

Abstract
Podoplanin overexpression has been reported in various cancers, however, the precise mechanism for podoplanin to promote tumor progression remains elusive. In the present study, podoplanin overexpression was found associated with invasiveness both in OSCC tissues and cell lines. Moreover, the cell invasiveness increased with forced podoplanin expression and decreased when podoplanin was knockdown, indicating podoplanin-mediated cell invasion during OSCC progression. To further identify the role of podoplanin in tumor invasion, cell spreading and immunofluorescence assay were performed firstly. It was found that podoplanin knockdown caused an impaired cell spreading with reduced filopodia and the premature assembly of stress fibers while podoplanin overexpression induced an increase in cellular protrusions and stress fibers with extensive parallel bundles. Then, pull-down assays revealed forced podoplanin expression increased Cdc42 activity and reduced RhoA activity while podoplanin knockdown decreased Cdc42 and increased RhoA markedly. Moreover, a hierarchy of crosstalk between RhoA and Cdc42 was confirmed in podoplanin-mediated cell motility. On the other hand, a significant correlation between podoplanin and MT1-MMP expression in OSCCs was found both in vivo and in vitro, co-located in invasive cells and cellular protrusions. Furthermore, our data showed MT1-MMP knockdown significantly blocked the upregulation of cell motility by forced podoplanin expression, indicating that MT1-MMP played a role in podoplanin-mediated tumor invasion. To further confirm the interaction between RhoA/Cdc42 complex, MT1-MMP and podoplanin, co-precipitation experiments were performed. Both the co-precipitation of podoplanin with MT1-MMP and the podoplanin-induced specific binding of MT1-MMP to Cdc42 were found, and immunofluorescence revealed the co-location of podoplanin, MT1-MMP and Cdc42 at the plasma membrane and filopodia induced an increase in cellular protrusion and stress fibers formation. Moreover, MT1-MMP inhibition could partly rescue the increase of Cdc42 activity caused by forced podoplanin expression. Taken together, our data demonstrated a hierarchy of crosstalk between RhoA and Cdc42 was involved in podoplanin-mediated cytoskeleton remodeling and invasion; the co-location and co-ordination of podoplanin, Cdc42 and MT1-MMP in the invadopodia might induce cytoskeleton remodeling, ECM degradation and tumor invasion, while podoplanin-induced EMT may not be indispensible during OSCC progression.
Keywords: Podoplanin, invasion, Cdc42/RhoA, MT1-MMP, oral squamous cell carcinoma
Introduction
Oral squamous cell carcinoma (OSCC) occupies approximately 90% of the head and neck cancers , the five-year survival rate of which ranges from 45-53% [1]. Local recurrence and metastasis would frequently appear after surgery resection [2]. Tumor cells spread beyond the resection margin that can’t be detected microscopically, containing cancer cells responsible for the recurrence and metastasis [3]. Therefore, the identification of specific biomarkers that characterize the invasion front will help to improve the prognosis of OSCC.
Podoplanin, a transmembrane sialomucin-like glycoprotein, plays an important role in the formation of lymphatic vascular system [4]. Overexpression of podoplanin has been reported in a variety of cancers, such as squamous cell carcinoma from skin [5], larynx [6], uterine cervix [7] and esophagus [8], colorectal adenocarcinomas [9] and germ cell tumor [10]. However, the precise mechanism for podoplanin to promote tumor progression remains elusive. Epithelial-to-mesenchymal transition (EMT) is a common phenomenon occurring in tumor invasion and metastasis, during which epithelial cancer cells lose the cell-cell tight junctions and change themselves into a mesenchymal elongated cellular shape [11]. Podoplanin has been reported to contribute to EMT-mediated cell invasion in some type of tumors [10,12], while it has also been demonstrated that podoplanin induces collective cell invasion in the absence of EMT [13]. Apart from EMT, invasive cancer cells escape from the primary tumor by forming invadopodia into the surrounding matrix, associated with concentrated matrix degradation and complex rearrangement of the actin cytoskeleton [14,15].
The actin cytoskeleton remodeling is considered as a key step for cell invasion and migration which allows the formation of cell protrusions that adhere to the extra-cellular matrix and generate intra-cellular forces for cell movement. It has been reported that podoplanin mediated the remodeling of the actin cytoskeleton via the downregulation of the activities of small Rho family GTPases [16]. On the other hand, podoplanin has also been found overexpressed in the invasive front of many tumors, located in the actin fiber rich cancer cell protrusions [12,16]. To degrade the extracellular matrix barrier, MMPs also localize at the leading edge of invasion and the cellular protrusions called invadopodia [17,18]. Moreover, it has been reported that dimerization of MT1-MMP may be regulated by Cdc42-driven cytoskeletal reorganization and MT1-MMP/Cdc42 dependent signaling regulates cell invasiveness into a three-dimensional matrix [19]. Taken together, we hypothesized podoplanin might promote tumor invasion through the regulation of cytoskeleton remodeling, and the crosstalk between podoplanin, Rho GTPases and MMPs may be involved in the podoplanin-mediated cell invasion during tumor progression.
Based on this information, the aim of this study was to investigate the co-ordination of podoplanin, Rho GTPases and MMPs in regulation of cytoskeleton remodeling and tumor invasion during OSCC progression.
Materials and methods
Cell lines and transfection
Three established OSCC cell lines, WSU-HN6, TCA83 and CAL27, were kindly donated by the Department of Central Laboratory, Peking University School and Hospital of Stomatology. WSU-HN6 and CAL27 cells were maintained in DMEM medium containing 10% fetal bovine serum (FBS) (Hyclone, Logan, UT, USA), and TCA83 was maintained in RPMI1640 medium supplemented with 10% FBS. All cells were incubated at 37°C in a 5% CO2 atmosphere.
WSU-HN6 cells were transfected with pCMV6-Entry empty vector or pCMN6-AC-GFP vector containing full-length podoplanin with Lipofectamine 2000 Reagent (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s instructions. G418-resistant transfectants were selected and GFP-tagged control WSU-HN6 cells (WSU-HN6/Mock) and GFP-tagged podoplanin-expressing WSU-HN6 cells (WSU-HN6/PDPN) were obtained. Western blotting was used to identify the successful transfection and the forced podoplanin expression in WSU-HN6/PDPN cells.
On-TARGET plus Human podoplanin siRNA-SMARTpool (L-017560-01-0005) and control siRNA (D-001810-10-05) were obtained from Dharmacon (Thermo Scientific, Lafayette, CO, USA). MT1-MMP, Cdc42 and RhoA siRNA regents were purchased from Ribo Bio (Ribo Bio, Guangzhou, China). Transfections with target siRNA or control siRNA were carried out in WSU-HN6, TCA83 and CAL27 cells using DharmaFECT transfection reagent according to the manufacturer’s instructions.
Invasion and cell spreading assay
The invasion of cells was analyzed using transwell system with 6.5-mm diameters and 8um pore sizes (Costar, Lowell, MA, USA), pre-coated with Matrigel (BD, San Jose, CA, USA). The trypsinized cells (1 × 104) were washed with PBS, resuspended in the serum-free medium, and placed in the upper chamber. Medium containing 20% FBS was placed in the lower chamber as a chemoattractant. After incubation for 48 h at 37°C, the non-invading cells were removed from the upper surface of the membrane using cotton tipped swabs, and then the cells in the lower surface of the membrane were fixed in 4% paraformaldehyde and stained with crystal violet. The invaded cells were counted in 6 random fields and photographed under a microscope (BX51, Olympus, Tokyo, Japan).
For cell spreading assay, the transfected cells in serum-free medium were seeded onto dishes that were coated with 500 ng/cm2 of collagen I and fibronectin (BD, San Jose, CA, USA) respectively. Cells were incubated at 37°C and monitored in regular intervals. Adherent cells were photographed with microscope (Eclipse TE2000-U, Nikon, Tokyo, Japan) under phase contrast and counted as flattened or round cells. All experiments were confirmed in three independent experiments.
Western blotting and cell immunofluorescence
For Western blotting, each lane was loaded with 30 µg of protein on 10% SDS-PAGE gels. The proteins were then transferred to PVDF membrane (Millipore, Bedford, MA, USA). The membrane was blocked with 5% skim milk for 1 h and then incubated overnight at 4°C with each primary antibody against its specific protein. The membrane were washed three times and incubated with a 1:10000 dilution of the secondary antibodies for 1 h and the protein bands were detected with ECL chemiluminescence (Cwbiotech, Beijing, China) according to the manufacturer’s instructions.
