

Abstract
Cancer epigenetics plays an important role in the pathogenesis of many cancers including gastric cancer. Histone deacetylases (HDACs) emerge as exciting therapeutic targets for cancer treatment and prevention. In this study, we identified DTWD1 as one of the 122 genes upregulated after treatment of trichostatin A (TSA) in two gastric cancer cell lines. Moreover, DTWD1 was downregulated in gastric cancer cell lines and primary gastric carcinoma tissues. It was further identified as the new target of p53. Then we revealed that HDAC3 downregulated DTWD1 by disrupting the interaction of p53 with DTWD1 promoter. Furthermore, DTWD1 functioned as a tumor suppressor by downregulating cyclin B1 expression to inhibit proliferation. In summary, as the new p53 target gene, DTWD1 was downregulated in gastric cancer by HDAC3 and acted as a novel tumor suppressor gene. Specific inhibitors of HDAC3 might be a new approach for gastric cancer treatment by activating DTWD1 expression.
Keywords: HDAC, DTWD1, p53, gastric cancer, tumor suppressor gene
Introduction
Gastric cancer is one of the most prevalent malignant tumors worldwide especially in East Asia [1]. Despite advances in the diagnosis and treatment, gastric cancer constitutes the second leading cause of cancer-related death worldwide [2]. The aberrant activation of oncogenes and the suppression of tumor suppressor genes contribute to the pathogenesis of gastric cancer. In the past decade, cancer epigenetics has attracted much more attention than ever before [3].
Histone acetylation and DNA methylation are the most important parts of epigenetics [4]. Histone deacetylases (HDACs) has emerged as a major enzyme in the epigenetic regulation of gene expression by catalyzing the removal of acetyl groups, modeling the structure of chromatin and inducing chromatin condensation and transcriptional repression [5]. This family of enzymes has been identified as four classes, namely Class I including HDAC1, HDAC2, HDAC3, and HDAC8, Class II including HDAC4, HDAC5, HDAC6, HDAC7, HDAC9, and HDAC10, Class III HDAC and Class IV such as HDAC11. Among them, the first two classes could be inhibited by trichostatin A (TSA) and widely investigated in human cancer development [6,7].
Increasing evidence suggested the importance of HDACs to human carcinogenesis. For example, HDAC1, 2 and 3 were reported to be upregulated in gastric cancers and indicated poor prognosis [8,9]. High expression of SIRT1,one of Class III HDACs, is closely correlated with progression and prognosis in gastric cancer patients [10]. By doing so, the balance between histone acetylation and deacetylation is disturbed in cancer, leading to abnormal expression of tumor suppressor genes and/or proto-oncogenes. Thus, HDACs appear to be a promising target for cancer prevention and treatment [11].
Currently, various HDAC inhibitors have been developed as novel anti-cancer agents by impairing cell differentiation or inducing apoptosis [12-15]. Being the selective inhibitor for the Class I and II HDACs, TSA was widely used to reactivate the expression of tumor suppressor genes in cancer cells [16-18]. In the present study, we aimed to identify novel tumor suppressor genes silenced via histone deacetylation in gastric cancer cells. By treating cells with TSA, we identified the new p53 target gene DTWD1 as a novel tumor suppressor gene in gastric cancer. On the other hand, p53-mediated DTWD1 expression was attenuated specially by HDAC3. In addition, DTWD1 inhibited tumor growth by reducing the expression of cyclin B1.
Materials and methods
Cells, antibodies, and plasmids
Human gastric epithelial cell line GES-1, six gastric cancer cell lines (AGS, SGC7901, MKN45, MKN28, BGC823, and NCI-N87), HCT116 and HEK293T cells were obtained from American Type Culture Collection (ATCC, Manassas, VA, USA). Unless specifically indicated, cells were cultured in DMEM or 1640 (Invitrogen) medium supplemented with 10% fetal bovine serum at 37°C with 5% CO2 and 95% humidity. Anti-HDAC 3 antibody was obtained from Epitomics (Burlingame, CA, USA). Anti-p53, anti-Myc-Tag and anti-GAPDH antibodies and CDK Ab Sampler kit were from Cell Signaling Technology (Boston, MA, USA). Anti-HDAC1 and anti-HDAC2 antibodies were from Millipore (Billerica, MA, USA). Trichostatin A (TSA) and sodium butyrate (SB) were purchased from Sigma (St. Louis, MO, USA). DTWD1 full-length open reading frame was amplified by PCR using the GoTaq Green Master Mix (Promega, Madison, WI, USA) with cDNA reverse transcripted from total RNA of GES-1 cells. The primers used are listed in the Table 1. The PCR product was cloned into the pMD-18T vector (Takara, Dalian, China). After sequence verification, the insert was sub-cloned into a mammalian expression vector pCMV-3Tag-7 (Agilent, La Jolla, CA, USA).
Table 1.
