

Abstract
Acute promyelocytic leukemia (APL) results from a blockade of granulocyte differentiation during the promyelocytic stage. As a fusion protein of promyelocytic leukemia (PML) and retinoic acid receptor-α (RARα), PML-RARα oncoprotein is degraded through the differentiation of all-trans retinoic acid (ATRA)-induced cells. Here reactive oxygen species (ROS) and high-mobility group box 1 (HMGB1) were proven essential for the differentiation of APL cells. A down-regulation of ROS by ROS quencher (NAC) blocked the differentiation of APL cell line NB4 while an over-expression of ROS by superoxide dismutase-1 (SOD1) RNA interference (RNAi) increased cell differentiation. HMGB1 was vital for the differentiation of ROS-mediated NB4 cells and its up-regulation promoted ATRA-induced autophagy and the degradation of PML-RARα. Furthermore, ATRA treatment elevated the levels of ROS, enhanced autophagic flux and thereby promoted cytosolic translocation of HMGB1. HMGB1 regulated the interactions between ubiquitin-binding adaptor protein p62/SQSTM and PML-RARα so as to affect the degradation of PML-RARα during ATRA-induced autophagy. Also a depletion of p62/SQSTM1 expression inhibited HMGB1-mediated PML-RARα degradation and cell differentiation. The overall results suggested that HMGB1 is an essential regulator of ROS-induced cell differentiation. And it may become a potential drug target for therapeutic intervention of APL.
Keywords: HMGB1, ROS, cell differentiation, autophagy
Introduction
As a subtype of acute myeloid leukemia (AML), acute promyelocytic leukemia (APL) was characterized by expansion and accumulation of leukemia cells during the promyelocytic stage of myelopoiesis [1,2]. The distinguishing feature of APL is the presence of a balanced reciprocal translocation involving RARα gene located on chromosome 17q21. In 95% of cases, RARα was conjugated with PML gene on chromosome 15q22 leading to PML/RARα gene fusion [3]. All-trans retinoic acid (ATRA), the first of two drugs capable of causing disease regression specifically in APL patients, induced a degradation of PML-RARα oncogene and a differentiation of leukemia cells and even clinical remission [4]. The capability of ATRA of activating disease regression through distinct molecular mechanisms and leading to PML/RARα degradation has been further underscored by recent studies. Thus a thorough understanding of ATRA activating the degradation of PML/RARα appears to represent a critical link for successful APL treatment.
As demonstrated by recent studies, the major mechanism of PML/RARα degradation included the activities of both proteasome and caspase pathways, although neither is sufficient for complete degradation [5]. Macroautophagy (hereinafter referred to as autophagy) is lysosome-mediated degradation of cytoplasmic constituents such as proteins and organelles, thus leading to cell renovation [6,7]. Playing important roles in bacterial, viral infections and neurodegenerative disorders, this cellular process is also essential for regulating cancer initiation and progression and determining the response of tumor cells to anticancer therapy [8-10]. It was also reported that autophagy could be activated by chemotherapeutic drugs in leukemia cell lines and contributed to therapy-induced degradation of PML/RARα [11]. However the underlying mechanisms have remained elusive. Previous studies have shown that ATRA-induced autophagy enhanced the differentiation of myeloid cells through p62/SQSTM1-mediated degradation of PML/RARα [12]. This degradation pathway may provide a new therapeutic option for APL.
As an evolutionarily conserved non-histone chromatin-binding protein, high-mobility group box 1 (HMGB1) plays significant roles in inflammatory diseases and cancer [13,14]. Under normal conditions, 90-95% of HMGB1 is located in nucleus where it serves as a DNA chaperone for maintaining nuclear homeostasis. When oxidative stress and ROS pathways are initiated by multiple cellular stressors (e.g. protein aggregates, radiation, chemotherapy & intracellular pathogens), HMGB1 is translocated to express on cell surface membranes, inside cytosol or diffuse into extracellular space [13,15]. And the expression of HMGB1 is obviously higher in leukemia cells and bone marrow mononuclear cells (BMMCs) derived from patients with primary and relapsing leukemia. Meanwhile, HMGB1 acts as a positive regulator of autophagy so that it plays an important role in leukemia pathogenesis and chemotherapy resistance [16,17].
HMGB1 is expressed abundantly in leukemia cell lines. And it plays important roles in leukemic tumorigenesis. However its role in cell differentiation of APL has been ill-defined. Here the authors demonstrated that ROS could regulate the translocation and release of HMGB1 and promoted the differentiation of APL cell (NB4). Furthermore, HMGB1 was essential for ROS-mediated cell differentiation. And the mechanism was partially due to p62/SQSTM1-mediated degradation of PML/RARα by HMGB1-mediated autophagy. It suggested that HMGB1 was a potential regulator of APL cell differentiation.
