

Abstract
Nitric oxide (NO) has been shown to be effective in cancer chemoprevention and therefore drugs that help generate NO would be preferable for combination chemotherapy or solo use. This study shows a new evidence of NO as a mediator of acute leukemia cell death induced by fisetin, a promising chemotherapeutic agent. Fisetin was able to kill THP-1 cells in vivo resulting in tumor shrinkage in the mouse xenograft model. Death induction in vitro was mediated by an increase in NO resulting in double strand DNA breaks and the activation of both the extrinsic and the intrinsic apoptotic pathways. Double strand DNA breaks could be reduced if NO inhibitor was present during fisetin treatment. Fisetin also inhibited the downstream components of the mTORC1 pathway through downregulation of levels of p70 S6 kinase and inducing hypo-phosphorylation of S6 Ri P kinase, eIF4B and eEF2K. NO inhibition restored phosphorylation of downstream effectors of mTORC1 and rescued cells from death. Fisetin induced Ca2+ entry through L-type Ca2+ channels and abrogation of Ca2+ influx reduced caspase activation and cell death. NO increase and increased Ca2+ were independent phenomenon. It was inferred that apoptotic death of acute monocytic leukemia cells was induced by fisetin through increased generation of NO and elevated Ca2+ entry activating the caspase dependent apoptotic pathways. Therefore, manipulation of NO production could be viewed as a potential strategy to increase efficacy of chemotherapy in acute monocytic leukemia.
Keywords: Fisetin, apoptosis, calcium, nitric oxide, leukemia, mTOR
Introduction
Numerous biological processes are mediated by nitric oxide (NO) generated constitutively within the cells. However, drugs and other stresses can also generate NO leading to cell death [1]. This property has generated substantial interest because of this awareness that NO can be used as a potent agent to induce cell death. Reports indicate that NO has a significant potential in killing cancer cells and inhibiting tumor growth [2]. It is now established that co-delivery of NO with chemotherapeutic drugs can enhance the suppression of tumors [3,4]. Therefore, drugs that generate NO are potentially of great usage for use as a combination therapy or for single use. NO can have dual roles either through activation or suppression of pathways that bring about cell survival or death [5]. Acute leukemia is due to uncontrolled expansion and differentiation of progenitor cells of the lymphoid or myeloid origin and is divided into acute myeloid leukemia (AML) when cells originate from myeloid progenitors and acute lymphoblastic leukemia (ALL) when the cells are of lymphoblastic origin. Although reports are available on cell death induced by chemically generated NO in these cells, data on chemotherapeutic agents acting through generation of NO is scarce and not well understood. The flavonoid fisetin, a tetrahydroxyflavone commonly found in fruits and vegetables like strawberry, apple, grape, onion, persimmon etc. is known to kill several types of cancer cells through its anti-proliferative, antioxidant and ROS generating activities [6,7]. This compound has been shown to suppress NO [8-10] but not inducing an increase, however, a complete picture of how fisetin aids in cell survival or death using NO as a mediator is not clear.
Nitric oxide mediated cell death occur through the process of apoptosis or autophagy [1,11]. While apoptosis is the major pathway leading to cell death [12], autophagy is a house keeping process involved in turnover of essential molecules through the lysosomal pathway [13] and balance between the two processes often determine cell fate [14]. The two primary apoptotic pathways recognized as the death receptor (extrinsic) and mitochondrial (intrinsic) pathways play critical roles in the working of chemotherapeutic regimens.
The present study provides new evidence for the involvement of NO as a mediator of AML cell death induced by fisetin. Fistein induced AML cell death through overproduction of cellular NO that caused double strand breaks and activated both the extrinsic and intrinsic pathway of apoptosis without any activation of autophagy. Fisetin inhibited phosphorylation of downstream effectors of the mTOR pathway leading to cell death that was reversed if NO generation was inhibited. Interestingly, an increase in intracellular Ca2+ occurred through L-type Ca2+ channels and inhibition of Ca2+ entry partially prevented cell death, but this Ca2+ increase was independent of the NO increase. Therefore, NO mediated cell death can be explored as a major mode for inducing AML cell apoptosis.
Materials and methods
Reagents
Fisetin (3,3’,4’,7-tetrahydroxyflavone,5-dexoyquercetin), propidium iodide (PI) and all other chemicals unless mentioned were obtained from Sigma-Aldrich (St. Louis, MO). Antibodies to caspase-8, caspase-3, FAS, BID, γH2AX (phosphor S139) and caspase inhibitors Z-VAD-FMK, Z-IETD-FMK, Z-DEVD-FMK, Z-LEHD-FMK and caspase substrates Ac-IETD-AFC, inhibitor Ac-IETD-CHO, Matrigel were from BD Biosciences (San Jose, CA). Antibodies against poly(ADP-ribose) polymerase (PARP), cytochrome C and caspase-9 were from Santa Cruz Biotechnology (Santa Cruz, CA) and antibodies against caspase-7, p70 S6 kinase, phosphor-S6 ribosomal protein (Ser240/244), phosphor-S6 ribosomal protein (Ser235/236), phospho-eIF4B (Ser422), phosphor-eEF2K were from Cell Signaling Technology (Beverly, MA). Fluo-3-acetoxymethyl ester (fluo-3-AM), 2’,7’-dichlorofluorescein diacetate (DCF-DA), 4-amino-5-methylamino-2’,7’-difluorofluorescein (DAF-FM), S-nitroso-N-acetylpenicillamine (SNAP), 2-(4-carboxyphenyl)-4,4,5,5-tetramethylimidazoline-l-oxyl-3-oxide (cPTIO), Hoechst 33342 and secondary anti-mouse IgG conjugated to Alexa Fluor 488 were obtained from Molecular Probes (Eugene, OR). G-Biosciences (St. Louis, MO) and Biological Industries (Kibbutz Beit Haemek, Israel) supplied CBX protein estimation kit and fetal bovine serum (FBS) and enhanced chemiluminescence system, respectively.
Cell culture
THP-1, K562 and U937 the monocytic leukemia cell lines, (ATCC, Manassas, VA) were maintained in Roswell Park Memorial Institute (RPMI)-1640 media (Sigma-Aldrich, St. Louis, MO), supplemented with 10% FBS. The cells were maintained at 37°C in 5% carbon dioxide and air. Fisetin treatment was given to these cells maintained in 24 well plates at a concentration of 2 × 105 ml-¹.
Cell viability measurement
For cell viability, MTT [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide] assay as described by Hansen et al. [15] was performed with slight modifications. Briefly, various treated cell groups were incubated in media containing MTT at 250 µg ml-¹ for 6 h at 37°C followed by solubilizing the crystals in lysis buffer (20% SDS in 50% dimethyl formamide) for 3 h at 37°C and OD was measured at 570 nm. Data were plotted against a standard curve prepared with known number of viable cells. For detection of PI positive cells, cells fixed in 70% ice-cold ethanol and stained with PI [propidium iodide] were analyzed by flow cytometry.
SDS-PAGE and Western blot
SDS-PAGE and Western blot was carried out as described previously [14]. Briefly, whole cell extracts (25-40 µg) prepared by mixing the cells with lysis buffer (0.125 M Tris, 4% SDS, 20% glycerol, and 10% 2-ME) were resolved in 12% SDS polyacrylamide gels and transferred onto nitrocellulose membranes. For all non-phospho antibodies, blocking was done in 5% nonfat dry milk and for all anti-phospho antibodies in 5% BSA in 0.05% PBS-Tween 20. The membranes were incubated with primary antibodies at required dilutions [anti-caspases-8,-9,-3,-7, anti-cytochrome C, anti-p70 S6 kinase, anti-phosphor-S6 anti-ribosomal protein (Ser240/244), anti-phosphor-S6 ribosomal protein (Ser235/236), anti-phospho-eIF4B (Ser422), anti-phosphor-eEF2K, (1:1,000), anti-γH2AX (phosphor S139), anti-PARP (1:2,000) and anti-Bid (1:5,000) and anti-actin (1:10,000)] in PBS-Tween-20 containing 5% non-fat dry milk and incubated overnight at 4°C. After secondary antibody (Jackson Immuno Research Laboratories Inc., West Grove, PA) incubation at 1:5,000-1:10,000 dilutions in 0.05% PBS-Tween-20 for 1 h at room temperature, protein bands were visualized on X-ray films using the enhanced chemiluminescence system.