Oligos used in the study
| Name | Sequence |
|---|---|
| Primers | |
| DTWD1 cloning | F: GGGATCCTCTCTCAATCCACCTAT |
| R: GCTCGAGCTAATGTGTAAGTTTTCCTGTTTCC | |
| DTWD1 RT-PCR | F: ATTCACTGATGAGCGATTC |
| R: ATCAGTATGGTAGTCTACCAG | |
| GAPDH RT-PCR | F: GGAGTCAACGGATTTGGT |
| R: GTGATGGGATTTCCATTGAT | |
| DTWD1 ChIP PCR | F: TTCCAGTTGGCTAAGACT |
| R: AACAACCCTCCCACTCTT | |
| DTWD1 promoter cloning | F1: GGAGCTCCTATTCTACTTTCCCTACCC |
| R1: AAGCTTGGAACACTATACAGACTCCC | |
| F2: GGAGCTCCTCTGGAGACCAGTGGTC | |
| F3: GGAGCTCTTTCGAGAACCGCTTCCC | |
| R2: GAAGCTTCTGACAAAGAATGTCCAC | |
| siRNAs | |
| HDAC1 | CCGGUCAUGUCCAAAGUAATT |
| UUACUUUGGACAUGACCGGTT | |
| HDAC2 | CCAGAACACUCCAGAAUAUTT |
| AUAUUCUGGAGUGUUCUGGTT | |
| HDAC3 | GAGCUUCCCUAUAGUGAAUTT |
| AUUCACUAUAGGGAAGCUCTT | |
| HDAC8 | GUCCCGGUUUAUAUCUAUATT |
| UAUAGAUAUAAACCGGGACTT | |
| p53 | CCACCAUCCACUACAACUATT |
| UAGUUGUAGUGGAUGGUGGTT |
RNA extraction and quantitative real-time RT-PCR
Total RNA was extracted using Trizol reagent (Invitrogen) according to the manufacturer’s instructions. RNA concentrations were quantified by NanoDrop 2000 (Nanodrop, Wilmington, Del, USA). Reverse transcription reaction was performed using 1 μg of total RNA with Quantscript RT kit (Tiangen Biotechnology, Beijing, China). The mRNA expression level was determined by quantitative real-time PCR using SYBR Green Master Mix Kit and ABI 7500 Real-Time PCR System (Applied Biosystems). Human glyceraldehyde-3-phosphate dehydrogenase was used as an internal control of RNA integrity. Primers used are listed in the Table 1.
SiRNAs and transfection
Four HDAC siRNAs were purchased from Qiagen (Hilden, Germany). P53 siRNA was synthesized by GenePharma (Shanghai, China). For siRNA transfection, cells were seeded overnight in 6-well plates (2-3×105/well) and transfected with siRNA duplexes (10 nM) using Lipofectamine™ RNAiMAX transfection reagent (Invitrogen) according to the manufacturer’s instructions. A negative control siRNA supplied by the manufacture was used as a control. For plasmid transfection, cells were seeded overnight in six-well plates (2-3×105/well) and 2 μg of plasmids were transfected with FuGENE HD (Roche Applied Science, Mannheim, Germany). Cells were harvested for RNA and protein extraction after 48-72 h for further analysis. The sequences of various siRNAs are listed in the Table 1.
Cell growth assay
Cell growth assay was performed with The CellTiter 96® AQueous Non-Radioactive Cell Proliferation Assay kit (MTS assay) (Promega). Briefly, 48 h after transfection, the transfected cells were transferred into a 96-well plate for 1 day. Then the cell growth was measured following the manufacturer’s instruction. In addition, SGC7901 lentivirus cells were measured manually after treatment with DOX. Samples were prepared in triplicates, and the cell viability was determined as the mean ± SD.
Western blotting
Cell lysates harvested were quantitated by Bio-Rad protein assay kit II (Bio-Rad Laboratories, Hercules, CA, USA). The boiled lysates were resolved by SDS-PAGE, transferred to PVDF membranes, and probed with the indicated primary antibodies, washed with TBS-T (0.01 M TRIS-HCl Buffer, 8.8 g/L NaCl, 0.1% Tween-20), then incubated with suitable HRP-conjugated second antibodies and autoradiographed with enhanced chemiluminescence (Millipore).
Colony formation assay
Anchorage-dependent growth of tumor cells was investigated by monolayer colony formation assay. SGC7901 cells and MKN28 cells were cultured overnight in a 12-well plate (1.0×105 cells per well) and transfected with pCMV-3Tag7-DTWD1 and control vector, using FuGENE HD (Roche Applied Science). Forty-eight hours later, the transfectants were replaced in triplicate and cultured for two weeks with complete RPMI 1640 medium containing hygromycin. Surviving colonies were stained with Gentian Violet after methanol fixation, and visible colonies (≥50 cells) were counted.
Chromatin immunoprecipitation
Chromatin Immunoprecipitation (ChIP) experiment was performed with the SimpleChIP™ Enzymatic Chromatin IP Kit (Cell Signaling Technology, Boston, MA, USA) according to the manufacturer’s instructions. Briefly, 1×107 cells were first fixed with 1% formaldehyde. Cells were then lysed and chromatin was harvested and fragmented using sonication. The chromatin was then subjected to immunoprecipitation using antibodies specific to p53 or mouse isotope IgG (1.5 μg) and HDAC3 or rabbit isotype IgG (1 μg). After immunoprecipitation, the protein-DNA cross-links were reversed and the DNA was purified. The enriched DNA sequence was then detected by quantitative real-time PCR with a pair of primers around the promoter region of DTWD1 gene. The primers used for the detection of ChIP-enriched DNA are listed in the Table 1.
In vivo tumorigenesis assay
Nude mice were purchased from Taconic and maintained under specific pathogen-free conditions. The mice were then randomly divided into 2 groups, and subcutaneous injected with SGC7901 cells (1×107) with DOX-inducible DTWD1 expression vector or control cells. Tumor volume was examined by caliper measurement every 3 days and calculated according to the following formula: V=π/6×f×(L×W)3/2, where V, volume (mm2); L, biggest diameter (mm); W, smallest diameter (mm), f (females) =1.58±0.01.