Material and methods
Reagents and antibodies
The antibodies to SOD1, actin and fibrillarin were purchased from Abcam (Cambridge, MA, USA); antibodies to HMGB1 from Cell Signaling Technology (Danvers, MA, USA) and Sigma (St Louis, MO, USA); antibodies to p62 and RARα from Santa Cruz Biotechnology (Santa Cruz, CA, USA); antibody to LC3 from Sigma (St Louis, MO, USA); antibody to CD11b from Invitrogen (Carlsbad, CA, USA); N-acetylcysteine (NAC), 3-methyladenine (3-MA) and ATRA from Sigma (St Louis, MO, USA).
Cell culture
As the sole genuine promyelocytic leukemia cell line, NB4 offer a unique in vitro model system for studying the cellular and molecular events involved in the proliferation and differentiation of normal and leukemic myelomonocytic cells [18]. NB4 cells (Xiangya School of Medicine Type Culture Collection, China) were cultured in RPMI 1640 with 10% heat-inactivated fetal bovine serum (FBS; Life Technologies, Grand Island, NY, USA) in 5% CO2 and 95% ambient air.
Gene transfection and RNAi
Transfection with human pEGFP-N1-HMGB1 (HMGB1 vectors) and HMGB1 shRNA vectors (a gift from Dr. Kang Rui, University of Pittsburgh, USA) and P62 shRNA, Atg5, SOD1 shRNA and control shRNA from Sigma (St Louis, MO, USA) were constructed with FuGENE HD Transfection Reagent (Roche Applied Science, Stockholm, Sweden) according to the manufacturer’s instructions.
Western blot
Cells were washed in phosphate buffer solution (PBS), collected, resuspended in lysis buffer (Beyotime, Beijing, China) and maintained on ice for 15 min. Cell extracts were cleared by microcentrifugation at 14,000 g for 30 min at 4°C. The whole cell lysate was separated by 8% (10%/12%) sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and electrophoretically transferred onto polyvinylidene difluoride (PVDF) blotting membrane (Beyotime, Beijing, China). The membrane was blocked with 5% non-fat dry milk in TBST (50 mM Tris pH 7.5, 100 mM NaCl, 0.15% Tween-20), incubated with diluted primary antibodies for 12 h at 4°C and washed thrice with TBST for 10 min. Then the membranes were incubated for 12 h at 4°C with different secondary antibodies and detected with enhanced chemiluminescence (ECL) reagents (Pierce, USA) after three rinses with TBST for 10 min. Membranes were exposed to X-ray film and the expressions of targeted proteins were quantified by detecting the specific bands on X-ray film. The BandScan 5.0 system was used to quantify and analyze each specific band of Western blot [15].
Immunoprecipitation
Cells were lysed at 4°C in ice-cold lysis buffer (50 mM Tris-HCl, Ph 7.4, containing 150 mM NaCl, 1% NP-40, 0.5% Na-deoxycholate, 0.1% SDS, protease inhibitor cocktail) and cell lysates were centrifuged (12 000 g , 10 min). The concentrations of proteins in supernatant were determined by bicinchoninic acid (BCA) assay. Prior to immunoprecipitation, samples containing equal amounts of proteins were pre-cleared with protein A or protein G agarose/sepharose (Santa Cruz, CA, USA) (4°C, 3 h) and subsequently incubated with various irrelevant IgG or specific antibodies (5 mg/mL) in the presence of protein A or G agarose/sepharose beads for 2 h or overnight at 4°C with gentle vortexing. After incubation, agarose/sepharose beads were rinsed thoroughly with PBS. And the proteins were eluted by boiling in 2 × SDS sample buffer prior to SDS-PAGE.
Immunocytochemistry
Cells were fixed in 4% formaldehyde for 30 min at room temperature before permeabilizing with 0.1% Triton X-100 (4°C, 10 min). Cells were saturated with PBS containing 2% BSA for 1 h at room temperature and processed for immunofluorescence with anti-HMGB1 antibody followed by Cy3 Ig (Cambridge, MA, USA). Between all incubation steps, cells were washed thrice for 3 min with PBS containing 0.2% BSA. Fluorescence signals were analyzed with fluorescence microscope (Olympus, Tokyo, Japan).
Determination of ROS generation
The intracellular alterations of reactive oxygen species (ROS) were determined by measuring the oxidative conversion of cell-permeable 2’, 7’-dichlorofluorescein diacetate (DCFH-DA) into fluorescent dichlorofluorescein (DCF) on a fluorospectrophotometer (F4000, Japan). In brief, NB4 cells with different treatments were collected, rinsed with D-Hank’s buffer and incubated with DCFH-DA at 37°C for 20 min. Then the DCF fluorescence of 20,000 cells was detected by fluorospectrophotometer at an excitation wavelength of 488 nm and an emission wavelength of 535 nm. The incremental production of ROS was expressed as a percentage of control [19].