Immunocytochemistry and annexin-V staining
Cells fixed in 4% formaldehyde were blocked with 3% normal goat serum containing 0.1% saponin at room temp for 30 min and subsequently incubated with primary antibody against γH2AX (phosphor S139) at 1:200 dilution for 1 h at 37°C followed by secondary antibody conjugated to Alexa Fluor 488 at 1:150 dilution for 1 h at the same temp. An inverted microscope [(TE-2000E) Nikon, Tokyo, Japan] equipped with a RetigaExi camera (Q-imaging) was used to acquire images (Media Cybernetics, Bethesda, MD). For the detection of apoptotic cell death, PI staining and phosphatidylserine exposure by Annexin V-labeling was conducted using Dead Cell Apoptosis Kit (Molecular Probes, Eugene, OR). Labeling was analyzed with a BD FACS Calibur (Becton-Dickinson, Franklin Lakes, NJ) flow cytometer equipped with a 488 nm air-cooled argon ion laser. Analysis was carried out using Flow Jo software (version 8.7.1). Mice were euthanized and dissected to remove tumors and various other organs, and were fixed in 4% formaldehyde and Bouin’s fluid for TUNEL and hematoxylin and eosin (H&E) staining for routine histology, respectively. Tissues were processed as per standard protocol.
Caspase activity assay
Caspase-8 activity in the treated and untreated cell lysates was assayed using the caspase-8 specific fluorescence peptide substrate Ac-IETD-AFC and the caspase-8 inhibitor Ac-IETD-CHO. Fluorescence from free AFC (7-amino-4-trifluoromethyl coumarin) was measured using a spectrofluorometer (PerkinElmer, Waltham, MA) with excitation wavelength of 400 nm and emission wavelength of 450-550 nm.
Measurement of ROS, NOS and cytosolic free Ca2+
Changes in intracellular free Ca2+, reactive oxygen species (ROS) and reactive nitrogen species (NOS) concentrations were monitored using the specific probes Fluo-3AM, DCF-DA and DAF-FM respectively as described previously [16]. Briefly, 106 cells ml-¹ was loaded in phenol free media without FBS and 0.5 µM Fluo-3AM with 0.5 µM pluronic acid F-127 (for well dispersal of the dye) and 250 µM of sulfinpyrazone (to prevent the leakage of dye) and time kinetics of Ca2+ changes was measured using a flow cytometer at 488 nm. Free ROS and NO were monitored at an excitation of 480 nm and emission of 520 nm respectively with a Fluostar Omega spectrofluorometer (BMG Technologies, Offenburg, Germany).
Xenograft tumors
All the animal studies were approved by Institutional Animal Ethics Committee of National Institute of Immunology, New Delhi. Approximately 5 × 106 THP-1 cells, suspended in phosphate-buffered saline, were mixed with Matrigel and implanted subcutaneously into the right axillary fossa of 5-6 week-old athymic nude mice [NIH(s) (nu/nu)]. Mice were housed under specific pathogen-free conditions. The tumor bearing animals (50-100 mm³) were divided randomly into two groups, one group treated with fisetin (2 mg/kg) intraperitoneally every alternative day for 12 days, and control group was treated with vehicle only. Tumor volumes were measured using Vernier calipers and calculated with the help of the equation: volume = (L × W²)/2 where L = length and W = width.
Statistical analysis
Data were analyzed by student’s unpaired t-test. The values were considered significantly different at p < 0.05. All experiments were repeated 3-5 times and data expressed as the mean ± SEM of several independent experiments.
Results
Fisetin affects survival of acute monocytic leukemia cells
To test the efficacy of fisetin to eliminate AML cells, both in vivo and in vitro systems were used. We generated xenograft tumors with THP-1 cells in nu/nu mice, grown till a volume of 50 mm2. These tumors were treated with fisetin (2 mg/kg/alternate day) and tumor sizes were measured at intervals till 32 days. As shown in Figure 1A-C, a significant difference in the sizes of the tumors were measured between fisetin treated and vehicle treated groups with the vehicle treated tumors being significantly larger in size in comparison to fisetin treated ones. TUNEL staining of tissue sections from the tumors for measuring DNA fragmentation [17] subsequent to treatment, showed a higher number of TUNEL positive cells in fisetin treated tumors confirming higher rates of cell death with apoptotic phenotype (Figure 1D). Bar graph in Figure 1E shows a significant difference in the number of cells with DNA fragmentation between the control and the fisetin treated group.
Figure 1.
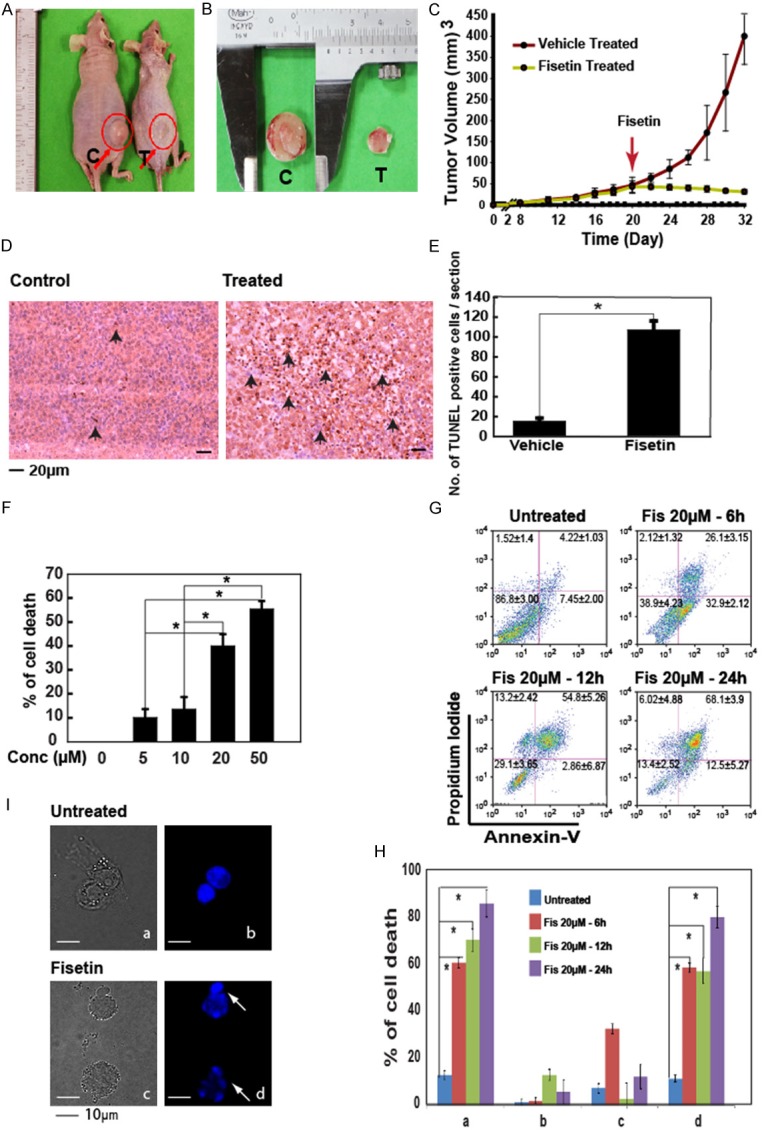
Fisetin induces apoptosis in THP-1 cells in-vivo and in-vitro. (A) Figure shows the effect of fisetin on tumors generated by THP-1 cells in nu/nu mice. C, control; T, treated. (B) A significant reduction in tumor size after 12 days of treatment was observed as measured with Vernier calipers. (C) Graph showing tumor volume changes within 12 days after initiation of fisetin treatment (marked by arrow). Data are mean ± SEM, n = 8. (D) TUNEL staining of the respective xenografted tumor sections show cells with fragmented DNA in fisetin treated group indicating increased apoptosis. (E) Bar graph represents the average number of TUNEL positive cells from a total of 10 tissue sections. P < 0.01. (F) THP-1 cells treated with fisetin at different concentrations for 12 h showing cytostasis at different doses. Data are ± SEM, *p < 0.05. n = 3. (G) Untreated cells or cells treated with 20 μM fisetin for 6, 12 and 24 h showing phosphatidylserine exposure. The upper right quadrant represents both annexin and PI positive cells (late apoptotic), the lower right quadrant shows only annexin positive cells (early apoptotic), the upper left quadrant only PI positive cells and lower left is unstained cells. (H) The bar graph shows comparative interpretation of data presented in (G). a, total cell death; b, necrotic cell death; c, early apoptotic cells; d, total apoptotic cells. The data are ± SEM of 3 experiments, *p < 0.05. (I) Analysis of nuclear morphology by fluorescence microscopy following Hoechst staining of nucleus of fisetin treated and untreated cells. The bar represents 10 µm.