Luciferase activity assay
About 2 kb sequence around the first exon region of DTWD1 was cloned by PCR. PCR products were inserted into pMD18T vector (Takala) for sequence validation. The correct inserts were subcloned into pGL2-promoter vector. HEK293T cells (1×105) seeded in 12-well plates were cotransfected with constructed luciferase reporter and pRL-TK-Renilla (Promega) conducts. 48 h after transfection, the activities of firefly luciferase was measured using the DualGlo luciferase assay system (Promega) as previously reported [19]. Relative luciferase activity was normalized with renilla luciferase activity and then compared with the control vector. The primers used are listed in the Table 1.
Immunohistochemistry
Anti-Ki 67 antibody was optimized for immunohistochemistry. Subsequently, an identical formalin-fixed and paraffin-embedded mice tumor was immunostained using microwave antigen retrieval in 0.01 M citrate buffer, pH 6.0. After washing, signal was detected using a suitable HRP-labeled second antibody with DAB as the chromogen (Dako, K3468). The staining results were judged by 2 independent pathologists in the department of pathology of our hospital. The positive staining was defined as uniform staining of more than 5% of 1,000 tumor cells and the staining of less than 5% of 1,000 tumor cells was defined as negative.
Lentivirus infection
For stable gene overexpression, pNL-EGFP and CMV plasmids along with packaging plasmid mix (Obio TechnologyMI0000780) were transfected into SGC7901 cells maintained in 10% FBS. 48 h after transfection, the supernatant fraction was harvested for viral titers determination. Approximately 1×109 virus particles were used for infection.
Statistical analysis
The experiments were repeated three times and representative results were shown as means ± SD. Asterisks indicated a statistically significant difference (p<0.05). Samples were compared between treatments with two-tailed Student’s t-test, and multiple groups were compared to single controls by one-way ANOVA using SPSS version 16.0 (SPSS/IBM, Chicago, Illinois).
Results
DTWD1 is upregulated by HDACi
Trichostatin A (TSA), the most frequently used inhibitor of HDAC, significantly inhibited the viability of gastric cancer cell lines including AGS, SGC7901 and MKN28 (Figure 1A). In our screen of TSGs suppressed by histone deacetylation, 605 genes in AGS and 3216 genes in SGC7901 were upregulated at least two folds after TSA treatment. Among them, 122 genes were overlapped within the two cell lines (Figure 1B). Our previous study suggested that TSA could activate expression of PUMA to promote cellular apoptosis and growth inhibition [13]. DTWD1 was one of the 122 overlapped genes not yet reported, thus we chose DTWD1 for further study. Indeed, we confirmed the upregulation of DTWD1 mRNA in gastric cancer cells treated with TSA (Figure 1C).
Figure 1.
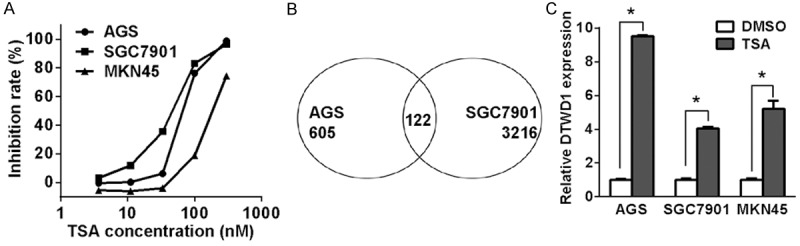
DTWD1 is upregulated by HDACi. A. The effect of various concentration of TSA on gastric cancer cells was determined by MTS assay. B. Profiling of genes upregulated in gastric cancer cells with treatment of TSA. C. DTWD1 expression in gastric cancer cells before and after TSA treatment was determined by real-time RT-PCR. Asterisks indicate statistical significance (p < 0.05, Student’s t test).
HDAC3 is responsible for DTWD1 suppression
To clarify which class of HDACs inhibited DTWD1 expression, we chose sodium butyrate (SB), which is a specific class I family of HDACs inhibitor, to narrow down the possible HDACs. We found that SB could upregulate the DTWD1 expression as similar with TSA (Figure 2A). Considering that class I family of HDACs was composed of HDAC1, HDAC2, HDAC3 and HDAC8, we then used siRNAs to knockdown individual HDACs respectively. As shown in Figure 2B and 2C, DTWD1 expression had a remarkable upregulation after knockdown of HDAC3 but not other HDACs, indicating that HDAC3 could have important relevance to DTWD1 expression. Indeed, we confirmed the direct interaction of HDAC3 with the promoter of DTWD1 by chromatin immunoprecipitation (Figure 2D), indicating that DTWD1 was a direct target of HDAC3.
Figure 2.