For immunofluorescence, cells were grown on coverslips, fixed with 4% paraformaldehyde for 30 min and then permeabilized with 0.1% Triton X 100 for 10min at room temperature. After incubation at 37°C for 30 min with blocking solution, cells were incubated with primary antibodies, and then a dilution of 1:1000 of either Alexa Fluo 488-conjugated or rhodamine-conjugated fluorescent antibodies (Cell Signaling Technology, Danvers, MA, USA) were incubated for 1 h at 37°C. Cytoskeleton stress fibres were visualized by staining with a 1:500 dilution of fluorescein isothiocyanate (FITC)-conjugated Phalloidin (Cytoskeleton, Denver, CO, USA).
Antibodies used were as follows: GAPDH (ZSGB-BIO, Beijing, China), podoplanin (Abcam, Cambridge, MA, USA), E-cadherin (Cell Signaling Technology), β-catenin (Cell Signaling Technology), CK 18 (ProteinTech Group, Chicago, IL, USA), Vimentin (Abcam), N-cadherin (R & D Systems, Minneapolis, MN, USA), Fibronectin (Abcam), MT1-MMP (Abcam), Cdc42 (Abcam). All experiments were confirmed in three independent experiments.
Quantitative Real-Time PCR (qRT-PCR) and RT-PCR
Total RNA was isolated from OSCC cells by the Tritrol reagent (Invitrogen) according to the manufacturer’s instructions. Then 2 μg of total RNA was reverse transcribed into cDNA using a RT-PCR system (Promega, Madison, WI, USA) and oligo (dT). The qRT-PCR was carried out using a 7500 real-time PCR system of Applied Biosystems with Fast Start Universal SYBR Green Master (Roche, Indianapolis, IN, USA) according to the manufacturer’s instructions. The relative mRNA expression was determined using the comparative Ct (ΔΔCt) method. The data is representative of three independent experiments. RT-PCR reactions were done using the Master-Mix RT-PCR kit (Promega) and the PCR products were visualized by UV transillumination of 1.5% agarose gels stained with ethidium bromide. The base sequences of the primers were showed in Table 1. All experiments were confirmed in three independent experiments.
Table 1.
Base sequence of the primers
| Primers (5’-3’) | ||
|---|---|---|
|
| ||
| sense | antisense | |
| podoplanin | TGACTCCAGGAACCAGCGAAG | GCGAATGCCTGTTACACTGTTGA |
| MMP-1 | CTGAAGGTGATGAAGCAGCC | AGTCCAAGAGAATGGCCGAG |
| MMP-2 | GCGACAAGAAGTATGGCTTC | TGCCAAGGTCAATGTCAGGA |
| MMP-3 | CTCACAGACCTGACTCGGTT | CACGCCTGAAGGAAGAGATG |
| MMP-7 | GTGGTCACCTACAGGATCGT | ACCATCCGTCCAGCGTTCAT |
| MMP-8 | ATGGACCAACACCTCCGCAA | GTCAATTGCTTGGACGCTGC |
| MMP-9 | CGCAGACATCGTCATCCAGT | GGATTGGCCTTGGAAGATGA |
| MMP-10 | GTCCTTCGATGCCATCAGCA | CTTGCTCCATGGACTGGCTA |
| MMP-11 | CAGGTGGCAGCCCATGAATT | GTACTGAGCACCTTGGAAGA |
| MMP-12 | CCACTGCTTCTGGAGCTCTT | GCGTAGTCAACATCCTCACG |
| MMP-13 | CTATGGTCCAGGAGATGAAG | AGAGTCTTGCCTGTATCCTC |
| MMP-14 | CAACACTGCCTACGAGAGGA | GTTCTACCTTCAGCTTCTGG |
| MMP-15 | GCATCCAGAACTACACGGAG | TACCGTAGAGCTGCTGGATG |
| MMP-16 | TGTACCTGACCAGACAAGAG | AGTGTCCATGGCTCATCTGA |
| MMP-17 | GACCTGTTTGCAGTGGCTGT | ACGATCTTGTGGTCGCTGGT |
| MMP-19 | CAGGCTCTCTATGGCAAGAA | GAGCTGCATCCAGGTTAGGT |
| MMP-20 | GACCAGACCACAATGAACGT | GTCCACTTCTCAGGATTGTC |
| GAPDH | GCACCGTCAAGGCTGAGAAC | TGGTGAAGACGCCAGTGGA |
Pull-down assays with PAK-GST-PBD and Rhotekin-RBD beads
RhoA/Cdc42/Rac1 activity was assessed by a pull-down assay according to the manufacturer’s instructions for RhoA/Cdc42/Rac1 Activation Assay Biochem Kit (Cytoskeleton, Denver, CO, USA). Transfected cells were harvested and lysed in buffer containing complete EDTA-free protease inhibitor cocktail tablets. Sample contain the same amount of protein (30 μg) from each cell was used for western blotting quantitation of total RhoA, Cdc42, and Rac1, and 300 ug total protein from each cell was added to 50 μg GST-Rhotekin-RBD beads (for RhoA activation assay) and 10ug GST-PAK-PBD beads (for Cdc42 and Rac1 activation assay), incubated at 4°C for 1 h. The GTP-bounded protein was collected and then detected by western blotting analysis. All experiments were confirmed in three independent experiments.
Co-immunoprecipitation (Co-IP)
Cells were harvested and lysed in RIPA buffer containing complete EDTA-free protease inhibitor cocktail tablets. Lysates were precleared with protein A/G-agarose beads (Santa Cruz Biotechnology, Santa Cruz, CA, USA) and incubated with indicated antibodies overnight at 4°C. The immunocomplexes were precipitated by protein A/G-agarose, washed three times with RIPA buffer and analyzed by western blotting analysis. Normal rabbit IgG served as a negative control. All experiments were confirmed in three independent experiments.
Tissue samples and immunohistochemistry
The total of 163 cases of formalin-fixed paraffin-embedded tissue samples was derived from the archives of the Department of Oral Pathology, Peking University School and Hospital of Stomatology for immunostaining, including 53 epithelial dysplasias, 110 invasive OSCCs (26 microinvasive OSCCs and 43 of which with nodal metastasis). Adjacent normal mucosa was used as normal control. This study was approved by the Ethical Committee of Peking University.
Four-micrometer-thick serial sections were cut from paraffin-embedded tissue blocks, deparaffinized in xylene and sequentially rehydrated through a graded ethanol series. Endogenous peroxidase activity was quenched by incubation with fresh 3% H2O2 in methanol for 20 min at room temperature, and then antigen retrieval was accomplished by 10% ethylene diaminetetra-acetic acid buffer (pH 9.0). The sections were incubated with antibodies against podoplanin and MT1-MMP (Abcam) at 4°C overnight and followed by incubated with the secondary antibodies for 30 min. The immunocomplexes were visua lized using liquid DAB + substrate + chromogen system (Zymed, San Francisco, CA, USA). Sections were lightly counterstained with Mayer’s hematoxylin and mounted. The immunostaining results were analyzed by two independent pathologists who were blinded for the information of each patient. Reactivity was determined according to the percentage of positive cells: up to 1% positive cells were scored as 0, 2-25% as 1, 26-50% as 2, 51-75% as 3, and over 75% as 4. Intensity was graded as follows: no signal (0), weak (1), moderate (2), and strong (3). A total score of 0 to 12 was finally calculated and graded as negative (-; score: 0-1), weak (+; 2-4), moderate (++; 5-8), and strong (+++; 9-12). The score of 0 to 6 was defined as low positive, and 7 to 12 was considered as high positive.
Statistical analysis
SPSS 13.0 (SPSS, Chicago, IL) was used to analyze all data. Chi-squared test or Student t test was used to compare the variables between groups. The survival rate of the patients was analyzed with Kaplan-Meier method and the difference between groups was analyzed with the log-rank test. All P-values < 0.05 was considered significant.