Next, we tested the efficacy of fisetin to kill cells in vitro. A dose dependent increase in arrest of metabolic activities at 12 h achieving above 50% cell death at 50 µM concentration was recorded (Figure 1F). Since 20 µM fisetin was at a range where sufficient viable cells were undergoing changes brought about by fisetin treatment, this dose was chosen for further studies. To check if the cells expressed apoptotic phenotype after exposure to fisetin, death was assessed by phosphatidylserine (PS) exposure to the outer leaflet of the cell [18]. Post fisetin treatment, a time dependent increase in the number of early and late apoptotic cells was observed (Figure 1G, 1H). At all hours, the cells with apoptotic phenotype were most abundant while cells dying by non-apoptotic means were significantly less (Figure 1G, 1H). Nuclear fragmentation, another distinct feature of apoptosis, where the nucleus splits into smaller bodies [19] was visible at 12 h in fisetin treated cells (Figure 1I). In summary, the above data from in vivo and in vitro tests confirmed that THP-1 acute monocytic leukemia cells were sensitive to fisetin treatment and most of the dying cells expressed an apoptotic phenotype.
Fisetin treatment resulted in increase of NO levels
Since most drugs generate ROS, we measured ROS production after fisetin treatment using DCF-DA, a dye that detects the total amount of ROS in the cell [20]. Although flavonoids frequently act through ROS [21], no ROS increase could be detected with DCF-DA after fisetin treatment (Figure 2A). However, when NO generation was measured using a NO sensitive dye DAF-FM, there was a significant increase in NO with increasing time of exposure (Figure 2B). This increase was sensitive to cPTIO (2-(4-Carboxyphenyl)-4,4,5,5-tetramethylimidazoline-l-oxyl-3-oxide), a cell-permeating NO scavenger, the addition of which during fisetin treatment brought down the NO levels (Figure 2B). Since NO production can signal for cell death [1], cell death was measured in the presence or absence of cPTIO, thus creating conditions of fisetin action in the presence or absence of NO. Presence of cPTIO was able to significantly reverse a seven times decrease in viability induced by fisetin treatment (Figure 2C). Bar graphs (Figure 2D) shows distribution of cells stained with annexin-V where it was evident that presence of cPTIO during fisetin treatment reduced early and late apoptotic cells as well as the cells dying by non-apoptotic means. The above experiments suggested a direct link between the reduction of NO and lowering of cell death.
Figure 2.

Fisetin induces reactive nitrogen species to induce cell death. (A) Figure shows generation of reactive oxygen species in response to fisetin as measured with fluorescence dye DCF-DA. Note that there was no ROS generation in fisetin treated cells. H2O2 was used as the positive control. (B) DAF-FM (sensor dye of NO) stained fisetin treated cells showing significant increase in NO as compared to control cells. cPTIO used as NO inhibitor shows attenuation of NO production. (C) Measurement of apoptotic death in fisetin treated cells in the presence and absence of the NO inhibitor cPTIO as measured by phosphatidylserine exposure, shows inhibition of cell death in the presence of cPTIO. The upper right quadrant represents both annexin and PI positive cells (late apoptotic), the lower right quadrant shows only annexin positive cells (early apoptotic), the upper left quadrant only PI positive cells and lower left is unstained cells. (D) Bar graph shows distribution of cells as achieved by AnnexinV staining shown in (C). Data are ± SEM, n = 4, *p < 0.05. (E) Cells stained with NO sensor dye DAF-FM shows an increase in NO in presence of NO generator SNAP. The cPTIO was used as a NO scavenger. (F) The bar graph represents cell death in the presence of SNAP and cPTIO. Data are ± SEM, n = 3, *p < 0.05.
To be sure that the above observations did not occur due to an interaction between cPTIO and fisetin, Mass Spectra of fisetin, cPTIO and the combination was recorded. Fisetin provided a single large peak at 286 and cPTIO provided two major peaks at 278 and 262 nm. In a mixture of both, all three peaks were present but no other product was found (Figure S1A) showing that the components did not react with each other. To further confirm this, absorbance spectra for fisetin, cPTIO and their combinations was measured. Fisetin and cPTIO show distinct spectra and the combination spectra resemble any one of them depending upon concentration but no change of pattern was noticed (Figure S1B).
To investigate if the THP-1 cells would respond to NO generated by other means, NO was induced in THP-1 cells by SNAP, an S-nitrosothiol which serves as a spontaneous NO donor under physiological conditions [22]. The NO generated by SNAP (Figure 2E) induced cell death and the amount of death was reduced when SNAP was given along with cPTIO (Figure 2F). This confirmed that NO was able to induce THP-1 cell death independent of the stimuli that generated it.
Fisetin treatment induced double strand DNA breaks
Flavonoids are known to inhibit the activity of topoisomerases and show genotoxic effects through catalytic inhibition of both topoisomerase I and II that could induce DNA double strand breaks [23]. To know if the effect of fisetin was due to its inhibitory effect on topoisomerase activity, DNA double strand breaks were identified by western blots and microscopy using gamma H2A-X phospho S139 antibody. As a positive control, etoposide, an inhibitor of topoisomerases was used. Fisetin treatment caused DNA double strand breaks in significant number of cells but the presence of cPTIO during fisetin exposure could inhibit the DNA breaks (Figure 3A-C). The cPTIO failed to inhibit etoposide induced double strand breaks (Figure 3A-C). The above data clearly suggests that fisetin causes DNA double strand breaks in the THP-1 cells primarily through the production of NO and do not act as a topoisomerase inhbitior as shown in other cell types.
Figure 3.

Fisetin induced nitric oxide causes DNA double strand breaks. A. Western blots of fisetin (20 µM for 6 h) treated cells in the presence (Group 4) or absence (Group 2) of NO inhibitor, cPTIO shows that scavenging of NO prevents fisetin induced double strand breaks. Etoposide (20 μM for 18 h) (Group 3) was used as a positive control and actin as the loading control. All western blots are representative of a minimum of 3 repeats. Bar graph shows relative expression of gamma H2A-X phospho S139. Data are ± SEM, n = 3, *p < 0.05. B. Number of cells positive for double strand break was measured using fluorescence microscopy. Groups are 1, control; 2, fisetin; 3, etoposide; 4, fisetin + cPTIO; 5, etoposide + cPTIO; 6, fisetin + cPTIO + etoposide. Data are ± SEM, n = 3. 150-200 cells were analyzed per assay. C. Representative microphotographs showing cells stained for DNA double strand breaks under different conditions.