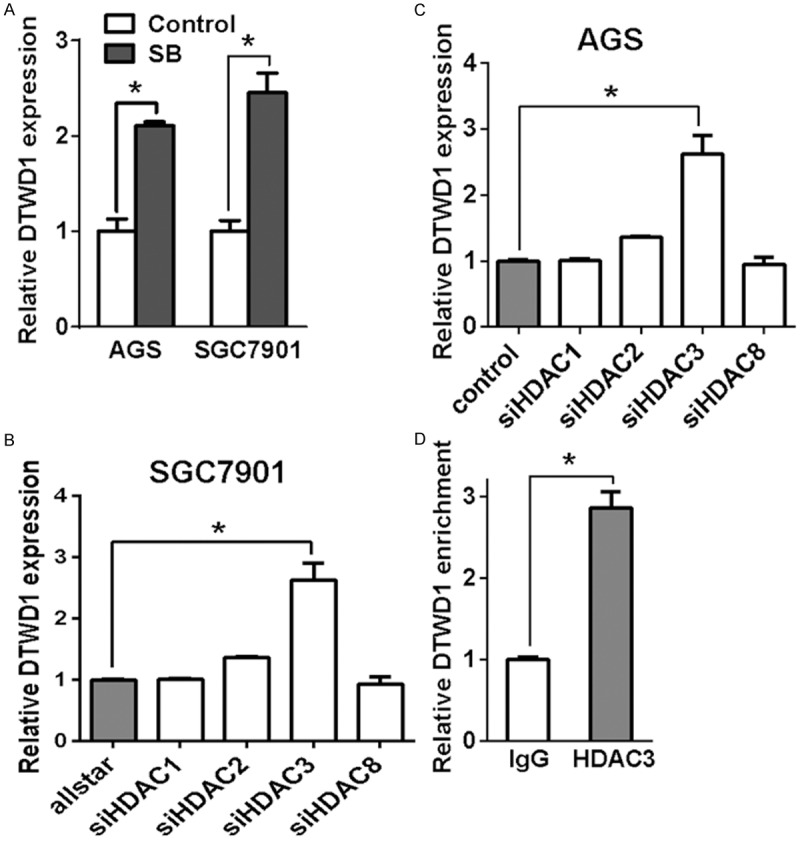
HDAC3 is responsible for DTWD1 suppression. A. DTWD1 expression in gastric cancer cells before and after SB treatment was determined by real-time RT-PCR. Asterisks indicate statistical significance (p < 0.05, Student’s t test). B and C. The effect of HDACs depletion on DTWD1 expression in AGS and SGC7901 were determined by real-time RT-PCR. Asterisks indicate statistical significance (p < 0.05, Student’s t test). D. The binding of HDAC3 to the DTWD1 gene was determined by ChIP assay.
DTWD1 is downregulated in gastric cancer
To clarify the relevance of DTWD1 to gastric carcinogenesis, we then detected the expression of DTWD1 in a panel of gastric cancer cells and GES-1, a non-tumor gastric epithelial cell line by real-time RT-PCR. As shown in Figure 3A, DTWD1 mRNA level was downregulated in most of the cell lines compared with GES-1. Moreover, the mRNA level of DTWD1 in primary gastric carcinoma tissues was remarkably downregulated when compared with its expression in adjacent non-tumor stomach tissues (n=19, p<0.05, Wilcoxin-matched pairs t test) (Figure 3B).
Figure 3.
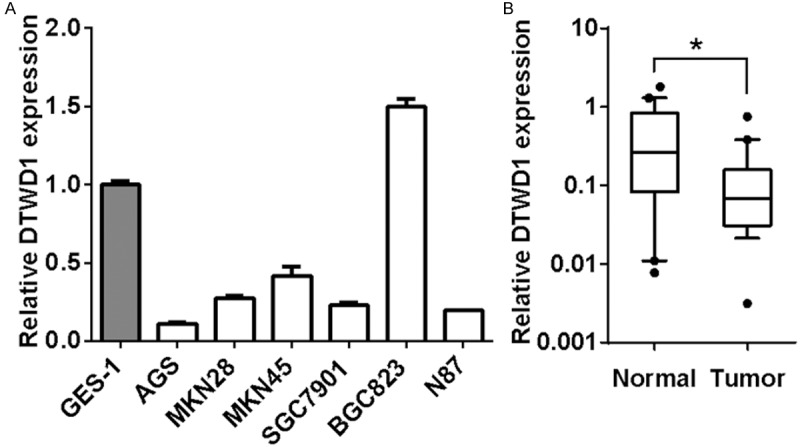
DTWD1 is downregulated in gastric cancer. A. DTWD1 expression in gastric cancer cells and GES-1 were determined by real-time RT-PCR. B. DTWD1 expression in primary gastric carcinoma tissues as well as adjacent non-tumor tissues was determined by real-time RT-PCR. The Asterisk indicates statistical significance (n = 19, p < 0.05, Wilcoxin-matched pairs t test).
DTWD1 is a target of p53
Next, we tried to define the minimal promoter of DTWD1 to clarify the regulation of DTWD1 transcription. We cloned different fragments of candidate DTWD1 promoter sequences including F1-R1, F1-R2, F2-R1 and F3-R2 and constructed them into the dual luciferase reporter vectors (Figure 4A). Compared with the pGL2 vector, the F3-R2 fragment exhibited the strongest transcriptional activity while F2-R1 was found little transcriptional activity (Figure 4B), indicating that F3-R2 fragment is essential for the transcription regulation of DTWD1 and could contain the binding sites for potential transcriptional factors. Indeed, we found a classical p53-binding site in the F3-R2 fragment [20]. Thus, we wondered whether p53 was the transcriptional factor responsible for DTWD1 transcription. Indeed, we found that p53 knock down could downregulate the DTWD1 mRNA level in GES-1 cells (Figure 4C) while overexpression of p53 upregulated the expression of DTWD1 in SGC7901 cells (Figure 4D). Moreover, we confirmed that p53 bound the promoter of DTWD1 by chromatin immunoprecipitation (Figure 4E). We further use the classical system of HCT116 p53+/+ and HCT116 p53-/- cells to further verify the regulation of DTWD1 by p53. Etoposide (VP16), an important chemotherapeutic agent to induce DNA damage [21] and p53 activity [22], stimulated DTWD1 level only in HCT116 p53+/+ but not HCT116 p53-/- cells (Figure 4F and 4G). These results demonstrated that p53 directly regulate DTWD1 transcription.