Results
Podoplanin promoted the invasion of OSCC cells
Western blotting and RT-PCR analysis showed that podoplanin expression was higher in CAL27 and TCA83 cells than in WSU-HN6 cells (Figure 1A), and by transwell assays, CAL27 and TCA83 cells showed 4.3 and 3.2 fold increase in gel invasion compared with WSU-HN6, respectively (Figure 1B). Furthermore, immunohistochemistry revealed podoplanin is consistently correlated with the invasiveness of OSCC. Although there was no statistically significant correlation between podoplanin status and age, gender, size or site, our results indicated that the intensity of podoplanin expression was enhanced as the severity of dysplasia increased (Tables 2 and 3). Furthermore, higher level of podoplanin in invasive OSCC cells with the metastasized cancer cells in lymph nodes showing higher podoplanin expression than the primary tumors (Figure 1C). Statistical analysis showed that the expression of podoplanin tended to be significantly associated with differentiation and nodal metastasis in 110 OSCC samples (Table 3). Five-year survival rate analysis also showed higher level of podoplanin expression significantly correlated to shorter survival (P = 0.012) (Figure 1D).
Figure 1.
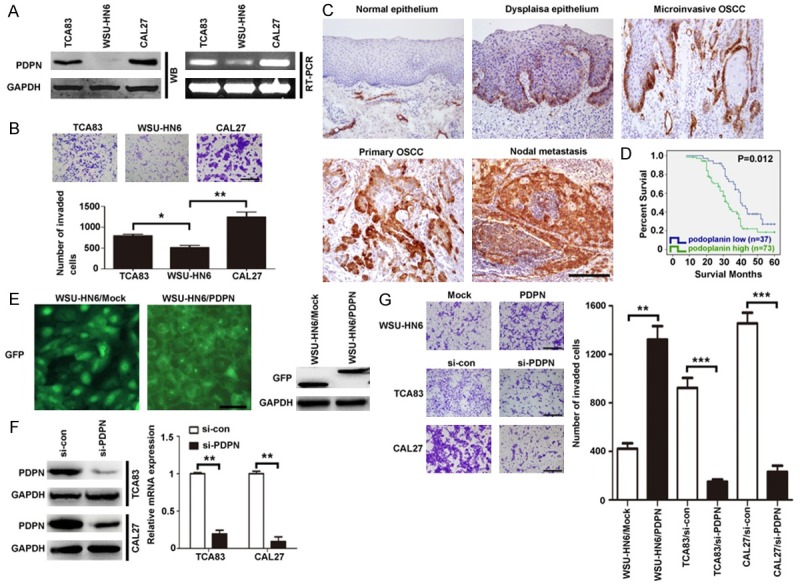
Podoplanin expression is positively associated with the invasiveness of OSCC cells both in vitro and in vivo. A. Expression of podoplanin in three OSCC cell lines. Equal amounts of proteins and cDNA from three OSCC cell lines were evaluated by western blot and RT-PCR. GAPDH was used as control. B. Invasion ability of three OSCC cell lines was accessed by transwell assay. 1 × 104 cells were seeded on the upper chamber and incubated for 48 h. Cells that invaded the membrane were then stained and counted. Scal bar = 400 μm. C. Representative photographs of immunostaining for podoplanin in normal epithelium, dysplasia epithelium, microinvasive OSCC, primary OSCC and nodal metastasis. Scal bar = 200 μm. D. Kaplan-Meier plots of podoplanin expression in 110 cases of OSCC patients. Overall survival rate was performed by log-rank test (immunoreactivity scores < or = 6 was ascribed to be low podoplanin expression, immunoreactivity scores > or = 7 was ascribed to be high podoplanin expression). P < 0.05 indicated significant differences between two groups. E. WSU-HN6 cells were stably transfected with pCMV6-Entry empty vector or pCMN6-AC-GFP vector containing full-length podoplanin. Western blot analysis revealed the expression of GFP-tagged podoplanin and control vector in WSU-HN6. GAPDH was used as control. Scale bar = 50 μm. F. TCA83 and CAL27 cells were treated with PDPN and control siRNA regents. After 24 h and 48 h, the expression of podoplanin was analyzed by qRT-PCR and western blotting, respectively. GAPDH was used as control. G. The invasion ability of each cell line was evaluated by transwell assay. WSU-HN6 with overexpressed podoplanin and TCA83 and CAL27 cells with podoplanin knockdown were subjected to the transwell assay. Scale bar = 400 μm. Experiments in A, B, F and G were performed in triplicates (n = 3). Error bars indicate SD; significance level as indicated: *P < 0.05, **P < 0.01, ***P < 0.001.
Table 2.
Association between podoplanin expression and clinicopathological parameters for 53 precancerous lesions
| Features | Number | Podoplanin expression level | P | |
|---|---|---|---|---|
|
| ||||
| Low positive | High positive | |||
| Age (years) | 0.407 | |||
| ≤ 59 | 26 | 20 | 6 | |
| > 59 | 27 | 18 | 9 | |
| Gender | 0.141 | |||
| Male | 20 | 12 | 8 | |
| Female | 33 | 26 | 7 | |
| Sites | 0.586 | |||
| buccal | 12 | 8 | 4 | |
| tongue | 32 | 22 | 10 | |
| gingival | 5 | 5 | 0 | |
| others | 4 | 3 | 1 | |
| Dysplasia | 0.021 | |||
| mild | 16 | 14 | 2 | |
| moderate | 20 | 16 | 4 | |
| severe/CIS | 17 | 8 | 9 | |
Table 3.
Association between podoplanin expression and clinicopathological parameters for 110 oral squamous cell carcinoma
| Features | Number | Podoplanin expression level | P | |
|---|---|---|---|---|
|
| ||||
| Low positive | High positive | |||
| Age (years) | 0.231 | |||
| ≤ 59 | 48 | 20 | 28 | |
| > 59 | 62 | 19 | 43 | |
| Gender | 0.461 | |||
| Male | 63 | 23 | 40 | |
| Female | 47 | 14 | 33 | |
| Sites | 0.688 | |||
| buccal | 21 | 7 | 14 | |
| tongue | 43 | 13 | 30 | |
| gingival | 26 | 8 | 18 | |
| others | 20 | 9 | 11 | |
| Differentiation | 0.014 | |||
| well | 30 | 16 | 14 | |
| moderate | 29 | 10 | 19 | |
| poor | 51 | 11 | 40 | |
| T stage | 0.978 | |||
| T1 | 27 | 10 | 17 | |
| T2 | 43 | 14 | 29 | |
| T3 | 19 | 6 | 13 | |
| T4 | 21 | 7 | 14 | |
| pN stage | 0.029 | |||
| pN- | 67 | 28 | 39 | |
| pN+ | 43 | 9 | 34 | |
To identify the role of podoplanin in the invasion of OSCC cells, we transfected a green fluorescence protein (GFP) and GFP-tagged podoplanin gene into WSU-HN6 cells, respectively. Western blotting revealed higher podoplanin expression was found in WSU-HN6/PDPN cells compared with WSU-HN6/Mock cells (Figure 1E). Meantime, it was observed that forced podoplanin expression increased the invasiveness of OSCC cells by more than 3 fold (Figure 1G). To further confirm the relationship between podoplanin expression and cell invasiveness, RNA interference (RNAi) strategy was used to knockdown podoplanin in two highly invasive OSCC cell lines, CAL27 and TCA83. The result demonstrated podoplanin siRNA strategy effectively suppressed podoplanin expression in OSCC cells (Figure 1F), and the invasiveness of CAL27 and TCA83 cells decreased significantly after podoplanin was knockdown (Figure 1G). Taken together, these results showed that podoplanin expression levels were positively associated with the invasion of OSCC cells.
Podoplanin might bypass EMT during OSCC progression
Accumulating evidences indicate that OSCC cells undergo EMT to increase invasiveness, changing from epithelial to a mesenchymal phenotype [20]. We hypothesize that podoplanin may regulate the mobility of OSCC cells by EMT. As a result, although forced podoplanin expression in WSU-HN6/PDPN cells seemed to induce the changing from epithelial to mesenchymal phenotype (Figure 2A), podoplanin knockdown in TCA83 cells did not affect the expression of EMT-related markers and even converted EMT phenotype in CAL27 cells (Figure 2B). These conflicting findings indicated that podoplanin induced phenotypic changes is dependent on cell type and may not be indispensable during OSCC progression.
Figure 2.