Fisetin induced NO production down regulates mTOR activity and causes activation of caspases
Since impaired mTOR signaling is known to mediate flavonoid action [24] and could be activated by NO [25], mTOR pathway components were checked for changes. Downstream components of mTOR signaling pathway revealed a significantly reduced basal level of p70S6 kinase upon fisetin treatment (Figure 4A). The p70S6 kinase is a protein known to modulate cell cycle progression and cell survival [26] and its levels were restored when fisetin treatment was given in the presence of cPTIO (Figure 4A). Fisetin reduced phosphorylation of S6 ribosomal protein (S6 Ri P), a component of 40S ribosomal subunit involved in the regulation of translation at Ser 235/236 and Ser 240/244 (Figure 4A). However, cPTIO treatment of the cells during fisetin exposure allowed Ser 235/236 and Ser 240/244 phosphorylation (Figure 4A) suggesting a role of NO in these events. Other downstream targets of mTOR, eukaryotic translation initiation factor 4B (eIF4B) and eukaryotic elongation factor 2 kinase (eEF2K) showed inhibition of phosphorylation after fisetin treatment that was reversed by cPTIO exposure (Figure 4A). Surprisingly, inhibition of mTOR did not lead to any changes in autophagy level as detected by autophagy efflux experiments and western blotting of key autophagy regulatory protein Beclin-1 (Figure S2).
Figure 4.

Nitric oxide down regulates mTOR pathway and activates caspases to promote apoptosis. A. Western blots of cells treated with fisetin (20 µM) for 6 h in the presence or absence of NO inhibitor cPTIO shows inhibition of mTOR downstream cascade. Graph shows relative expression of the proteins as measured by densitometry. B. Blots show cleavage of pro-caspase 8 and Bid. The bar graphs below the blots show relative expression levels. C. Blot shows cleavage of pro-caspase 7 on fisetin treatment. Bar graph represents relative expression levels of the proteins. D. Blots show cleavage of pro-caspase 9, 3 and PARP. Bar graph represents relative expression levels of the proteins. All western blots are quantified and numbers on Y-axis represents relative expression level compared to control (corrected by actin). Data are ± SEM, n = 3, *p < 0.05. E. THP-1 cells were pre-incubated for 30 min with caspase inhibitors (CI) (Z-DEVD-FMK, Z-LEHD-FMK, Z-IETD-FMK and Z-VAD-FMK) for caspases-3, -7, -9, -8 and all-caspases respectively shows phosphatidylserine exposure measured by Annexin V-PI staining through flow-cytometry. The upper right quadrant represents both annexin and PI positive cells (late apoptotic), the lower right quadrant shows only annexin positive cells (early apoptotic), the upper left quadrant only PI positive cells and lower left is unstained cells. Note that caspase-8 inhibition worked best in terms of prevention of cell killing.
Since most of the apoptotic deaths occur through caspase dependent pathways, caspase activation was tested in the presence or absence of the NO inhibitor cPTIO. Significant activation of initiator caspase-8 was observed after fisetin treatment but this could be prevented in the presence of cPTIO (Figure 4B). Bid, a connector protein between caspase-8 of the receptor mediated death pathway and capsae-9 of the mitochondrial pathway of death was cleaved by fisetin and the cleavage was prevented in the presence of cPTIO (Figure 4B). Executioner caspases-7 and -3 were also cleaved and the cleavage could be inhibited by scavenging of the NO (Figure 4C, 4D). PARP cleavage (Figure 4D) confirmed the functional outcome of the cleavages of caspases-3 and -7, as it is a substrate for caspase-3 and -7 and a hallmark of activation of these enzymes. Initiator caspase-9 activation was also visible (Figure 4D). To look at the functional outcome of caspase activation, caspase inhibition experiments were carried out with inhibitors for caspases-8, -9, -3. Caspase-8 inhibitor Z-IETD-FMK (CI8) was most effective in reducing cell death as compared to the terminal caspase inhibitor, Z-DEVD-FMK (CI3) (Figure 4E). Caspase-9 inhibitor Z-LEHD-FMK (CI9) was least effective (Figure 4E). These experiments suggested that cell death was primarily mediated through caspase activation post fisetin exposure. Since caspase-8 inhibition was most effective in reducing cell death, enzymatic assay was used to further confirm the inhibition of caspase-8 activity as shown in Figure S3A. It was evident from the above data that inhibition of NO production was directly linked to the suppression of caspase activation because cleavages of caspase-7, -8, -3 and -9 were prevented significantly by scavenging of NO.
Fisetin altered Ca2+ levels and activated caspases
Since changes in the intracellular Ca2+ are known to be associated with activation of apoptosis, cellular free Ca2+ was monitored after exposure to fisetin. Within 60 seconds of addition of fisetin there was a significant increase in detectable free Ca2+ as compared to the basal values and this elevation in level was sustained till the time followed (300 sec) (Figure 5A). Preincubation with nifedipine, a L-type Ca2+ channel blocker prior to fisetin treatment reduced Ca2+ increase suggesting L-type channels as the main port of entry for Ca2+ from the extracellular millieu. To investigate the biological significance of the Ca2+ influx, caspase activation and cell death was checked in the presence or absence of nifedipine. Cleavages of procaspases-7, -9 and -3 were partially inhibited in the presence of nifedipine (Figure 5B-D) while cleavage of procaspase-8 to its fully cleaved active product (23 kDa) was totally prevented by nifedipine (Figure 5D). BID, the connector between extrinsic and intrinsic pathway was cleaved by fisetin and this cleavage was partially prevented in the presence of nifedipine (Figure 5D). Caspase-8 activity assay using Ac-IETD-AFC as substrate confirmed decreased activity in the presence of nifedipine (Figure 5E). Importantly, nifedipine addition had a major functional consequence in terms of reduction of fisetin induced cell death to a significant proportion (Figure 5F), suggesting that prevention of Ca2+ entry abrogated the activation of caspases. From the above data it is apparent that fisetin induced cell death involved activation of caspases preceded by a change in Ca2+ levels. Interestingly, presence of cPTIO did not interfere with Ca2+ entry suggesting that NO was not involved in changing Ca2+ levels (Figure S3B). Inhibition of Ca2+ entry using nifedipine did not interfere with NO increase (Figure 2B).
Figure 5.