Figure 4.
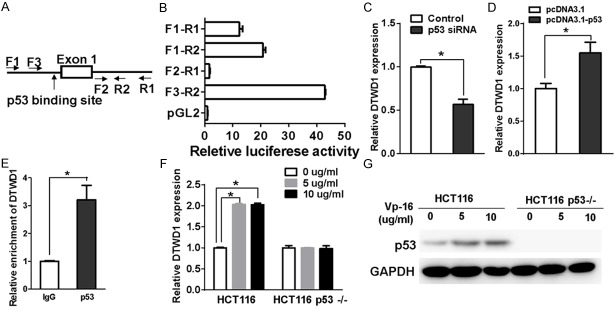
DTWD1 is a target of p53. (A) Four fragments of candidate DTWD1 promoter including F1-R1, F1-R2, F2-R1 and F3-R2 were cloned to construct luciferase reporter system. (B) The effect of luciferase reporter driven by different DTWD1 fragments in (A) was determined by luciferase activity assay. (C) The effect of p53 depletion on DTWD1 expression in GES-1 was determined by real-time RT-PCR. The asterisk indicates statistical significance (p < 0.05, Student’s t test). (D) The effect of p53 overexpression on DTWD1 expression in SGC7901 was determined by real-time RT-PCR. The asterisk indicates statistical significance (p < 0.05, Student’s t test). (E) The binding of p53 to the DTWD1 gene was determined by ChIP assay. The effect of VP16 on DTWD1expression (F) and p53 expression (G) in HCT116 or HCT116 p53-/- cells were determined by real-time RT-PCR and Western blotting, respectively. Asterisks indicate statistical significance (p < 0.05, Student’s t test).
HDAC3 regulates p53-mediated DTWD1 expression
Next, we wondered how HDAC3 regulated p53-mediated DTWD1 transcription. First, we found that the p53 protein level exhibited a remarkable rise after treatment of TSA (Figure 5A). Chromatin immunoprecipitation also showed that the DTWD1 DNA level enriched by p53 was elevated after treatment of TSA (Figure 5B). Moreover, knock down of p53 could also attenuate the TSA-promoted DTWD1 mRNA expression (Figure 5C), confirming the importance of p53 in DTWD1 transcription. However, knock down of HDAC3 did not upregulate the p53 protein expression (Figure 5D) although the interaction of p53 with DTWD1 promoter was enhanced (Figure 5E). In addition, upregulation of DTWD1 expression by HDAC3 knock down could be reversed by p53 depletion (Figure 5F). All of these results suggested that HDAC3 regulated DTWD1 expression by interfering the interaction of p53 with DTWD1 promoter.
Figure 5.

HDAC3 regulates p53-mediated DTWD1 expression. A. The effect of TSA on p53 expression in SGC7901 was determined by Western blotting. B. The effect of TSA on the binding of p53 to DTWD1 gene was determined by ChIP assay. The asterisk indicates statistical significance (p < 0.05). C. The effect of TSA and p53 depletion on DTWD1 expression was determined by real-time RT-PCR in SGC7901. The asterisk indicates statistical significance (p < 0.05, Student’s t test). D. The effect of HDAC3 depletion on p53 and HDAC3 expression in SGC7901 cells was determined by Western blotting. E. The effect of HDAC3 depletion on the binding of p53 to DTWD1 gene was determined by ChIP assay. The asterisk indicates statistical significance (p < 0.05). F. The effect of HDAC3 depletion and p53 depletion on DTWD1 expression in SGC7901 cells was determined by real-time RT-PCR. The asterisk indicates statistical significance (p < 0.05, Student’s t test).
DTWD1 functions as a tumor suppressor by regulating cyclin B1
To clarify the biological relevance of DTWD1, we explored the effect of DTWD1 on the in vitro and in vivo growth of gastric cancer cells. Ectopic expression of DTWD1 inhibited the anchorage-dependent growth of gastric cell lines (Figure 6A and 6B). To further determine the long-term effect of DTWD1 on gastric cancer, we engineered SGC7901 cells to express DTWD1 in a doxycycline (DOX)-inducible manner via lentivirus infection. The growth of SGC7901 in vitro was inhibited in the present of DOX (Figure 6C). In accordance with the in vitro result, in vivo tumor growth was significantly impaired upon DOX-induced DTWD1 expression (Figure 6D).
Figure 6.