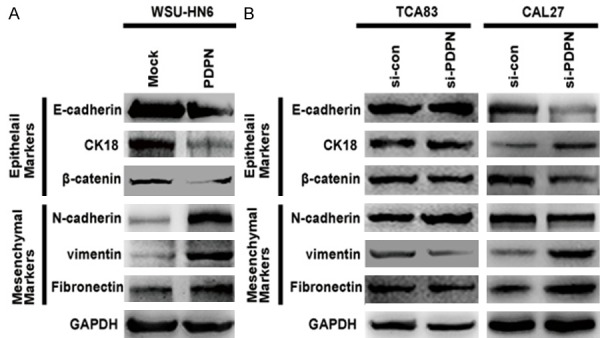
Podoplanin alters the EMT phenotype of OSCC cells. A. The expressions of EMT related epithelial markers (E-cadherin, CK 18 and β-catenin) and mesenchymal markers (N-cadherin, vimentin and Fibronectin) were examined by western blotting in WSU-HN6 cells with forced podoplanin expression. GAPDH was used as control. B. The expression of EMT related epithelial markers (E-cadherin, CK 18 and β-catenin) and mesenchymal markers (N-cadherin, vimentin and Fibronectin) were examined by western blotting in TCA83 and CAL27 cells with podoplanin-silencing. Experiments in A and B were performed in triplicates (n = 3).
Podoplanin regulated cytoskeleton remodeling and cell motility via Rho GTPases
Apart from EMT, invasive cancer cells escape from the primary tumor by forming invadopodia into the surrounding matrix, associated with concentrated matrix degradation and complex rearrangement of the actin cytoskeleton. To functionally assess the ability of OSCC cells to interact with extracellular matrix (ECM) substrates, the transfected OSCC cells were seeded onto culture dishes coated with ECM substrates (Collagen I and Fibronectin), and cell spreading assays were performed. Both the podoplanin-silenced cells and the control counterpart did not spread at 0.5 h post-transfection, seeded in culture dishes precoated with Type I collagen and Fibronectin. However, 12 h after seeding, the spreading area of mock-transfected cells was much larger than those of poplanin-silenced cells (Figure 3A). Meantime, it was found that WSU-HN6/PDPN cells with podoplanin overexpression began to spread at a very early stage, compared with WSU-HN6/Mock cells (Figure 3A). These results suggested that podoplanin affected the spreading of OSCC cells on both ECM substrates. Since polymerization and depolymerization of cytoskeleton lead to morphologic changes associated with cell motility and the formation of filopodia affects cell spreading, the actin cytoskeleton was stained with fluorescein isothiocyanate (FITC)-conjugated phalloidin in transfected cells to further investigate the role of podoplanin in cytoskeleton reconstruction. Immunofluorescence images revealed that podoplanin overexpression in WSU-HN6/PDPN cells induced an increase in cellular protrusion and stress fibers which appeared as extensive parallel bundles and were densely stained in a well-organized manner. However, these parallel bundles were disrupted with the stress fibers loosely organized when podoplanin was knockdown in TCA83 and CAL27 cells, and the number and size of filopodia decreased (Figure 3A).
Figure 3.
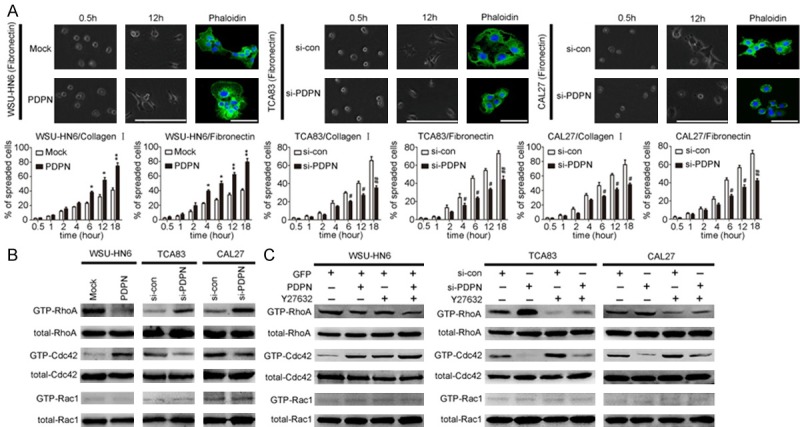
Podoplanin meditates the actin cytoskeleton remodeling through RhoA/Cdc42/Rac1 pathway in OSCC cells. A. Cell spreading assays with extracellular matrix (collagen I and Fibronectin) were performed in all the transfected OSCC cells, and then the transfected OSCC cells were stained with fluorescein isothiocyanate (FITC)-conjugated phalloidin to reveal filamentous actin stress fibres (green). Scale bar = 100 μm (phase); Scale bar = 100 μm (immunofluorescence). B. The activities and expressions of Rho family small GTPases were analyzed by Rho family pull-down assays in Podoplanin-transfected OSCC cells. C. Podoplanin-transfected OSCC cells were treated with small molecule inhibitors of Rho-associated kinase (Y27632), and then the activities and expressions of Rho family small GTPases were analyzed by Rho family pull-down assays. Experiments in A-C were performed in triplicates (n = 3); error bars indicate SD; *P < 0.05 and **P < 0.01 vs. Mock group; #P < 0.05 and ##P < 0.01 vs. si-con group.
RhoA, Cdc42, and Rac1 are most characterized members of Rho GTPases which belong to Ras superfamily and play a pivotal role in both cell spreading and cytoskeleton remodeling [21,22]. To determine whether podoplanin affect the status of RhoA/Cdc42/Rac1, GTP-bound RhoA/Cdc42/Rac1 was investigated by pull-down assay. The level of active GTP-Cdc42 was found increased significantly with RhoA activity reduced markedly in WSU-HN6/PDPN cells, compared with the WSU-HN6/Mock cells (Figure 3B). Concordantly, Cdc42 activity decreased and RhoA activity increased significantly in TCA83 and CAL27 cells with podoplanin knockdown (Figure 3B). However, the status of Rac1 was not affected in all transfected OSCC cells.
To further confirm a hierarchy of crosstalk between RhoA and Cdc42 was involved in podoplanin-regulated morphology and motility of OSCC cells, the transfected OSCC cell were treated with small molecule inhibitors of Rho-associated kinase (Y27632). It was found that inhibition of RhoA markedly increased Cdc42 activity in in all the transfected OSCC cells (Figure 3C). Moreover, RhoA inhibition further enhanced the upregulation of active GTP-Cdc42 which was caused by podoplanin overexpression in WSU-HN6/PDPN cells, and meantime, inhibition of RhoA almost rescued the downregulation of active GTP-Cdc42 by podoplanin knockdown in TCA83 and CAL27 cells (Figure 3C). We also used RNAi strategy to knockdown RhoA or Cdc42 in all the transfected OSCC cells and control counterpart, respectively. It was observed that RhoA knockdown augmented the upregulation of cell spreading areas caused by overexpressed podoplanin in WSU-HN6/PDPN cells and meanwhile rescued the downregulation of cell spreading areas in TCA83 and CAL27 cells with podoplanin knockdown (Figure 4A). In addition, transwell assays further confirmed the crosstalk between RhoA and Cdc42 and their opposing roles in podoplanin-mediated cell motility during OSCC progression (Figure 4B).
Figure 4.
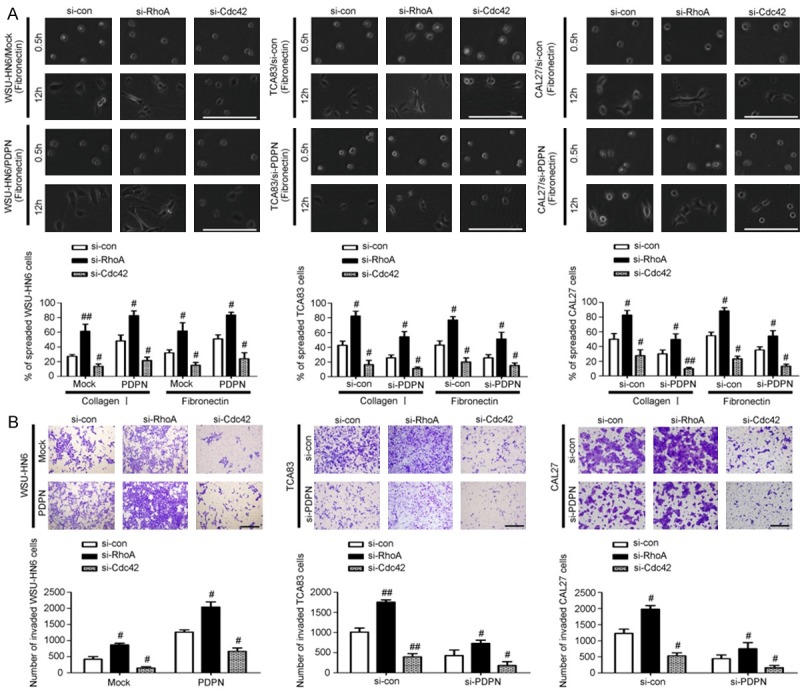
A hierarchy of crosstalk between RhoA and Cdc42 is involved in podoplanin-induced OSCC cell spreading and invasion. A. Cell spreading assays with extracellular matrix (collagen I and Fibronectin) were performed in WSU-HN6 cells with forced podoplanin expression and TCA83 and CAL27 cells with podoplanin-silencing when treated with RhoA siRNA and Cdc42 siRNA regents. Scale bar = 100 μm. B. Transwell assays together with RNAi were performed in WSU-HN6 cells with forced podoplanin expression and TCA83 and CAL27 cells with podoplanin-silencing which revealed Rho A and Cdc42 have opposing roles in cell invasion. Scale bar = 400 μm. Experiments in A and B were performed in triplicates (n = 3); error bars indicate SD; #P < 0.05 and ##P < 0.01 vs. si-con group.