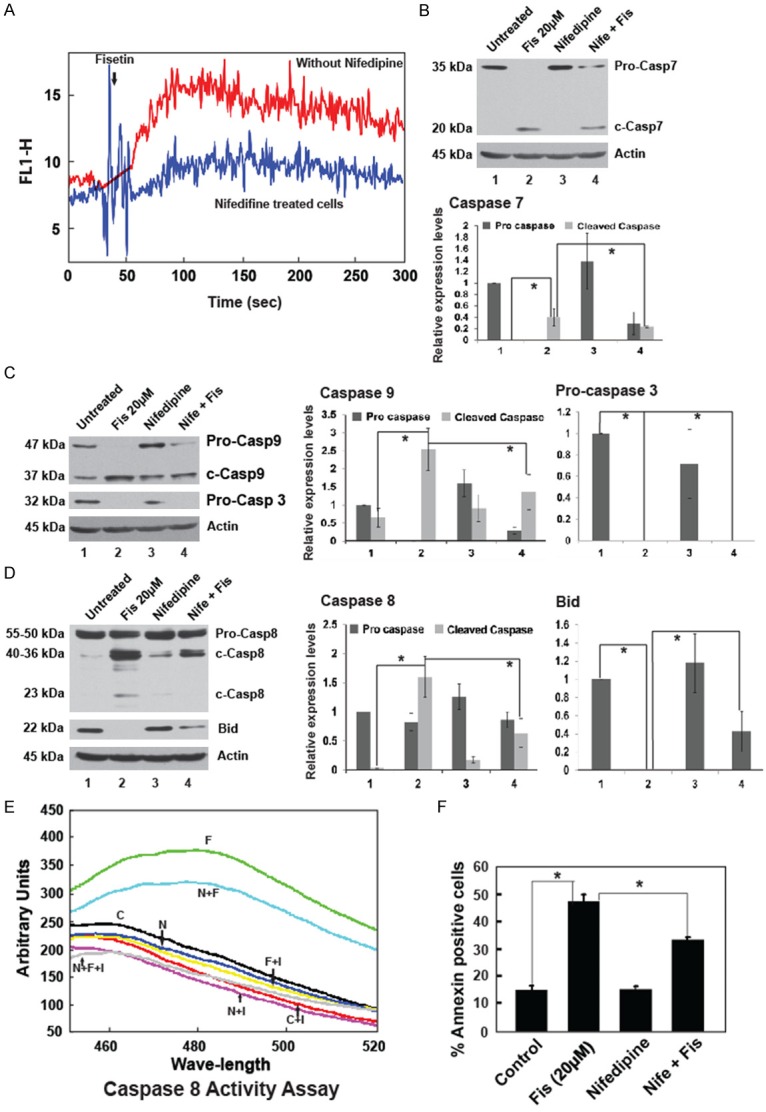
Activation of caspases is partially dependent on Ca2+. (A) Time kinetics of the intracellular Ca2+ level after fisetin treatment measured for fluorescence in cells stained with Fluo-3AM showing an increase in intracellular Ca2+. Note that nifedipine (10 µM), an L-type Ca2+ channel blocker could block Ca2+ entry. (B) Blot shows cleavage of pro-caspase 7 on fisetin treatment in the presence and absence of nifedipine. The bar graph represents relative expression of the protein. (C) Blots show cleavage of pro-caspase 9 and 3. The bar graphs represent relative expression of the proteins. (D) Blots shows cleavage of pro-caspase 8 and Bid. The bar graphs represent relative expression of the proteins. All western blots are quantified and numbers on Y-axis represents relative expression level compared to control (corrected by actin). Results are ± SEM, n = 3, *p < 0.05. (E) Caspase-8 enzyme activity of cells treated for 4 h with fisetin with or without nifedipine. (F) Annexin stained fisetin treated cells (20 µM for 12 h) with or without nifedipine pre-incubation showing a significant reduction in apoptotic cell death. The bars represent mean ± SEM of 3 independent experiments, *p < 0.05.
Fisetin induced apoptosis in other leukemic cell lines is mediated through NO
Since fisetin was able to induce apoptosis of THP-1 cells through production of NO, we were further interested to know whether it is a cell specific phenomenon or not. To test this hypothesis, we treated two other human monocytic cell lines, U937 cells that are lymphoma cells of myeloid origin and K562, another human mylegenous leukemia cell line to fisetin. Fisetin induced NO production in both cell lines in the same time frame as that of the THP-1 cells and this NO could be scavenged by cPTIO (Figure 6A). Pre-incubation of the cells with NO scavenger cPTIO reduced fisetin induced cell death significantly (Figure 6A). The dose of 50 µM of fisetin induced cell death in these two cell lines, around 96% and 40% in U937 and K562 cell lines respectively (Figure 6B). The above data confirmed that fisetin could induce apoptosis in several leukemia cell lines through the generation of NO.
Figure 6.
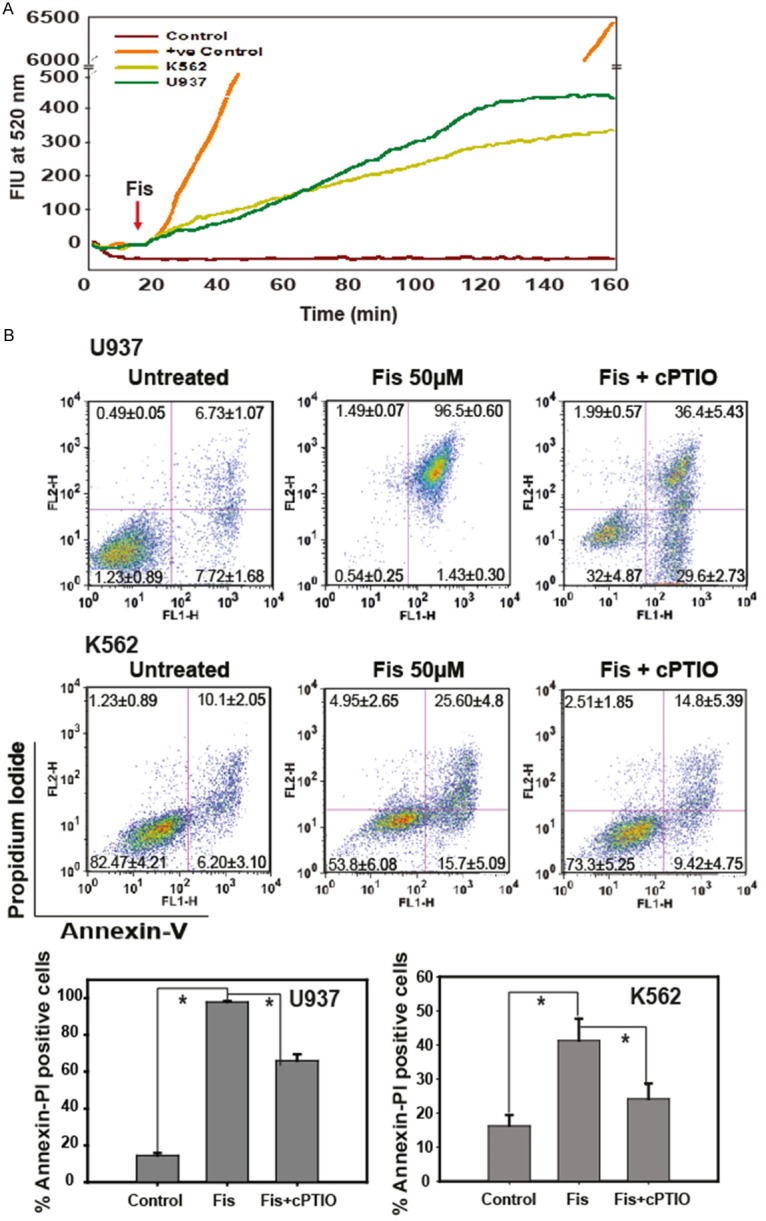
Fisetin induces NO mediated apoptosis in U937 and K562 cell lines. A. K562 and U937 cells stained with DAF-FM shows an increase in NO level on fisetin (20 µM) treatment. B. Apoptotic death 24 h after fisetin (50 µM) treatment in U937 and K562 cell line in the presence and absence of the NO inhibitor cPTIO was measured by Annexin V-PI staining using flow-cytometry. Fisetin causes a significant cell death in both cell lines which is reversed in presence of cPTIO. The upper right quadrant represents both annexin and PI positive cells (late apoptotic), the lower right quadrant shows only annexin positive cells (early apoptotic), the upper left quadrant only PI positive cells and lower left is unstained cells. Bar graphs represent total no of Annexin V-PI positive cells with ± SEM, n = 3, *p < 0.05.