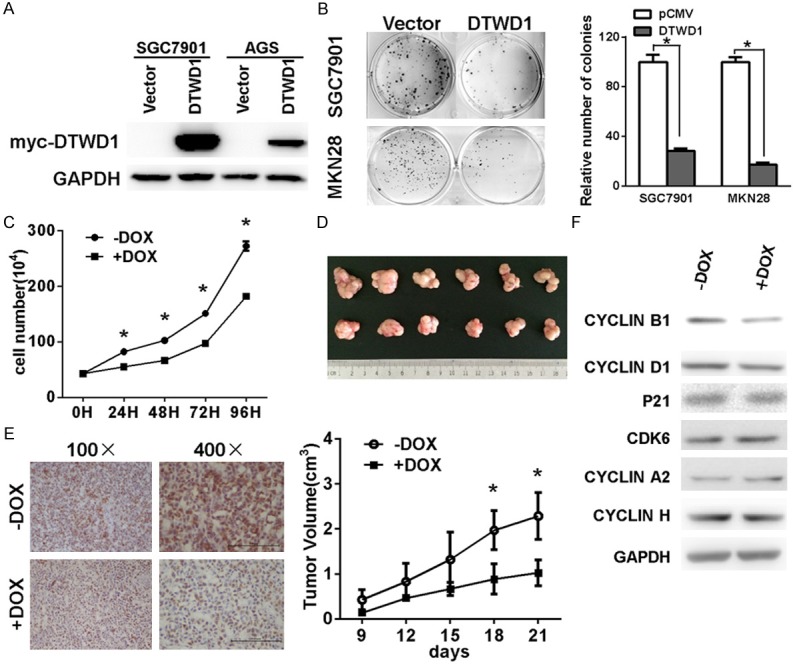
DTWD1 functions as a tumor suppressor by regulating cyclin B1. The effect of ectopic expression of DTWD1 (A) on cell growth was determined by colony formation assay (B) and growth curve assay (C). Asterisks indicate statistical significance (p < 0.05). The effect of DTWD1 expression on the growth of gastric cancer cells in vivo was analyzed by inoculating SGC7901 cells into nude mice. The growth of tumors and immunohistochemistry staining of mice tumors were shown in (D) (student’s t-test, P < 0.05) and (E). The effect of DOX-induced DTWD1 expression on expression of cyclin B1, p21, CDK6, cyclin D1, cyclin A2 and cyclin H in DOX-induced DTWD1 SGC7901 cells were determined by Western blotting (F).
Then, we aimed to explore the mechanism underlying the tumor-suppressing role of DTWD1. No significant apoptosis were found after DOX treatment (data not shown). However, immunohistochemistry analysis showed much lower expression of Ki 67 in tumor cells treated with DOX (Figure 6E), indicating that DTWD1 impaired proliferation rather than activated apoptosis. Thus, we investigated the effect of DTWD1 on the expression of several important regulators related with cell cycle progression and found the expression of cyclin B1 was the only one affected by DTWD1 expression (Figure 6F). Collectively, all of these data demonstrated that DTWD1 played as a tumor suppressor by regulating the expression of cyclin B1.
Discussion
Despite recent success toward discovery of more effective anticancer drugs, gastric cancer remains a huge threat to human health. There is emerging evidence that epigenetics plays a key role in the initiation and progression of gastric cancer. Epigenetic regulators such as histone deacetylases (HDACs) play an important role in the expression of many genes critical to the pathogenesis of many types of cancers [23-25]. Thus, HDACs are being investigated as a therapeutic target for the clinical intervention of human cancers.
In this study, we demonstrated that DTWD1 was upregulated in gastric cancer cells treated with HDAC inhibitors. Interestingly, DTWD1 could be upregulated by inhibitors of HDACs such as TSA in two independent ways (Figure 5A and 5D). Since acetylation of p53 abrogated Mdm2-mediated repression to stabilize p53 protein level, TSA could upregulate p53 expression probably through the alteration of posttranslational modifications of p53 [26,27]. Therefore, TSA could elevate the expression of DTWD1 through increasing protein level of p53. In addition, HDAC3 regulated p53-mediated DTWD1 expression independent of p53 stabilization, probably through modeling the structure of chromatin to control the interaction of transcription factors with DTWD1. Knock down of HDAC3 relaxed the chromatin condensation thus made the DTWD1 promoter more accessible for the binding of transfection factors. Therefore, HDAC3 could serve as a promising target in clinical gastric cancer treatment with limited side effects. In fact, different HDACis had been applied in clinical trials with FDA approval and exerted an remarkable co-anticancer therapeutic effect combining with chemotherapy drugs, photodynamic therapy, even autophagy inhibitors [28-30].
Jamie M. Hearnes et al combined chromatin immunoprecipitation (ChIP) with a yeast-based assay to screen the genome for p53 binding sites screened genes and reported that DTWD1 gene could be the target of p53 [20]. Indeed, we confirmed that p53 directly interact with DTWD1 gene and positively regulate DTWD1 transcription. When treated with VP16, DNA damage induced p53 stabilization and upregulated DTWD1 expression (Figure 4F and 4G). However, VP16 failed to induce DTWD1 expression once p53 was absent (Figure 4F and 4G). Certainly, we could not exclude other potential transcription factors responsible for DTWD1 expression. For example, Anne Laurencon et al found that DTWD1 could be a target gene of RFX through a genome wide computer screen [31].
In addition, the mechanism underlying the tumor-suppressing role of DTWD1 needed to be investigated further. In our study, we uncovered that DTWD1 expression downregulated the expression of cyclin B1 but not other cyclins or CDKs. Since cyclin B1 activation of CDK1 is required for the progression of cells through the G2 phase, DTWD1 could induce cell cycle arrest at G2 phase by downregulation of cyclin B1. Indeed, less Ki 67 expression but not caspase activation were found in tumors formed by DTWD1-expressing cells (Figure 6E and data not shown). However, the mechanism how cyclin B1 was regulated by DTWD1 was still unknown.