Podoplanin regulated cell motility and invasion via MT1-MMP
As mentioned above, overexpression of podoplanin was found in invasive OSCC cells, which promoted cell motility and invasion by regulating the actin cytoskeleton remodeling including forming invadopodia into the surrounding matrix (Figures 1G and 3A). To degrade the extracellular matrix (ECM) barrier, MMPs also localizes at the leading edge of invasion and the cellular protrusions called invadopodia. Therefore, we hypothesized MMPs might be involved in the podoplanin-mediated cytoskeleton rearrangement and tumor invasion. Firstly, we scanned mRNA expressions of MMP families in podoplanin-transfected OSCC cells by RT-PCR. Among the sixteen MMPs examined, MT1-MMP (also known as MMP-14) expression was found not only significantly increased in WSU-HN6/PDPN cells with podoplanin overexpression but also decreased markedly in CAL27 and TCA83 cells with podoplanin-knockdown, (Figure 5A), which was then confirmed at protein level by western blotting analysis (Figure 5B).
Figure 5.
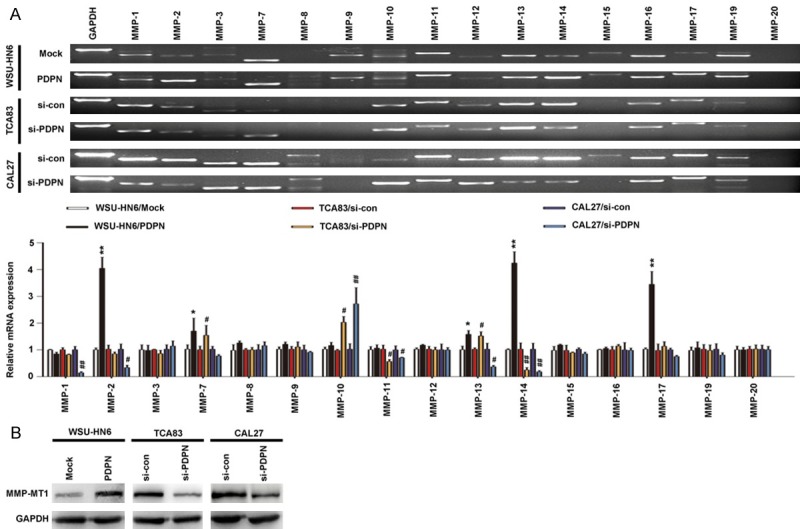
Podoplanin regulates the expression of MT1-MMP (also known as MMP-14) in OSCC cells. A. Relative expression of the indicated MMPs was determined by RT-PCR (upper panel), quantified (lower panel). B. The expression of MT1-MMP was examined by western blotting in podoplanin-transfected OSCC cells. GAPDH was used as control. Experiments in A and B were performed in triplicates (n = 3); error bars indicate SD; *P < 0.05 and **P < 0.01 vs. Mock group; #P < 0.05 and ##P < 0.01 vs. si-con group.
To identify the role of MT1-MMP in podoplanin-mediated cell motility and invasion, both tissue inhibitor of MT1-MMP (TIMP2) and RNAi strategy were used to suppress MT1-MMP in WSU-HN6/PDPN cells with forced podoplanin expression and the control WSU-HN6/Mock cells. Cell spreading assay showed that the spreading of podoplanin-transfected OSCC cells was also suppressed when MT1-MMP was knockdown (Figure 6A), and meantime, the invasion of WSU-HN6/PDPN cells was significantly blocked by MT1-MMP knockdown (Figure 6B). All these data demonstrated that MT1-MMP played an important role in the process of podoplanin- meditated cell invasion.
Figure 6.
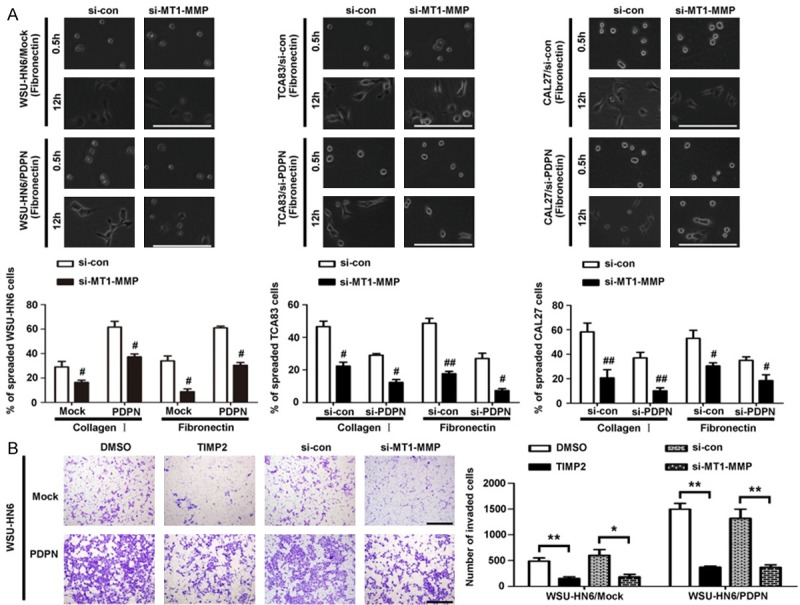
MT1-MMP is required for podoplanin-induced OSCC cell spreading and invasion. A. Cell spreading assays with extracellular matrix (collagen I and Fibronectin) were performed in all the transfected OSCC cells, treated with MT1-MMP siRNA regents. Scale bar = 100 μm; #P < 0.05 and ##, P < 0.01 vs. si-con group. B. WSU-HN6 cells with forced podoplanin expression were subjected to transwell assay in the presence of TIMP2, MT1-MMP siRNA and the control siRNA. Cells that invaded the membrane were then stained and counted. Scale bar = 400 μm; *P < 0.05 and **P < 0.01. Experiments in A and B were performed in triplicates (n = 3); error bars indicate SD.
Overexpression of podoplanin and MT1-MMP in invasive cells was observed in OSCC tissues and related to shorter survival
Expressions of podoplanin and MT1-MMP were examined in different stages of OSCC tumorigenesis, including dysplasia, microinvasive OSCCs, primary tumors of invasive OSCC as well as the nodal metastasis. Although no evident signal was found in normal epithelium, while high expression of podoplanin and MT1-MMP was found confined to the basal layer in some dysplasia and much more intensive immunostaining for both podoplanin and MT1-MMP was found in most of the microinvasive OSCC cells as well as the invasion front of primary invasive OSCCs (Figure 7A). Moreover, the metastasized cancer cells in lymph nodes showed higher podoplanin and MT1-MMP expression than primary tumors (Figure 7A). As mentioned above, our data showed high level of podoplanin expression indicated poor prognosis in OSCC (Figure 1D). When combined with the expression of podoplanin and MT1-MMP, significantly poorer survival rates were found in the patients with both podoplanin and MT1-MMP high expression (MT1-MMP high/podoplanin high) compared with those with low MT1-MMP and high podoplanin expression (MT1-MMP low/podoplanin high) (P = 0.002) (Figure 7B). Collectively, our data indicated the crosstalk between MT1-MMP and podoplanin might be involved in podoplanin-mediated tumor invasion and metastasis, which played a pivotal role during OSCC progression.
Figure 7.