Discussion
NO donors and NO mimetics have been shown to be effective in cancer chemoprevention [27] and therefore, drugs that help generate NO could be more useful for combination therapies or for solo use [1]. Dietary flavonoids, now recognized as potential candidates for developing chemotherapeutic agents offer several promising candidates and fisetin is one of such candidates [10,24,28]. Our previous studies have shown that fisetin enhances the cytotoxic effects of cisplatin in embryonal carcinoma cells, thereby establishing its potential as a possible candidate for combination therapies [14]. Fisetin can either increase or decrease NO depending on the cell type [29,30], however, how the NO precipitates cell death is not well understood.
Using the model cell line THP-1, an acute monocytic leukemia cell line, we show that fisetin is able to generate NO that in turn creates double strand breaks leading to activation of the mTORC1 pathway and activation of death inducing caspases. NO has been reported to either prevent or induce apoptosis depending on its concentration in a given cell type and the existing oxidative milieu [1,2]. Since in our experiments, scavenging of NO resulted in inhibition of cell death, a distinct link between NO elevation and the process of cell death induced by fisetin was established. This is in contrast to reports of fisetin action mediated through suppression of NO in multiple cancer cell types [29,10]. While correlation of NO to cell survival could be established by the observed reduction of cell death with NO inhibition, causality to cell death was established through other evidences. These included increase of cell death when NO was generated in untreated cells through an NO generator and reduction of death when NO generated by this process was reduced. Clearly, THP-1 cells were susceptible to damage induced by increased NO, whether it was generated by fisetin or by other sources. This supports observations in human promyeloid leukemic cells using unrelated agents where NO could be generated by fucoidin, a polysaccharide [31]. The susceptibility of other monocytic leukemia cell lines like U937 and K562 cells to fisetin generated NO suggested that NO could be the primary mediator of cell death in monocytic leukemia cells. As there was no involvement of ROS in fisetin action as found in other cell types [32], RNS appears to be the primary mediator of cellular injury leading to cell death.
Presence of DNA double strand breaks post fisetin treatment suggested two possibilities, one was the direct influence of NO to create the double strand breaks or the breakage was due to the topoisomerase inhibitory action of the fisetin [32]. Since the DNA double strand breaks were reduced when NO was scavenged, involvement of NO in inducing the breaks was clearly indicated. This observation was supported by data from different treatments in other cell types where NO has been shown to induce DNA double strand breaks [33,34]. A correlation can be drawn from observations on other cancer cell types where inhibition of mTOR was shown to make cells more sensitive to DNA damage [35]. In our studies, since phosphorylation of the downstream components of the mTOR pathway like S6 Ri P kinase, eIF4B and eEF2K was prevented after fisetin treatment, the susceptibility to the DNA damage could be a result of this inhibition.
Fisetin has been shown to activate multiple caspases [28] but not through the activation of NO. Activation of both the intrinsic and extrinsic pathway of apoptosis by fisetin shows a characteristic effect of flavonoids of involvement of multiple pathways. Since a forced inhibition of fisetin generated NO reduced caspase cleavage, it was a direct correlation of NO to the events of caspase activation. This was further validated by blocking of caspase activation by suitable caspase inhibitors during fisetin treatment where cell survival increased. NO influences caspase activation in some cell types like SK-HEP-1 hepatocellular carcinoma and HL-60 human promyelocytic leukemic cells, where caspase-3 activation is also involved [36,37]. In HeLa cells, the human cervical adenocarcinoma cells, fisetin exposure show both caspase-8 and caspase-3 activation [28]. In this study, the activation of caspase-7 and -9 along with caspase-8 and -3 shows involvement of additional caspases in THP-1 cells. The ability of fisetin to kill THP-1 cells in vitro through apoptosis was corroborated in vivo by significant xenograft tumor shrinkage accompanied by the presence of TUNEL positive cells within the tumors when tumors were treated with fisetin. Fisetin also acts through different mechanisms like inhibition of COX2 as in HT-29 human colon carcinoma cells [38].
Through the evolution of multicellular organisms, Ca2+ ion has emerged as a crucial intracellular signaling molecule [39]. Not only does this ion facilitate signaling, under circumstances of cellular stress initiate cell death process by a variety of mechanisms. The increase in intracellular Ca2+ in response to fisetin indicated a pro-apoptotic event as Ca2+ is known to mediate major apoptotic changes [40]. These studies show a distinct elevation of intracellular Ca2+ preceding the NO increase induced by fisetin, however, NO elevation was not related to an increase in Ca2+ because in the absence of Ca2+ increase there was no hindrance to NO increase. As multiple Ca2+ channels could be involved in the influx [41], different inhibitors for Ca2+ channels like pimozide, nifedipine and verapamil were used and nifedipine, an L-type Ca2+ channel blocker was able to block the increase suggesting primary involvement of L-type Ca2+ channels. Effects of flavonoids on Ca2+ homeostasis are not unknown and flavonoids such as quercetin, a natural polyphenolic compound can induce major changes through Ca2+ in neuronal cells [42]. Sylibin induces apoptosis in human chronic myeloid leukemia K562 cells through increase in Ca2+ [43]. Therefore, the changes brought about by fisetin in the Ca2+ homeostasis of the THP-1 cells also contributed to the activation of caspases and cell death. Although NO production was post Ca2+ influx, we tested if fisetin generated NO inhibition would have any effect on Ca2+ increase because NO dependency of Ca2+ is not uncommon [44], but as there was no change in the influx the NO changes appeared independent of Ca2+ changes.
In multiple cellular systems, NO is known to exert its functions in association with mTOR [45]. It is interesting that inhibition of mTOR, the checkpoint protein kinase in mammals [46,47] by NO did not result in any change in autophagy in these studies. This is not surprising because rapamycin, a well-known inhibitor of mTOR can induce apoptotic death in HL-60 promyelocytic leukemia cells [48] or a close derivative RAD001 that can enhance cisplatin induced death without initiating autophagy [49]. On the contrary, existing reports in the literature show the ability of fisetin to induce autophagic cell death by inhibiting both the mTORC1 and mTORC2 pathways in PC-3 human prostate cancer cells [24].
In summary, this study provides a new possibility that fisetin can be evaluated as a possible therapeutic agent for acute monocytic leukemia as it activates multiple pathways of death. This is important because in cancer cells some pathways can be deactivated, therefore, any agent that tends to initiate multiple death pathways is preferred. The ability to execute cell death activities through NO generation provides a possible means of improving efficacy through enhancement of NO induced by the drug. Therefore, given the efficacy of fisetin in killing monocytic leukemia cells, fisetin can be considered as a possible chemotherapeutic agent.
Acknowledgements
This work was supported by core grant and Center for Molecular Medicine grant to National Institute of Immunology from the Department of Biotechnology, New Delhi and J.C. Bose fellowship (Department of Science and Technology, New Delhi) to CS and AS. Technical assistance from Mr. G.S. Neelaram is acknowledged.
Disclosure of conflict of interest
None.