Measurements of HMGB1 release
NB4 cells with different treatments (NAC pretreatment or SOD1 shRNA transfection) were cultured with ATRA for 24, 48 and 72 h. The release of HMGB1 into cell culture supernatants was evaluated with ELISA kits from Shino-Test Corporation (Sagamihara-shi, Kanagawa, Japan) according to the manufacturer’s instructions.
Electron microscopy
NB4 cells were collected and fixed in 2.5% glutaraldehyde for at least 3 h. Then the cells were treated with 2% paraformaldehyde at room temperature for 60 min, 0.1% glutaraldehyde in 0.1 M sodium cacodylate for 2 h post-fixed with 1% OsO4 for 1.5 h, after a second washing, dehydrated with graded acetone and finally embedded in Quetol 812. Ultrathin sections were observed under Hitachi H7500 electron microscope (Tokyo, Japan).
Morphological evaluations of differentiation
NB4 cells with different treatments were cultured with ATRA for 48 h. Then the cells were resuspended in PBS after rinsing and fixed on slide. Differentiation of NB4 cells was determined by morphological observations after staining with Wright-Giemsa staining solution.
Measurement of CD11b expression
Cell surface differentiation antigen (CD11b) was measured by flow cytometry. Briefly, NB4 cells with different treatments were cultured with ATRA for 24, 48 and 72 h. Cells were incubated with antibody against CD11b for 30 min at room temperature. IgG was used as an isotype control for calibrating threshold parameters. Finally, the cells were resuspended in PBS after rinsing and assayed with flow cytometry.
Nitroblue tetrazolium (NBT) reduction assay
NB4 cells with different treatments were cultured with ATRA for 24, 48 and 72 h. Then each aliquot of cell suspension was mixed with an equal volume of RPMI-1640 medium containing 0.2% NBT (St Louis, MO, USA) and 2 μg/mL 12-O-tetradecanoylphorbol-13-acetate (TPA) and incubated in darkness for 30 min at 37°C. Cells were seeded in 96-well plate with 100 μL of dimethyl sulfoxide (DMSO). After gentle vortexing for 20 min, the 96-well plates were measured for absorbance at 570 nm (A570) on a Thermo Scientific Multiskan Ascent.
Statistical analysis
Quantitative data were presented as means ± standard deviation. Data were analyzed with indicated statistical methods using GraphPad Prism (Version 5.04). For calculation of the P value, parameters of two-tailed, 95% confidence interval were used for all analysis. A P-value < 0.05 was considered significant.
Results
ROS regulates the differentiation of NB4 cells
Involved in intracellular signal transduction and gene expression as an important signaling molecule in cells, ROS play important roles in cell proliferation, differentiation and death [20]. ROS production might be induced by multiple stimuli, such as nutrient starvation, mitochondrial toxins, hypoxia and chemotherapeutics [21]. As a protective strategy of cells or tissues against oxidative stress, superoxide dismutase (SOD) family members, catalase or NAC neutralize ROS under physiological conditions [22]. ATRA is a widely used differentiating agent. For determining the role of ROS regulating the differentiation of NB4 cells, the authors firstly examined whether ATRA at therapeutic doses (1 μM) induced ROS production in NB4 cells. In the present study, it was found that a treatment of ATRA induced ROS production whereas a pretreatment with NAC decreased ROS production in NB4 cells. In contrast, a knockdown of SOD1 by RNAi increased ATRA-induced ROS production (Figure 1A), suggesting that ATRA was valid stimulus for ROS production in NB4 cells.
Figure 1.
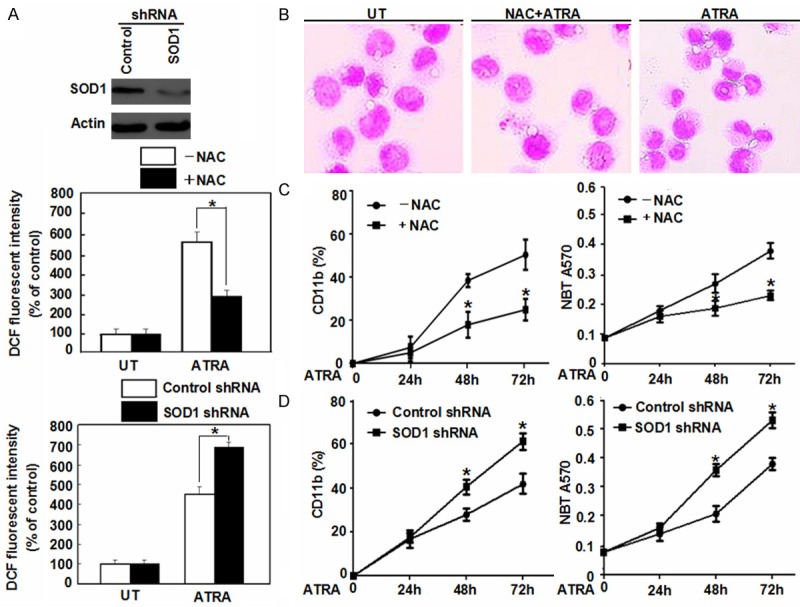
Effects of ROS on the differentiation of NB4 cells. A. NB4 cells were pretreated NAC (2 mM) or SOD1 RNAi and then ATRA (1 μM) for 72 h. ROS production was assessed by measuring the fluorescent intensity of DCF on a fluorescence plate reader. The incremental production of ROS was expressed as a percentage of control. *P < 0.05 (n = 4, Student’s t-test). UT, untreated group. B. Morphological features of NB4 cells as indicated after a 72 h ATRA (1 μM) treatment with or without NAC (2 mM) pretreatment by Wright-Giemsa stain (100 × magnification). C. NB4 cells were treated with ATRA (1 μM) for 24-72 h with or without NAC (2 mM) pretreatment. And then the cell differentiation was observed by CD11b expression and NBT reduction. *P < 0.05 (n = 3, Student’s t-test). D. After a 48 h transfection with SOD1 shRNA, the cells were treated with ATRA (1 μM) for 24-72 h. And then cell differentiation was observed by CD11b expression and NBT reduction. *P < 0.05 (n = 3, Student’s t-test).