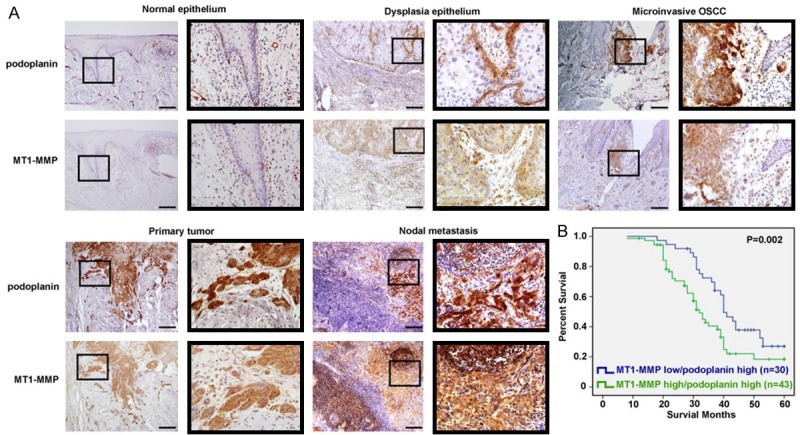
Overexpression of podoplanin and MT1-MMP in OSCC tissues is related to shorter survival. A. Representative photographs of immunostaining for podoplanin and MT1-MMP in normal epithelium, dysplasia epithelium, microinvasive OSCC, primary OSCC and nodal metastasis. Small and large black frames indicate the original and the magnified areas, respectively. Scal bar = 100 μm. B. The Kaplan-Meier estimate of 5-year survival rate in 73 OSCC patients according to podoplanin and MT1-MMP expression.
Podoplanin, Cdc42 and MT1-MMP colocalized in the invadopodia and coordinated in the regulation of cell invasion during OSCC progression
Cell migration or invasion involves the coordination between cytoskeletal reorganization, cell adhesion and interaction with ECM, as well as proteinase activities and ECM degradation. As mentioned above, our data have shown that a hierarchy of crosstalk between RhoA and Cdc42 was involved in podoplanin-regulated cytoskeletal remodeling and cell invasion (Figures 3 and 4) and MT1-MMP also played an important role in the process of podoplanin-meditated cell invasion (Figures 5 and 6). In addition, it has been reported recently that dimerization of MT1-MMP may be mainly regulated by Cdc42-driven cytoskeletal reorganization and MT1-MMP/Cdc42 dependent signaling regulates cell invasiveness into a three-dimensional matrix by HT1080 cells [23]. In order to detect the precise relationship between RhoA/Cdc42 complex, MT1-MMP and podoplanin, co-precipitation experiments were performed. When the lysates of the transfected cells were incubated with GST-PAK-PBD beads that specifically bound to GTP-Cdc42 (activated Cdc42), endogenous MT1-MMP was found specifically co-precipitated with GTP-Cdc42, and this co-association of MT1-MMP and GTP-Cdc42 was increased by podoplanin overexpression and decreased by podoplanin knockdown (Figure 8A). This podoplanin-induced binding of MT1-MMP to activated Cdc42 was specific as no co-precipitation of MT1-MMP with RhoA was found when lysates were incubated with GST-Rhotekin-RBD beads (Figure 8B). Furthermore, immunofluorescence images revealed that podoplanin overexpression in WSU-HN6/PDPN cells induced an increase in cellular protrusion and stress fibers and the collocation of MT1-MMP and GTP-Cdc42 was found to be distributed at the plasma membrane and filopodia of WSU-HN6/PDPN cells (Figure 8A). However, no co-precipitation of podoplanin with either RhoA or Cdc42 was found (Figure 8A and 8B). Meantime, the co-precipitation of MT1-MMP with podoplanin was also found and this co-association was increased by podoplanin overexpression and decreased by podoplanin knockdown (Figure 8C). Moreover, immunofluorescence images also revealed that the collocation of MT1-MMP and podoplanin was distributed at the plasma membrane and filopodia of WSU-HN6/PDPN cells (Figure 8C). Since the co-association of MT1-MMP with podoplanin and Cdc42 was identified respectively and no co-precipitation of podoplanin with Cdc42 was found, tissue inhibitor of MT1-MMP (TIMP2) was used to further determine the role of MT1-MMP in podoplanin-driven upregulation of Cdc42 activity. Although MT1-MMP knockdown did not affect Cdc42 activity directly, it could partly rescue the marked increase of GTP-Cdc42 caused by podoplanin overexpression (Figure 8D).
Figure 8.
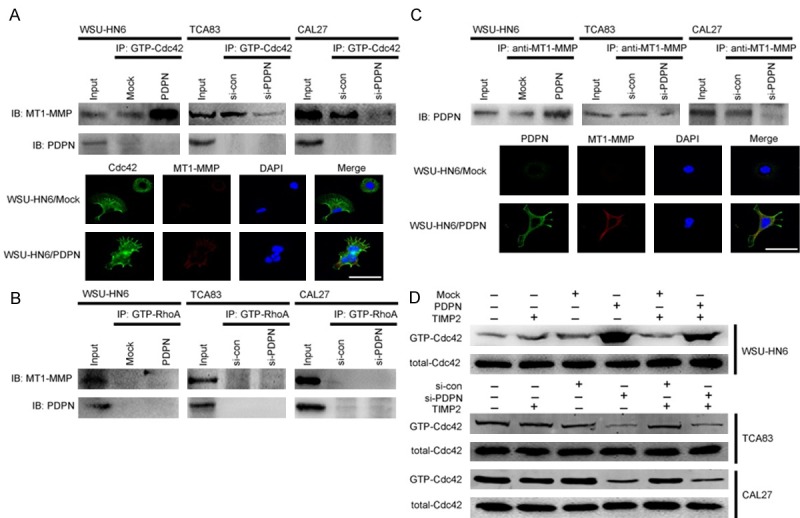
Interaction between podoplanin, MT1-MMP and Cdc42. A. The interaction between GTP-Cdc42 with podoplanin and MT1-MMP was examined in WSU-HN6 cells with forced podoplanin expression and TCA83 and CAL27 cells with podoplanin-silencing. Immunoprecipitation was performed with anti-MT1-MMP and anti-podoplanin antibodies, and normal rabbit immunoglobulin G served as a control (upper panel). Immunofluorescent staining for Cdc42 (green) and MT1-MMP (red) in WSU-HN6 cells with forced podoplanin expression. The nucleus was stained with 4, 6-diamidino-2-phenylindole (DAPI; blue). Scale bar = 100 μm (lower panel). B. The interaction between GTP-RhoA with podoplanin and MT1-MMP was examined in WSU-HN6 cells with forced podoplanin expression and TCA83 and CAL27 cells with podoplanin-silencing. Immunoprecipitation was performed with anti-MT1-MMP and anti-podoplanin antibodies, and normal rabbit immunoglobulin G served as a control. C. The interaction between podoplanin with MT1-MMP was examined in WSU-HN6 cells with forced podoplanin expression and TCA83 and CAL27 cells with podoplanin-silencing. Immunoprecipitation was performed with an anti-podoplanin antibody, and normal rabbit immunoglobulin G served as a control (upper panel). Immunofluorescent staining for podoplanin (green) and MT1-MMP (red) in WSU-HN6 cells with forced podoplanin expression. The nucleus was stained with 4, 6-diamidino-2-phenylindole (DAPI; blue). Scale bar = 100 μm (lower panel). D. WSU-HN6 cells with forced podoplanin expression and TCA83 and CAL27 cells with podoplanin-silencing were treated with TIMP2. The expression of GTP-Cdc42 in podoplanin-transfected OSCC cells was analyzed by Rho family pull-down assay. Experiments in A-D were performed in triplicates (n = 3).
Discussion
Podoplanin overexpression has been reported in a variety of cancers [5-10]. Consistently, our present study demonstrated high level of podoplanin in most of the microinvasive OSCC cells and the outer edge of the invasive OSCCs, together with higher podoplanin expression in the nodal metastases than the primary tumors. Moreover, CAL27 and TCA83 cell lines with high podoplanin expression showed more invasiveness than WSU-HN6 cells with low podoplanin expression, indicating podoplanin expression is associated with tumor invasion and metastasis.