Supporting Information
References
- 1.Baek MW, Seong KJ, Jeong YJ, Kim GM, Park HJ, Kim SH, Chung HJ, Kim WJ, Jung JY. Nitric oxide induces apoptosis in human gingival fibroblast through mitochondria-dependent pathway and JNK activation. Int Endod J. 2014;48:287–97. doi: 10.1111/iej.12314. [DOI] [PubMed] [Google Scholar]
- 2.Sang J, Chen Y, Tao Y. Nitric oxide inhibits gastric cancer cell growth through the modulation of the Akt pathway. Mol Med Rep. 2011;4:1163–1167. doi: 10.3892/mmr.2011.535. [DOI] [PubMed] [Google Scholar]
- 3.Azizzadeh B, Yip HT, Blackwell KE, Horvath S, Calcaterra TC, Buga GM, Ignarro LJ, Wang MB. Nitric oxide improves cisplatin cytotoxicity in head and neck squamous cell carcinoma. Laryngoscope. 2001;111:1896–1900. doi: 10.1097/00005537-200111000-00004. [DOI] [PubMed] [Google Scholar]
- 4.Frederiksen LJ, Sullivan R, Maxwell LR, Macdonald-Goodfellow SK, Adams MA, Bennett BM, Siemens DR, Graham CH. Chemosensitization of cancer in vitro and in vivo by nitric oxide signaling. Clin Cancer Res. 2007;13:2199–2206. doi: 10.1158/1078-0432.CCR-06-1807. [DOI] [PubMed] [Google Scholar]
- 5.Hirst D, Robson T. Nitric oxide in cancer therapeutics: interaction with cytotoxic chemotherapy. Curr Pharm Des. 2010;16:411–420. doi: 10.2174/138161210790232185. [DOI] [PubMed] [Google Scholar]
- 6.Lee WR, Shen SC, Lin HY, Hou WC, Yang LL, Chen YC. Wogonin and fisetin induce apoptosis in human promyeloleukemic cells, accompanied by a decrease of reactive oxygen species, and activation of caspase 3 and Ca(2+)-dependent endonuclease. Biochem Pharmacol. 2002;63:225–236. doi: 10.1016/s0006-2952(01)00876-0. [DOI] [PubMed] [Google Scholar]
- 7.Jang KY, Jeong SJ, Kim SH, Jung JH, Kim JH, Koh W, Chen CY, Kim SH. Activation of reactive oxygen species/AMP activated protein kinase signaling mediates fisetin-induced apoptosis in multiple myeloma U266 cells. Cancer Lett. 2012;319:197–202. doi: 10.1016/j.canlet.2012.01.008. [DOI] [PubMed] [Google Scholar]
- 8.Lyu SY, Park WB. Production of cytokine and NO by RAW 264.7 macrophages and PBMC in vitro incubation with flavonoids. Arch Pharm Res. 2005;28:573–581. doi: 10.1007/BF02977761. [DOI] [PubMed] [Google Scholar]
- 9.Wang L, Tu YC, Lian TW, Hung JT, Yen JH, Wu MJ. Distinctive antioxidant and antiinflammatory effects of flavonols. J Agric Food Chem. 2006;54:9798–9804. doi: 10.1021/jf0620719. [DOI] [PubMed] [Google Scholar]
- 10.Bhat TA, Nambiar D, Pal A, Agarwal R, Singh RP. Fisetin inhibits various attributes of angiogenesis in vitro and in vivo--implications for angioprevention. Carcinogenesis. 2012;33:385–393. doi: 10.1093/carcin/bgr282. [DOI] [PubMed] [Google Scholar]
- 11.He H, Feng YS, Zang LH, Liu WW, Ding LQ, Chen LX, Kang N, Hayashi T, Tashiro S, Onodera S, Qiu F, Ikejima T. Nitric oxide induces apoptosis and autophagy; autophagy down-regulates NO synthesis in physalin A-treated A375-S2 human melanoma cells. Food Chem Toxicol. 2014;71:128–135. doi: 10.1016/j.fct.2014.06.007. [DOI] [PubMed] [Google Scholar]
- 12.Schultz DR, Harrington WJ Jr. Apoptosis: programmed cell death at a molecular level. Semin Arthritis Rheum. 2003;32:345–369. doi: 10.1053/sarh.2003.50005. [DOI] [PubMed] [Google Scholar]
- 13.Hale AN, Ledbetter DJ, Gawriluk TR, Rucker EB 3rd. Autophagy: regulation and role in development. Autophagy. 2013;9:951–972. doi: 10.4161/auto.24273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tripathi R, Samadder T, Gupta S, Surolia A, Shaha C. Anticancer activity of a combination of cisplatin and fisetin in embryonal carcinoma cells and xenograft tumors. Mol Cancer Ther. 2011;10:255–268. doi: 10.1158/1535-7163.MCT-10-0606. [DOI] [PubMed] [Google Scholar]
- 15.Hansen MB, Nielsen SE, Berg K. Re-examination and further development of a precise and rapid dye method for measuring cell growth/cell kill. J Immunol Methods. 1989;119:203–210. doi: 10.1016/0022-1759(89)90397-9. [DOI] [PubMed] [Google Scholar]
- 16.Mishra DP, Pal R, Shaha C. Changes in cytosolic Ca2+ levels regulate Bcl-xS and Bcl-xL expression in spermatogenic cells during apoptotic death. J Biol Chem. 2006;281:2133–2143. doi: 10.1074/jbc.M508648200. [DOI] [PubMed] [Google Scholar]
- 17.Taatjes DJ, Sobel BE, Budd RC. Morphological and cytochemical determination of cell death by apoptosis. Histochem Cell Biol. 2008;129:33–43. doi: 10.1007/s00418-007-0356-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ashman RF, Peckham D, Alhasan S, Stunz LL. Membrane unpacking and the rapid disposal of apoptotic cells. Immunol Lett. 1995;48:159–166. doi: 10.1016/0165-2478(95)02471-9. [DOI] [PubMed] [Google Scholar]
- 19.Liu X, Li P, Widlak P, Zou H, Luo X, Garrard WT, Wang X. The 40-kDa subunit of DNA fragmentation factor induces DNA fragmentation and chromatin condensation during apoptosis. Proc Natl Acad Sci U S A. 1998;95:8461–8466. doi: 10.1073/pnas.95.15.8461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kuznetsov AV, Kehrer I, Kozlov AV, Haller M, Redl H, Hermann M, Grimm M, Troppmair J. Mitochondrial ROS production under cellular stress: comparison of different detection methods. Anal Bioanal Chem. 2011;400:2383–2390. doi: 10.1007/s00216-011-4764-2. [DOI] [PubMed] [Google Scholar]
- 21.Matsuo M, Sasaki N, Saga K, Kaneko T. Cytotoxicity of flavonoids toward cultured normal human cells. Biol Pharm Bull. 2005;28:253–259. doi: 10.1248/bpb.28.253. [DOI] [PubMed] [Google Scholar]
- 22.Kumari S, Sammut IA, Giles GI. The design of nitric oxide donor drugs: s-nitrosothiol tDodSNO is a superior photoactivated donor in comparison to GSNO and SNAP. Eur J Pharmacol. 2014;737:168–176. doi: 10.1016/j.ejphar.2014.05.012. [DOI] [PubMed] [Google Scholar]
- 23.Sudan S, Rupasinghe HP. Flavonoid-Enriched Apple Fraction AF4 Induces Cell Cycle Arrest, DNA Topoisomerase II Inhibition, and Apoptosis in Human Liver Cancer HepG2 Cells. Nutr Cancer. 2014;66:1237–1246. doi: 10.1080/01635581.2014.951733. [DOI] [PubMed] [Google Scholar]
- 24.Suh Y, Afaq F, Khan N, Johnson JJ, Khusro FH, Mukhtar H. Fisetin induces autophagic cell death through suppression of mTOR signaling pathway in prostate cancer cells. Carcinogenesis. 2010;31:1424–1433. doi: 10.1093/carcin/bgq115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Brix B, Mesters JR, Pellerin L, Johren O. Endothelial cell-derived nitric oxide enhances aerobic glycolysis in astrocytes via HIF-1alpha-mediated target gene activation. J Neurosci. 2012;32:9727–9735. doi: 10.1523/JNEUROSCI.0879-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fingar DC, Richardson CJ, Tee AR, Cheatham L, Tsou C, Blenis J. mTOR controls cell cycle progression through its cell growth effectors S6K1 and 4E-BP1/eukaryotic translation initiation factor 4E. Mol Cell Biol. 2004;24:200–216. doi: 10.1128/MCB.24.1.200-216.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Al-Sa’doni HH, Ferro A. S-nitrosothiols as nitric oxide-donors: chemistry, biology and possible future therapeutic applications. Curr Med Chem. 2004;11:2679–2690. doi: 10.2174/0929867043364397. [DOI] [PubMed] [Google Scholar]