For determining the regulation of ROS on the differentiation of APL cells, NB4 cells were pretreated with NAC and cell differentiation was judged by morphological observations. Co-treatment of NAC and ATRA for 72 h inhibited the level of cell maturation and was comparable to that seen with ATRA alone (Figure 1B). Furthermore, a pretreatment of NAC reduced both the ATRA-induced expression of cell differentiation marker CD11b by flow cytometry and functional differentiation by NBT reduction assay (Figure 1C). In contrast, a depletion of SOD1 expression increased ATRA-induced CD11b expression and NBT reduction (Figure 1D). These data suggested that elevated ROS level is essential for ATRA-induced differentiation of APL cells.
HMGB1 is essential for ROS-mediated cell differentiation
HMGB1 serves some important functions in a variety of biological processes, ranging from gene transcription, DNA repair, cell differentiation to tumor progression [13]. For determining whether HMGB1 is a direct activator of ROS-mediated cell differentiation, NB4 cells were treated with HMGB1 gene transfection and RNAi. After transfection, the NB4 cells showed an obvious increase of cell differentiation while a co-treatment of NAC failed to completely block this increase of cell maturation compared with the control group as judged by morphological observations, CD11b expression and NBT reduction assay (Figure 2A). On the other hand, the suppressed expression of HMGB1 by RNAi inhibited ATRA-induced cell differentiation and a depletion of SOD1 expression did not reverse HMGB1 RNAi-induced decrease of cell maturation (Figure 2B), suggesting a critical role for HMGB1 as a direct regulator of cell differentiation.
Figure 2.
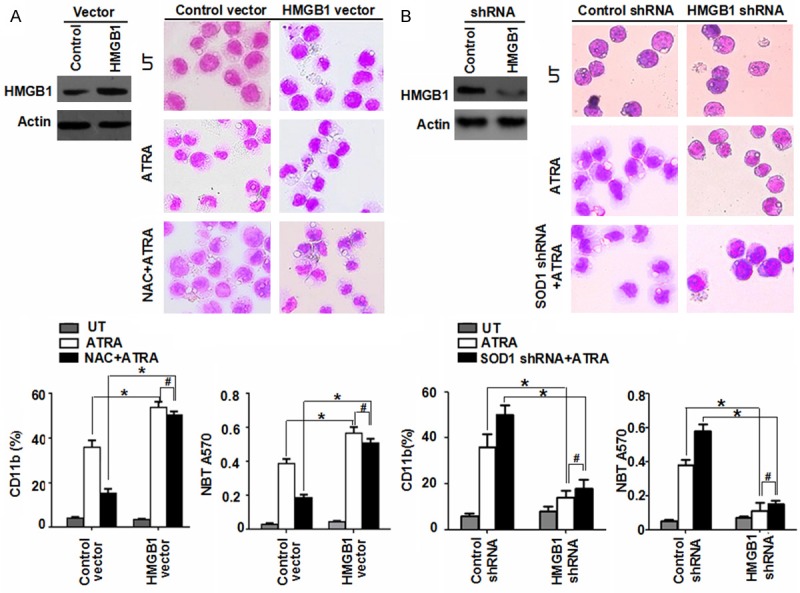
HMGB1 is essential for ROS-mediated cell differentiation. A. NB4 cells with HMGB1 transfection were treated with ATRA (1 μM) for 72 h with or without NAC (2 mM) pretreatment. Then cell differentiation was observed by cytomorphology (100 × magnification), CD11b expression and NBT reduction. *P < 0.05, #P > 0.05 (n = 3, Student’s t-test). UT, untreated group. B. A knockdown HMGB1 by RNAi in NB4 cells and then ATRA (1 μM) for 72 h with or without SOD1 depletion. And cell differentiation was observed by cytomorphology (100 × magnification), CD11b expression and NBT reduction. *P < 0.05, #P > 0.05 (n = 3, Student’s t-test). UT, untreated group.