Tumor invasion is considered as the first step of metastasis which may be the result of a continuously repetitive steps involving the morphologic changes and forming of cell protrusions, interaction with the extracellular matrix (ECM), degradation of ECM and moving forward of tumor cells. This process underlies almost all forms of cancer cell invasion, while the molecular mechanism may vary from one invasion pattern to another, such as so-called single cell invasion and collective cell invasion [24]. Epithelial-to-mesenchymal transition (EMT) is important in embryogenesis as well as carcinogenesis and mainly responsible for single cell invasion, during which cancer cells not only change their shapes and adhesive properties, but also lose the expression of epithelial markers and acquire mesenchymal signatures. Alternatively, cancer cells migrate as cohorts, that is, so-called collective cell invasion which involves the coordination between cytoskeletal reorganization, cell adhesion and interaction with ECM, proteinase activities and ECM degradation [24]. Podoplanin has been reported to contribute to EMT-mediated cell motility and promote single cell invasion in some type of tumors [23,24], whereas accumulating evidences demonstrate podoplanin induces collective cell invasion in the absence of EMT [19]. To date, the functional contribution of podoplanin to tumor progression has remained elusive. In the present study, forced podoplanin expression in OSCC cells significantly increased the invasiveness, and podoplanin knockdown markedly decreased the invasiveness of OSCC cells. Although forced podoplanin expression in WSU-HN6/PDPN cells seemed to induce the changing from epithelial to mesenchymal phenotype, podoplanin knockdown in TCA83 cells did not affect the expression of EMT-related markers and even converted EMT in CAL27 cells. These conflicting findings incorporating with previous studies [25] indicated that podoplanin induced phenotypic changes is dependent on cell type and may not be indispensable during OSCC progression.
Apart from EMT, invasive cancer cells escape from the primary tumor by forming invadopodia into the surrounding matrix, associated with concentrated matrix degradation and complex rearrangement of the actin cytoskeleton. To determine the involvement of podoplanin in cytoskeletal remodeling and interaction with ECM substrates, the transfected OSCC cells were seeded onto culture dishes coated with ECM substrates (Collagen I and Fibronectin), and cell spreading assays were performed. It was found that podoplanin knockdown caused an impaired cell spreading on both Collagen I and Fibronectin substrate. Meantime, WSU-HN6/PDPN cells with podoplanin overexpression began to spread at a very early stage compared with WSU-HN6/Mock cells. Since the formation of filopodia affects cell spreading significantly, the actin cytoskeleton was then stained with fluorescein isothiocyanate (FITC)-conjugated phalloidin to further investigate the role of podoplanin in cytoskeleton reconstruction. Immunofluorescence images revealed that podoplanin overexpression in WSU-HN6/PDPN cells induced an increase in cellular protrusions and stress fibers which appeared as extensive parallel bundles and were densely stained in a well-organized manner. However, podoplanin-silencing resulted in reduced filopodia and the premature assembly of stress fibers, which demonstrated the involvement of podoplanin in cytoskeleton reconstruction.
RhoA, Cdc42, and Rac1 are most characterized members of Rho GTPases which play a pivotal role in both cell spreading and cytoskeletal rearrangement [21,22]. In the present study, pull-down assays revealed forced podoplanin expression increased Cdc42 activity and reduced RhoA activity significantly. Moreover, podoplanin knockdown decreased Cdc42 activity and increased RhoA activity markedly. Cdc42 is considered as the major contributor to functional filopodia formation, and the active Cdc42 promotes cancer cell migration and invasion [26,27]. Although a positive role of RhoA in cell migration and invasion has been reported in many cell systems [28], the decrease in cell motility due to constitutive activation of RhoA has also been described. Ispanovic et al demonstrated the role of RhoA and Cdc42 was opposite in the process of regulating the endothelial sprouting [29]. El-Sibai et al also relieved that inhibition of RhoA enhanced the formation of cellular protrusion [30]. To further confirm a hierarchy of crosstalk between RhoA and Cdc42 was involved in podoplanin-regulated cell motility, the transfected OSCC cells were treated with small molecule inhibitors of Rho-associated kinase and siRNA. As a result, RhoA inhibition markedly increased Cdc42 activity which further enhanced the upregulation of Cdc42 activity caused by podoplanin overexpression and rescued the downregulation of Cdc42 activity by podoplanin knockdown. Furthermore, cell spreading and transwell assays further confirmed a hierarchy of crosstalk between RhoA and Cdc42 was involved in podoplanin-mediated cell motility during OSCC progression.
Recently it has been reported that certain MMPs may directly (via modulation of the extracellular environment) or indirectly (through initiation of vascular regression) affect tumor progression [31,32], and to degrade ECM barrier, MMPs also localize at the leading edge of invasion and the cellular protrusions [20,21]. In the present study, a significant positive correlation between podoplanin and MT1-MMP expression during OSCC progression was found both in vivo and in vitro, and the co-location of podoplanin and MT1-MMP was observed in the invading cells and the cellular protrusions. Furthermore, our data showed MT1-MMP knockdown significantly blocked the upregulation of cell motility by forced podoplanin expression in OSCC cell lines, which indicated that MT1-MMP played a role in the podoplanin-mediated cytoskeleton rearrangement and tumor invasion.
As mentioned above, tumor invasion involves the coordination between cytoskeletal reorganization, interaction with ECM, proteinase activities and ECM degradation. Our data showed podoplanin promoted cytoskeletal remodeling and cell invasion through up-regulating Cdc42 activity together with RhoA inhibition. Meanwhile, MT1-MMP co-located with podoplanin in the invadopodia and also played an important role in the process of podoplanin-meditated cell invasion. Recently, it has been reported that dimerization of MT1-MMP may be mainly regulated by Cdc42-driven cytoskeletal reorganization and MT1-MMP/Cdc42 dependent signaling regulates cell invasiveness into a three-dimensional matrix by HT1080 cells [23]. It has also been demonstrated that high levels of RhoA and stress fiber formation contribute to a non-proteolytic endothelial phenotype which disrupted the MT1-MMP molecules on the cell surface and activation of Cdc42 causes the same effect as RhoA inhibition which increases MT1-MMP cell surface location by increasing exocytosis of MT1-MMP from intracellular stores to the plasma membrane [29]. To further confirm the interaction between RhoA/Cdc42 complex, MT1-MMP and podoplanin during tumor invasion, co-precipitation experiments were performed. Both the co-precipitation of podoplanin with MT1-MMP and the podoplanin-induced specific binding of MT1-MMP to activated Cdc42 were found, while no co-association of podoplanin with Cdc42, podoplanin with RhoA or RhoA with MT1-MMP was found. Furthermore, immunofluorescence images revealed that the co-location of podoplanin, MT1-MMP and Cdc42 expression at the plasma membrane and filopodia induced an increase in cellular protrusion and stress fibers formation. Moreover, MT1-MMP inhibition could partly rescue the increase of Cdc42 activity caused by forced podoplanin expression although it seemed not to affect Cdc42 activity directly.
In summary, we demonstrated a hierarchy of crosstalk between RhoA and Cdc42 were involved in podoplanin-mediated cell motility and the co-location and co-ordination of podoplanin, Cdc42 and MT1-MMP in the invadopodia might induce cytoskeleton remodeling, ECM degradation and tumor invasion during OSCC progression. In addition, a significant positive correlation between podoplanin and MT1-MMP expression in OSCCs was found both in vivo and in vitro, which also indicated a shorter survival for OSCC patients.
Acknowledgements
This study was supported by the National Nature Science Foundation of China (No. 30973338).
Disclosure of conflict of interest
None.