In summary, we found that DTWD1 is a new target gene of p53 and downregulated by HDAC3 in gastric cancer. DTWD1 functioned as a novel tumor suppressor in gastric cancer by regulating cyclin B1. Specific inhibitors of HDAC3 might be novel anti-cancer treatment approaches with limited side effects by specifically activating DTWD1.
Acknowledgements
This work was supported by Ministry of Education (20110101110137), the National Natural Science Foundation of China (81372178), Natural Science Foundation of Zhejiang Province (LR12H16001) Department of Sci-ence and Technology in Zhejiang Province (2012C37105) and Key program of Science and Technology in Zhejiang Province (2012C13014-4).
Disclosure of conflict of interest
None to disclose.
References
- 1.Hartgrink HH, Jansen EP, van Grieken NC, van de Velde CJ. Gastric cancer. Lancet. 2009;374:477–490. doi: 10.1016/S0140-6736(09)60617-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ushijima T, Sasako M. Focus on gastric cancer. Cancer Cell. 2004;5:121–125. doi: 10.1016/s1535-6108(04)00033-9. [DOI] [PubMed] [Google Scholar]
- 3.Taby R, Issa JP. Cancer epigenetics. CA Cancer J Clin. 2010;60:376–392. doi: 10.3322/caac.20085. [DOI] [PubMed] [Google Scholar]
- 4.Swami M. Epigenetics: Demethylation links cell fate and cancer. Nat Rev Cancer. 2010;10:740. doi: 10.1038/nrc2948. [DOI] [PubMed] [Google Scholar]
- 5.Marks P, Rifkind RA, Richon VM, Breslow R, Miller T, Kelly WK. Histone deacetylases and cancer: causes and therapies. Nat Rev Cancer. 2001;1:194–202. doi: 10.1038/35106079. [DOI] [PubMed] [Google Scholar]
- 6.Gray SG, Ekstrom TJ. The human histone deacetylase family. Exp Cell Res. 2001;262:75–83. doi: 10.1006/excr.2000.5080. [DOI] [PubMed] [Google Scholar]
- 7.Bieliauskas AV, Pflum MK. Isoform-selective histone deacetylase inhibitors. Chem Soc Rev. 2008;37:1402–1413. doi: 10.1039/b703830p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Song J, Noh JH, Lee JH, Eun JW, Ahn YM, Kim SY, Lee SH, Park WS, Yoo NJ, Lee JY, Nam SW. Increased expression of histone deacetylase 2 is found in human gastric cancer. APMIS. 2005;113:264–268. doi: 10.1111/j.1600-0463.2005.apm_04.x. [DOI] [PubMed] [Google Scholar]
- 9.Mutze K, Langer R, Becker K, Ott K, Novotny A, Luber B, Hapfelmeier A, Gottlicher M, Hofler H, Keller G. Histone deacetylase (HDAC) 1 and 2 expression and chemotherapy in gastric cancer. Ann Surg Oncol. 2010;17:3336–3343. doi: 10.1245/s10434-010-1182-1. [DOI] [PubMed] [Google Scholar]
- 10.Noguchi A, Kikuchi K, Zheng H, Takahashi H, Miyagi Y, Aoki I, Takano Y. SIRT1 expression is associated with a poor prognosis, whereas DBC1 is associated with favorable outcomes in gastric cancer. Cancer Med. 2014;3:1553–1561. doi: 10.1002/cam4.310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kim HJ, Bae SC. Histone deacetylase inhibitors: molecular mechanisms of action and clinical trials as anti-cancer drugs. Am J Transl Res. 2011;3:166–179. [PMC free article] [PubMed] [Google Scholar]
- 12.Cornago M, Garcia-Alberich C, Blasco-Angulo N, Vall-Llaura N, Nager M, Herreros J, Comella JX, Sanchis D, Llovera M. Histone deacetylase inhibitors promote glioma cell death by G2 checkpoint abrogation leading to mitotic catastrophe. Cell Death Dis. 2014;5:e1435. doi: 10.1038/cddis.2014.412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Feng L, Pan M, Sun J, Lu H, Shen Q, Zhang S, Jiang T, Liu L, Jin W, Chen Y, Wang X, Jin H. Histone deacetylase 3 inhibits expression of PUMA in gastric cancer cells. J Mol Med (Berl) 2013;91:49–58. doi: 10.1007/s00109-012-0932-x. [DOI] [PubMed] [Google Scholar]
- 14.Yoshida M, Kijima M, Akita M, Beppu T. Potent and specific inhibition of mammalian histone deacetylase both in vivo and in vitro by trichostatin A. J Biol Chem. 1990;265:17174–17179. [PubMed] [Google Scholar]
- 15.Newmark HL, Lupton JR, Young CW. Butyrate as a differentiating agent: pharmacokinetics, analogues and current status. Cancer Lett. 1994;78:1–5. doi: 10.1016/0304-3835(94)90023-x. [DOI] [PubMed] [Google Scholar]
- 16.Lee SH, Kim J, Kim WH, Lee YM. Hypoxic silencing of tumor suppressor RUNX3 by histone modification in gastric cancer cells. Oncogene. 2009;28:184–194. doi: 10.1038/onc.2008.377. [DOI] [PubMed] [Google Scholar]