HMGB1-induced autophagy promoted the degradation of PML/RARα in NB4 cells
As a direct molecular target of ATRA in human myeloid cells, PML/RARα oncoprotein mediated differentiation. PML-RARα degradation is not only mediated by the proteasome and caspase pathways, but also through autophagy [5,11]. During autophagy, LC3 is processed post-translationally into soluble LC3-I and subsequently converted into membrane-bound LC3-II correlated with the number of autophagosomes [23]. For determining whether HMGB1 is a direct activator of autophagic flux, LC3 conversion (LC3-I to LC3-II) was detected by immunoblotting. Similar to classical autophagic stimuli (e.g. rapamycin), ATRA treatment triggered an induction of LC3-II (Figure 3A). Transfection with HMGB1 plasmid and with ATRA for 72 h significantly increased the expression of LC3-II in NB4 cells (Figure 3A). Furthermore, the authors evaluated the expression of SQSTM1/sequestosome 1 (p62), a long-lived scaffolding protein bound to LC3 serving as a selective substrate of autophagy [24]. Up-regulated HMGB1 expression increased LC3-II protein levels, but decreased the expression of p62 compared with the control group (Figure 3A). Moreover, LC3 and p62 accumulation after an over-expression of HMGB1 was exaggerated in leukemia cells after treatments with lysosomal protease inhibitor E64d and pepstatin A (Figure 3A). These data suggested that elevated LC3-II was not due to a decreased degradation of lapidated LC3, but rather an increased autophagic flux. Meanwhile, HMGB1-promoted autophagy was substantiated by electron microscopy, the most convincing and standard method of detecting autophagy [23]. NB4 cells treated with HMGB1 gene transfection plus ATRA for 72 h exhibited more autophagosomes when compared with the vector group (Figure 3B).
Figure 3.
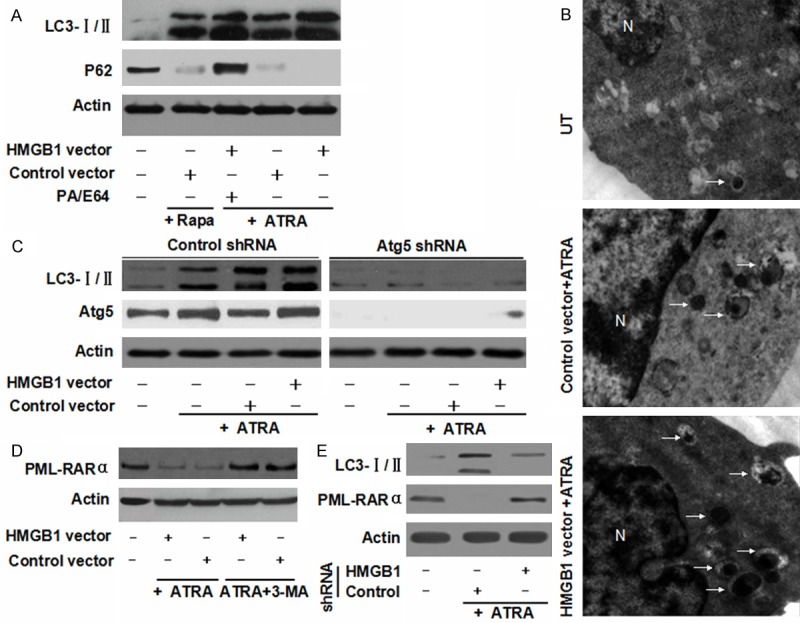
HMGB1-induced autophagy promoted the degradation of PML/RARα in NB4 cells. A. NB4 cells were pretreated with HMGB1 transfection and then treated with ATRA (1 μM) or rapamycin (Rapa, 100 nM) for 72 h with or without pepstatin A (PA, 10 μM) and E64D (10 μM) pretreatment. And the levels of LC3-I/II and p62 were assayed by Western blot; B. Ultrastructural features with HMGB1 transfection for ATRA (1 μM) treatment. The number of visible autophagosomes treated with HMGB1 transfection was higher compared with the control group (20,000 × magnification). White arrows, autophagosomes; N, nuclear; UT, untreated group. C. After a 48 h transfection of Atg5 shRNA, the indicated cells were treated with ATRA (1 μM) for 48 h with or without HMGB1 transfection. And the level of LC3-I/II level was assayed by Western blot; D. NB4 cells with HMGB1 transfection were treated with ATRA (1 μM) for 72 h with or without 3-methyladenine (3-MA, 10 mM) pretreatment. And PML-RARα was assayed by Western blot; E. NB4 cells with HMGB1 shRNA transfection were treated with ATRA (1 μM) for 72 h. And the levels of PML-RARα and LC3-I/II were assayed by Western blot.