- 28.Ying TH, Yang SF, Tsai SJ, Hsieh SC, Huang YC, Bau DT, Hsieh YH. Fisetin induces apoptosis in human cervical cancer HeLa cells through ERK1/2-mediated activation of caspase- 8-/caspase-3-dependent pathway. Arch Toxicol. 2012;86:263–273. doi: 10.1007/s00204-011-0754-6. [DOI] [PubMed] [Google Scholar]
- 29.Goh FY, Upton N, Guan S, Cheng C, Shanmugam MK, Sethi G, Leung BP, Wong WS. Fisetin, a bioactive flavonol, attenuates allergic airway inflammation through negative regulation of NF-kappaB. Eur J Pharmacol. 2012;679:109–116. doi: 10.1016/j.ejphar.2012.01.002. [DOI] [PubMed] [Google Scholar]
- 30.Taubert D, Berkels R, Klaus W, Roesen R. Nitric oxide formation and corresponding relaxation of porcine coronary arteries induced by plant phenols: essential structural features. J Cardiovasc Pharmacol. 2002;40:701–713. doi: 10.1097/00005344-200211000-00008. [DOI] [PubMed] [Google Scholar]
- 31.Jin JO, Song MG, Kim YN, Park JI, Kwak JY. The mechanism of fucoidan-induced apoptosis in leukemic cells: involvement of ERK1/2, JNK, glutathione, and nitric oxide. Mol Carcinog. 2010;49:771–782. doi: 10.1002/mc.20654. [DOI] [PubMed] [Google Scholar]
- 32.Bensasson RV, Zoete V, Jossang A, Bodo B, Arimondo PB, Land EJ. Potency of inhibition of human DNA topoisomerase I by flavones assessed through physicochemical parameters. Free Radic Biol Med. 2011;51:1406–1410. doi: 10.1016/j.freeradbiomed.2011.06.021. [DOI] [PubMed] [Google Scholar]
- 33.Rothkamm K, Burdak-Rothkamm S. Ionizing radiation-induced DNA strand breaks and gamma-H2AXgamma-H2AX foci in cells exposed to nitric oxide. Methods Mol Biol. 2011;704:17–25. doi: 10.1007/978-1-61737-964-2_2. [DOI] [PubMed] [Google Scholar]
- 34.Han W, Chen S, Yu KN, Wu L. Nitric oxide mediated DNA double strand breaks induced in proliferating bystander cells after alpha-particle irradiation. Mutat Res. 2010;684:81–89. doi: 10.1016/j.mrfmmm.2009.12.004. [DOI] [PubMed] [Google Scholar]
- 35.Shen C, Oswald D, Phelps D, Cam H, Pelloski CE, Pang Q, Houghton PJ. Regulation of FANCD2 by the mTOR pathway contributes to the resistance of cancer cells to DNA double-strand breaks. Cancer Res. 2013;73:3393–3401. doi: 10.1158/0008-5472.CAN-12-4282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kim PK, Kwon YG, Chung HT, Kim YM. Regulation of caspases by nitric oxide. Ann N Y Acad Sci. 2002;962:42–52. doi: 10.1111/j.1749-6632.2002.tb04054.x. [DOI] [PubMed] [Google Scholar]
- 37.Chen YC, Shen SC, Lee WR, Lin HY, Ko CH, Shih CM, Yang LL. Wogonin and fisetin induction of apoptosis through activation of caspase 3 cascade and alternative expression of p21 protein in hepatocellular carcinoma cells SK-HEP-1. Arch Toxicol. 2002;76:351–359. doi: 10.1007/s00204-002-0346-6. [DOI] [PubMed] [Google Scholar]
- 38.Suh Y, Afaq F, Johnson JJ, Mukhtar H. A plant flavonoid fisetin induces apoptosis in colon cancer cells by inhibition of COX2 and Wnt/EGFR/NF-kappaB-signaling pathways. Carcinogenesis. 2009;30:300–307. doi: 10.1093/carcin/bgn269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mukherjee SB, Das M, Sudhandiran G, Shaha C. Increase in cytosolic Ca2+ levels through the activation of non-selective cation channels induced by oxidative stress causes mitochondrial depolarization leading to apoptosis-like death in Leishmania donovani promastigotes. J Biol Chem. 2002;277:24717–24727. doi: 10.1074/jbc.M201961200. [DOI] [PubMed] [Google Scholar]
- 40.Pimentel AA, Benaim G. [Ca2+ and sphingolipids as modulators for apoptosis and cancer] . Invest Clin. 2012;53:84–110. [PubMed] [Google Scholar]
- 41.Cao YQ, Tsien RW. Different relationship of N- and P/Q-type Ca2+ channels to channel-interacting slots in controlling neurotransmission at cultured hippocampal synapses. J Neurosci. 2010;30:4536–4546. doi: 10.1523/JNEUROSCI.5161-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wu SN, Chiang HT, Shen AY, Lo YK. Differential effects of quercetin, a natural polyphenolic flavonoid, on L-type calcium current in pituitary tumor (GH3) cells and neuronal NG108-15 cells. J Cell Physiol. 2003;195:298–308. doi: 10.1002/jcp.10244. [DOI] [PubMed] [Google Scholar]
- 43.Ju HQ, Wang SX, Xiang YF, Liu Z, Liu JY, Chen ZP, Zeng FL, Xia M, Liu ZH, Xing GW, Wang SY, Wang YF. BJ-B11, a novel Hsp90 inhibitor, induces apoptosis in human chronic myeloid leukemia K562 cells through the mitochondria-dependent pathway. Eur J Pharmacol. 2011;666:26–34. doi: 10.1016/j.ejphar.2011.05.020. [DOI] [PubMed] [Google Scholar]
- 44.Haines RJ, Corbin KD, Pendleton LC, Eichler DC. Protein kinase Calpha phosphorylates a novel argininosuccinate synthase site at serine 328 during calcium-dependent stimulation of endothelial nitric-oxide synthase in vascular endothelial cells. J Biol Chem. 2012;287:26168–26176. doi: 10.1074/jbc.M112.378794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kim JA, Jang HJ, Martinez-Lemus LA, Sowers JR. Activation of mTOR/p70S6 kinase by ANG II inhibits insulin-stimulated endothelial nitric oxide synthase and vasodilation. Am J Physiol Endocrinol Metab. 2012;302:E201–E208. doi: 10.1152/ajpendo.00497.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kim DH, Sarbassov DD, Ali SM, King JE, Latek RR, Erdjument-Bromage H, Tempst P, Sabatini DM. mTOR interacts with raptor to form a nutrient-sensitive complex that signals to the cell growth machinery. Cell. 2002;110:163–175. doi: 10.1016/s0092-8674(02)00808-5. [DOI] [PubMed] [Google Scholar]
- 47.Janus A, Robak T, Smolewski P. The mammalian target of the rapamycin (mTOR) kinase pathway: its role in tumourigenesis and targeted antitumour therapy. Cell Mol Biol Lett. 2005;10:479–498. [PubMed] [Google Scholar]
- 48.Hosoi H, Dilling MB, Shikata T, Liu LN, Shu L, Ashmun RA, Germain GS, Abraham RT, Houghton PJ. Rapamycin causes poorly reversible inhibition of mTOR and induces p53-independent apoptosis in human rhabdomyosarcoma cells. Cancer Res. 1999;59:886–894. [PubMed] [Google Scholar]
- 49.Shi Y, Frankel A, Radvanyi LG, Penn LZ, Miller RG, Mills GB. Rapamycin enhances apoptosis and increases sensitivity to cisplatin in vitro. Cancer Res. 1995;55:1982–1988. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.