References
- 1.Warnakulasuriya S. Global epidemiology of oral and oropharyngeal cancer. Oral Oncol. 2009;45:309–316. doi: 10.1016/j.oraloncology.2008.06.002. [DOI] [PubMed] [Google Scholar]
- 2.Kreppel M, Drebber U, Eich HT, Dreiseidler T, Zoller JE, Muller RP, Scheer M. Combined-modality treatment in advanced oral squamous cell carcinoma: Primary surgery followed by adjuvant concomitant radiochemotherapy. Strahlenther Onkol. 2011;187:555–560. doi: 10.1007/s00066-010-2245-8. [DOI] [PubMed] [Google Scholar]
- 3.Pena Murillo C, Huang X, Hills A, McGurk M, Lyons A, Jeannon JP, Odell E, Brown A, Lavery K, Barrett W, Sherriff M, Brakenhoff R, Partridge M. The utility of molecular diagnostics to predict recurrence of head and neck carcinoma. Br J Cancer. 2012;107:1138–1143. doi: 10.1038/bjc.2012.213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Breiteneder-Geleff S, Soleiman A, Kowalski H, Horvat R, Amann G, Kriehuber E, Diem K, Weninger W, Tschachler E, Alitalo K, Kerjaschki D. Angiosarcomas express mixed endothelial phenotypes of blood and lymphatic capillaries: podoplanin as a specific marker for lymphatic endothelium. Am J Pathol. 1999;154:385–394. doi: 10.1016/S0002-9440(10)65285-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Erovic BM, Neuchrist C, Kandutsch S, Woegerbauer M, Pammer J. CD9 expression on lymphatic vessels in head and neck mucosa. Mod Pathol. 2003;16:1028–1034. doi: 10.1097/01.MP.0000089777.58000.B2. [DOI] [PubMed] [Google Scholar]
- 6.Rodrigo JP, Garcia-Carracedo D, Gonzalez MV, Mancebo G, Fresno MF, Garcia-Pedrero J. Podoplanin expression in the development and progression of laryngeal squamous cell carcinomas. Mol Cancer. 2010;9:48. doi: 10.1186/1476-4598-9-48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dumoff KL, Chu CS, Harris EE, Holtz D, Xu X, Zhang PJ, Acs G. Low podoplanin expression in pretreatment biopsy material predicts poor prognosis in advanced-stage squamous cell carcinoma of the uterine cervix treated by primary radiation. Mod Pathol. 2006;19:708–716. doi: 10.1038/modpathol.3800580. [DOI] [PubMed] [Google Scholar]
- 8.Rahadiani N, Ikeda J, Makino T, Tian T, Qiu Y, Mamat S, Wang Y, Doki Y, Aozasa K, Morii E. Tumorigenic role of podoplanin in esophageal squamous-cell carcinoma. Ann Surg Oncol. 2010;17:1311–1323. doi: 10.1245/s10434-009-0895-5. [DOI] [PubMed] [Google Scholar]
- 9.Kato Y, Fujita N, Kunita A, Sato S, Kaneko M, Osawa M, Tsuruo T. Molecular identification of Aggrus/T1alpha as a platelet aggregation-inducing factor expressed in colorectal tumors. J Biol Chem. 2003;278:51599–51605. doi: 10.1074/jbc.M309935200. [DOI] [PubMed] [Google Scholar]
- 10.Schacht V, Dadras SS, Johnson LA, Jackson DG, Hong YK, Detmar M. Up-regulation of the lymphatic marker podoplanin, a mucin-type transmembrane glycoprotein, in human squamous cell carcinomas and germ cell tumors. Am J Pathol. 2005;166:913–921. doi: 10.1016/S0002-9440(10)62311-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Friedl P, Wolf K. Tumour-cell invasion and migration: diversity and escape mechanisms. Nat Rev Cancer. 2003;3:362–374. doi: 10.1038/nrc1075. [DOI] [PubMed] [Google Scholar]
- 12.Martin-Villar E, Scholl FG, Gamallo C, Yurrita MM, Munoz-Guerra M, Cruces J, Quintanilla M. Characterization of human PA2.26 antigen (T1alpha-2, podoplanin), a small membrane mucin induced in oral squamous cell carcinomas. Int J Cancer. 2005;113:899–910. doi: 10.1002/ijc.20656. [DOI] [PubMed] [Google Scholar]
- 13.Wicki A, Christofori G. The potential role of podoplanin in tumour invasion. Br J Cancer. 2007;96:1–5. doi: 10.1038/sj.bjc.6603518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Linder S. The matrix corroded: podosomes and invadopodia in extracellular matrix degradation. Trends Cell Biol. 2007;17:107–117. doi: 10.1016/j.tcb.2007.01.002. [DOI] [PubMed] [Google Scholar]
- 15.Sakurai-Yageta M, Recchi C, Le Dez G, Sibarita JB, Daviet L, Camonis J, D’Souza-Schorey C, Chavrier P. The interaction of IQGAP1 with the exocyst complex is required for tumor cell invasion downstream of Cdc42 and RhoA. J Cell Biol. 2008;181:985–998. doi: 10.1083/jcb.200709076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wicki A, Lehembre F, Wick N, Hantusch B, Kerjaschki D, Christofori G. Tumor invasion in the absence of epithelial-mesenchymal transition: podoplanin-mediated remodeling of the actin cytoskeleton. Cancer Cell. 2006;9:261–272. doi: 10.1016/j.ccr.2006.03.010. [DOI] [PubMed] [Google Scholar]
- 17.Weaver AM. Invadopodia: specialized cell structures for cancer invasion. Clin Exp Metastasis. 2006;23:97–105. doi: 10.1007/s10585-006-9014-1. [DOI] [PubMed] [Google Scholar]
- 18.Mori H, Tomari T, Koshikawa N, Kajita M, Itoh Y, Sato H, Tojo H, Yana I, Seiki M. CD44 directs membrane-type 1 matrix metalloproteinase to lamellipodia by associating with its hemopexin-like domain. EMBO J. 2002;21:3949–3959. doi: 10.1093/emboj/cdf411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Itoh Y, Palmisano R, Anilkumar N, Nagase H, Miyawaki A, Seiki M. Dimerization of MT1-MMP during cellular invasion detected by fluorescence resonance energy transfer. Biochem J. 2011;440:319–326. doi: 10.1042/BJ20110424. [DOI] [PubMed] [Google Scholar]
- 20.Krisanaprakornkit S, Iamaroon A. Epithelial-mesenchymal transition in oral squamous cell carcinoma. ISRN Oncol. 2012;2012:681469. doi: 10.5402/2012/681469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hall A. Rho GTPases and the control of cell behaviour. Biochem Soc Trans. 2005;33:891–895. doi: 10.1042/BST20050891. [DOI] [PubMed] [Google Scholar]
- 22.Ridley AJ. Rho GTPases and cell migration. J Cell Sci. 2001;114:2713–2722. doi: 10.1242/jcs.114.15.2713. [DOI] [PubMed] [Google Scholar]
- 23.Fisher KE, Sacharidou A, Stratman AN, Mayo AM, Fisher SB, Mahan RD, Davis MJ, Davis GE. MT1-MMP- and Cdc42-dependent signaling co-regulate cell invasion and tunnel formation in 3D collagen matrices. J Cell Sci. 2009;122:4558–4569. doi: 10.1242/jcs.050724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yilmaz M, Christofori G, Lehembre F. Distinct mechanisms of tumor invasion and metastasis. Trends Mol Med. 2007;13:535–541. doi: 10.1016/j.molmed.2007.10.004. [DOI] [PubMed] [Google Scholar]
- 25.Nakashima Y, Yoshinaga K, Kitao H, Ando K, Kimura Y, Saeki H, Oki E, Morita M, Kakeji Y, Hirahashi M, Oda Y, Maehara Y. Podoplanin is expressed at the invasive front of esophageal squamous cell carcinomas and is involved in collective cell invasion. Cancer Sci. 2013;104:1718–1725. doi: 10.1111/cas.12286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yamazaki D, Kurisu S, Takenawa T. Regulation of cancer cell motility through actin reorganization. Cancer Sci. 2005;96:379–386. doi: 10.1111/j.1349-7006.2005.00062.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sahai E, Marshall CJ. RHO-GTPases and cancer. Nat Rev Cancer. 2002;2:133–142. doi: 10.1038/nrc725. [DOI] [PubMed] [Google Scholar]
- 28.Jeong KJ, Park SY, Cho KH, Sohn JS, Lee J, Kim YK, Kang J, Park CG, Han JW, Lee HY. The Rho/ROCK pathway for lysophosphatidic acid-induced proteolytic enzyme expression and ovarian cancer cell invasion. Oncogene. 2012;31:4279–4289. doi: 10.1038/onc.2011.595. [DOI] [PubMed] [Google Scholar]
- 29.Ispanovic E, Serio D, Haas TL. Cdc42 and RhoA have opposing roles in regulating membrane type 1-matrix metalloproteinase localization and matrix metalloproteinase-2 activation. Am J Physiol Cell Physiol. 2008;295:C600–610. doi: 10.1152/ajpcell.00460.2007. [DOI] [PubMed] [Google Scholar]
- 30.El-Sibai M, Pertz O, Pang H, Yip SC, Lorenz M, Symons M, Condeelis JS, Hahn KM, Backer JM. RhoA/ROCK-mediated switching between Cdc42- and Rac1-dependent protrusion in MTLn3 carcinoma cells. Exp Cell Res. 2008;314:1540–1552. doi: 10.1016/j.yexcr.2008.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Berrier AL, Yamada KM. Cell-matrix adhesion. J Cell Physiol. 2007;213:565–573. doi: 10.1002/jcp.21237. [DOI] [PubMed] [Google Scholar]
- 32.Egeblad M, Werb Z. New functions for the matrix metalloproteinases in cancer progression. Nat Rev Cancer. 2002;2:161–174. doi: 10.1038/nrc745. [DOI] [PubMed] [Google Scholar]