Autophagy-related (Atg) gene products are the major proteins involved in the process of autophagy. Atg5 is an autophagy-regulatory gene essential for ATRA-induced autophagy in NB4 cells [12,25]. To further characterizing the role of HMGB1 regulated autophagy in NB4 cell, a target-specific shRNA against Atg5 gene was used. A depletion of Atg5 expression in NB4 cells inhibited ATRA-induced LC3 conversion despite HMGB1 gene transfection (Figure 3C). Moreover, it was found that HMGB1 gene transfection with ATRA treatment induced a degradation of PML-RARα in NB4 cells (Figure 3D). Autophagy inhibitor (e.g., 3-MA) significantly blocked ATRA-induced degradation of PML-RARα (Figure 3D). Consistent with this finding, a knockdown of HMGB1 decreased the degradation of PML-RARα and the conversion of LC3 (Figure 3E). These data suggested that PML-RARα degradation was mediated by HMGB1-induced autophagy.
ROS are essential for HMGB1 translocation and enhanced autophagy
Autophagy is an intracellular lysosomal degradation process induced under stress conditions. ROS regulate autophagy in cell survival, death, development and many human diseases [26]. To evaluate the relationship between ROS and autophagy in NB4 cells, LC3 conversion (LC3-I to LC3-II) were detected at different ROS levels. A knockdown of SOD1 expression by RNAi increased ATRA-induced LC3 conversion (Figure 4A). In contrast, NAC inhibited ATRA-induced LC3-II expression (Figure 4A), suggested that ROS regulated the autophagy of NB4 cells.
Figure 4.
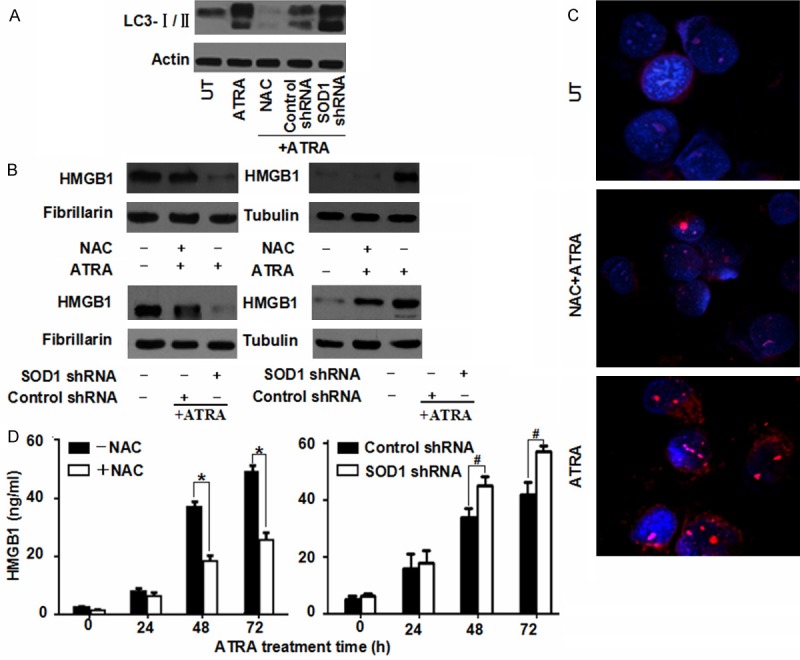
ROS are required for HMGB1 translocation and enhanced autophagy. A. NB4 cells were pretreated NAC (2 mM) or SOD1 RNAi and then treated with ATRA (1 μM) for 72 h. And the level of LC3-I/II was assayed by Western blot. UT, untreated group. B. NB4 cells were pretreated with NAC (2 mM) or SOD1 RNAi and then ATRA (1 μM) for 72 h. And the nuclear/cytosolic HMGB1 expression was assayed by Western blot; C. NB4 cells were treated with ATRA (1 μM) for 72 h with or without NAC (2 mM) pretreatment and then immunostained with HMGB1-specifc antibody (red) and Hoechst 33342 (blue) (400 × magnification). D. NB4 cells were pretreated with NAC (2 mM) or SOD1 RNAi and then ATRA (1 μM) for 24-72 h. And the release of HMGB1 was analyzed by ELISA. Cell viability of control was set as 100%. *P < 0.05, #P < 0.05 (n = 4, Student’s t-test).