- 17.Tu Z, Li H, Ma Y, Tang B, Tian J, Akers W, Achilefu S, Gu Y. The enhanced antiproliferative response to combined treatment of trichostatin A with raloxifene in MCF-7 breast cancer cells and its relevance to estrogen receptor beta expression. Mol Cell Biochem. 2012;366:111–122. doi: 10.1007/s11010-012-1288-9. [DOI] [PubMed] [Google Scholar]
- 18.Catalano MG, Fortunati N, Pugliese M, Marano F, Ortoleva L, Poli R, Asioli S, Bandino A, Palestini N, Grange C, Bussolati B, Boccuzzi G. Histone deacetylase inhibition modulates E-cadherin expression and suppresses migration and invasion of anaplastic thyroid cancer cells. J Clin Endocrinol Metab. 2012;97:E1150–1159. doi: 10.1210/jc.2011-2970. [DOI] [PubMed] [Google Scholar]
- 19.Zhang S, Jiang T, Feng L, Sun J, Lu H, Wang Q, Pan M, Huang D, Wang X, Wang L, Jin H. Yin Yang-1 suppresses differentiation of hepatocellular carcinoma cells through the downregulation of CCAAT/enhancer-binding protein alpha. J Mol Med (Berl) 2012;90:1069–1077. doi: 10.1007/s00109-012-0879-y. [DOI] [PubMed] [Google Scholar]
- 20.Hearnes JM, Mays DJ, Schavolt KL, Tang L, Jiang X, Pietenpol JA. Chromatin immunoprecipitation-based screen to identify functional genomic binding sites for sequence-specific transactivators. Mol Cell Biol. 2005;25:10148–10158. doi: 10.1128/MCB.25.22.10148-10158.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gantchev TG, Hunting DJ. Enhancement of etoposide (VP-16) cytotoxicity by enzymatic and photodynamically induced oxidative stress. Anticancer Drugs. 1997;8:164–173. doi: 10.1097/00001813-199702000-00007. [DOI] [PubMed] [Google Scholar]
- 22.Karpinich NO, Tafani M, Rothman RJ, Russo MA, Farber JL. The course of etoposide-induced apoptosis from damage to DNA and p53 activation to mitochondrial release of cytochrome c. J Biol Chem. 2002;277:16547–16552. doi: 10.1074/jbc.M110629200. [DOI] [PubMed] [Google Scholar]
- 23.Halkidou K, Gaughan L, Cook S, Leung HY, Neal DE, Robson CN. Upregulation and nuclear recruitment of HDAC1 in hormone refractory prostate cancer. Prostate. 2004;59:177–189. doi: 10.1002/pros.20022. [DOI] [PubMed] [Google Scholar]
- 24.Zhang Z, Yamashita H, Toyama T, Sugiura H, Ando Y, Mita K, Hamaguchi M, Hara Y, Kobayashi S, Iwase H. Quantitation of HDAC1 mRNA expression in invasive carcinoma of the breast*. Breast Cancer Res Treat. 2005;94:11–16. doi: 10.1007/s10549-005-6001-1. [DOI] [PubMed] [Google Scholar]
- 25.Zhu P, Martin E, Mengwasser J, Schlag P, Janssen KP, Gottlicher M. Induction of HDAC2 expression upon loss of APC in colorectal tumorigenesis. Cancer Cell. 2004;5:455–463. doi: 10.1016/s1535-6108(04)00114-x. [DOI] [PubMed] [Google Scholar]
- 26.Roy S, Packman K, Jeffrey R, Tenniswood M. Histone deacetylase inhibitors differentially stabilize acetylated p53 and induce cell cycle arrest or apoptosis in prostate cancer cells. Cell Death Differ. 2005;12:482–491. doi: 10.1038/sj.cdd.4401581. [DOI] [PubMed] [Google Scholar]
- 27.Tang Y, Zhao W, Chen Y, Zhao Y, Gu W. Acetylation is indispensable for p53 activation. Cell. 2008;133:612–626. doi: 10.1016/j.cell.2008.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Colarossi L, Memeo L, Colarossi C, Aiello E, Iuppa A, Espina V, Liotta L, Mueller C. Inhibition of Histone Deacetylase 4 Increases Cytotoxicity of Docetaxel in Gastric Cancer Cells. Proteomics Clin Appl. 2014;8:924–931. doi: 10.1002/prca.201400058. [DOI] [PubMed] [Google Scholar]
- 29.Mahalingam D, Mita M, Sarantopoulos J, Wood L, Amaravadi RK, Davis LE, Mita AC, Curiel TJ, Espitia CM, Nawrocki ST, Giles FJ, Carew JS. Combined autophagy and HDAC inhibition: a phase I safety, tolerability, pharmacokinetic, and pharmacodynamic analysis of hydroxychloroquine in combination with the HDAC inhibitor vorinostat in patients with advanced solid tumors. Autophagy. 2014;10:1403–1414. doi: 10.4161/auto.29231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ye RR, Tan CP, He L, Chen MH, Ji LN, Mao ZW. Cyclometalated Ir(III) complexes as targeted theranostic anticancer therapeutics: combining HDAC inhibition with photodynamic therapy. Chem Commun (Camb) 2014;50:10945–10948. doi: 10.1039/c4cc05215c. [DOI] [PubMed] [Google Scholar]
- 31.Laurencon A, Dubruille R, Efimenko E, Grenier G, Bissett R, Cortier E, Rolland V, Swoboda P, Durand B. Identification of novel regulatory factor X (RFX) target genes by comparative genomics in Drosophila species. Genome Biol. 2007;8:R195. doi: 10.1186/gb-2007-8-9-r195. [DOI] [PMC free article] [PubMed] [Google Scholar]