016/j.addr.2009.05.010. [DOI] [PubMed] [Google Scholar]