HMGB1 protein is both a nuclear DNA binding factor and a secreted protein. And its activities are determined by its intracellular localization and posttranslational modifications [27]. Chemotherapeutics promoted a translocation of HMGB1 from nucleus into cytosol and resulted in enhanced autophagy after sustained cellular stress [15]. To evaluating whether HMGB1 translocation was regulated by ROS during ATRA-induced autophagy, the authors pretreated NB4 cells with either NAC incubation or SOD1 RNAi treatment. It was found that HMGB1 was predominantly located in nucleus under normal conditions, whereas it was noticeably high in cytosol under ATRA stimuli (Figure 4B). A pretreatment of NAC in NB4 cells blocked HMGB1 translocation from nucleus into cytosol (Figure 4B, 4C). In contrast, a knockdown of SOD1 expression increased HMGB1 translocation (Figure 4B). Consistent with this finding, a pretreatment of NAC inhibited HMGB1 release into supernatant with ATRA incubation while a down-regulation of SOD1 by RNAi increased HMGB1 release (Figure 4D). The overall results suggested that ROS are required for HMGB1 translocation and sustained autophagy in APL cells.
HMGB1-mediated interaction between p62 and PML-RARα regulated the differentiation of NB4 cells
Autophagic degradation of polyubiquitinated protein aggregates is important for cell survival. And p62 has been shown to be an autophagy receptor acting as a link between ubiquitination and autophagic machinery [24]. Previous studies have demonstrated that PML-RARα is a polyubiquitinated protein. Under basal conditions, PML-RARα co-immunoprecipitated with p62 and became degraded through autophagy [5,12]. To characterizing the mechanism of HMGB1-induced autophagy regulating the differentiation of NB4 cells, the authors firstly explored whether HMGB1 regulated the interaction between p62 and PML-RARα under ATRA treatment by co-immunoprecipitation (Co-IP). It was found that, under ATRA treatment, P62 and PML-RARα co-immunoprecipitated with each other in control group and this interaction was significantly blocked by HMGB1 RNAi (Figure 5A), suggesting a regulatory role of HMGB1 in regulation for p62/PML-RARα complex.
Figure 5.
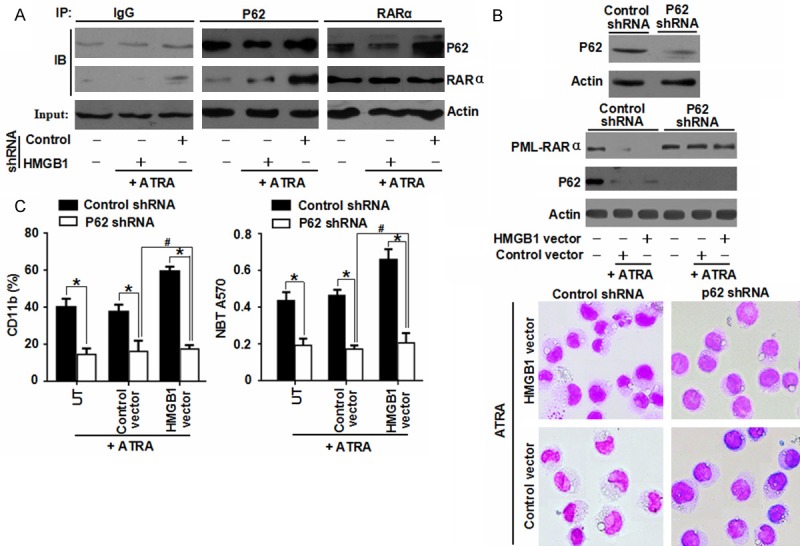
HMGB1-mediated interaction between p62 and PML-RARα regulated the differentiation of NB4 cells. A. NB4 cells with HMGB1 shRNA transfection were treated with ATRA (1 μM) for 72 h. And the protein expression levels were assayed by Co-IP or Western blot; B, C. After a 48 h transfection with p62 shRNA, NB4 cells were treated with ATRA (1 μM) for 72 h with or without HMGB1 transfection. And then the level of PML-RARα was assayed by Western blot. In parallel, cell differentiation was observed by cytomorphology, CD11b expression and NBT reduction. *P < 0.05, #P > 0.05 (n = 3, Student’s t-test).
Furthermore, for determining the potential regulatory role for p62/PML-RARα complex in cell differentiation, the target-specific shRNA against P62 and HMGB1 plasmid were transfected into NB4 cells. A knockdown of p62 reduced ATRA-induced PML-RARα protein degradation and cell maturation based on morphological assay, expression of myeloid differentiation marker CD11b and functional differentiation (Figure 5B, 5C). Moreover, an over-expression of HMGB1 did not promote PML-RARα protein degradation and cell differentiation after a depletion of P62 expression (Figure 5B, 5C). These data indicated that HMGB1 regulated the differentiation of NB4 cells through a p62/PML-RARα-dependent pathway.
Discussion
As a family of chemically active molecules containing free radicals, ROS are involved in modulating biological cell functions, cell signaling and homeostasis. In normal mammalian hematopoietic systems, there is a low level of ROS in hematopoietic stem cells (HSCs) during the regulation of stem cell pluripotency, proliferation and differentiation [28]. Leukemia cells are intrinsically under oxidative stress and thus more vulnerable to further stress. Major activated oncogenes, such as Bcr/Abl, Ras and c-myc, have been found to correlate with enhanced ROS production [29,30]. For identifying the genes associated with leukemia cell differentiation, Yang et al [31] reported that ectopic over-expressions of mda-7/IL-24 and IL-24 delE5 induced ROS production in leukemia cells, contributing to the differentiation of leukemia cells. The molecular mechanisms underlying ROS production and APL differentiation have been incompletely elucidated. In the present study, it was found that a novel function of ROS was regulating cell differentiation by HMGB1-mediated autophagy in APL cells. Also HMGB1 was found to be a direct mediator of P62/PML-RARα interaction. Therefore mechanistic insights might be gained into the role of HMGB1 in regulating cell differentiation.
HMGB1 protein is both a nuclear DNA binding factor and a secreted protein. And its activities are determined by its intracellular localization and posttranslational modifications. Endogenous over-expression of HMGB1, seen in many (if not all) tumor cells, accelerated cell cycle progression, but became down-regulated during aging, suggesting a critical role in development and cancer [32,33]. In leukemia cells, the expression of endogenous HMGB1 in myeloid cells was higher than that in lymphoid cells and correlated with the differentiation stage of these cells [34,35]. Exogenous HMGB1 is secreted by activated macrophages, mature dendritic cells and natural killer cells in responses to injury, infection or other inflammatory stimuli [36]. It was also released from leukemia cell lines after chemotherapy-induced cytotoxicity [10]. In chronic lymphocytic leukemia (CLL), CLL cells passively released HMGB1. And the timing and concentrations of HMGB1 in medium were associated with differentiation of nurse-like cells (NLCs) [37]. HMGB1-mediated NLC differentiation involved internalization of both receptor for advanced glycation end products (RAGE) and Toll-like receptor-9 (TLR9) [37]. However, the function of endogenous HMGB1 in response to leukemia cells was previously unknown. In the present study, an over-expression of HMGB1 in leukemia cells by gene transfection promoted the differentiation of NB4 cells. In contrast, suppressed expression of HMGB1 by RNA interference inhibited the maturation of NB4 cells. Nevertheless, HMGB1-mediated cell differentiation was not completely affected by the levels of ROS. It suggested a strong influence of endogenous HMGB1 on the differentiation of APL cells.
Furthermore, HMGB1-mediated autophagy regulated APL cell differentiation potentially via controlling the degradation of PML-RARα. PML-RARα is directly targeted and degraded by two effective therapeutic agents for APL, i.e. ATRA and arsenic trioxide (As2O3) [4]. The degradation of PML-RARα protein promoted the differentiation of APL cells. It was recently reported that both ATRA and As2O3 induced autophagy via the mammalian target of rapamycin (mTOR) pathway in APL cells. And autophagic degradation contributed significantly to basal turnover as well as therapy-induced proteolysis of PML-RARα protein [11]. Our previous studies have shown that endogenous HMGB1 acted as an intrinsic regulator of autophagy in leukemia cells through the mTOR pathway [16]. The present study has shed insights into the role of endogenous HMGB1 in regulating autophagy in leukemia cells. However, exact mechanisms regarding endogenous HMGB1-mediated autophagy in response to APL cell differentiation remain largely unknown. It was found that an over-expression of HMGB1 by gene transfection increased ATRA-induced NB4 cell autophagy and promoted the degradation of PML-RARα protein. In contrast, a knockdown of HMGB1 expression or pharmacological inhibition of autophagy (e.g. 3-MA) inhibited ATRA-induced PML-RARα protein degradation. These results suggested that endogenous HMGB1-mediated autophagy was important for APL cell differentiation.
ROS function as signaling molecules of regulating both cell survival and death through various pathways. Compared with normal cells, both ROS and autophagy are altered in cancer cells. On one hand, ROS could induce autophagy through several distinct mechanisms involving catalase activation of Atg4 and disturbances in mETC [38]. On the other hand, defective autophagy increased oxidative stress in tumor cells [39]. Here it was found that pharmacological inhibition of ROS production inhibited ATRA-induced autophagy, whereas a knockdown of SOD1 expression promoted ATRA-induced autophagy. It suggested that ROS signals were required for sustained autophagy in APL cells. Recently, Kang et al reported that HMGB1 was an autophagy sensor in the presence of oxidative stress [40]. Hydrogen peroxide (H2O2) and loss of SOD1-mediated oxidative stress promoted the cytosolic expression of HMGB1 and an extracellular release in fibroblasts and cancer cells [15]. Furthermore, NAC, a ROS quencher, dose-dependently inhibited starvation and rapamycin-induced autophagy and HMGB1 translocation [15]. It suggested that HMGB1 translocation was correlated well with cell autophagy. Our experimental data also suggested that ATRA-induced ROS generation might regulate HMGB1 translocation in APL cells. A pre-treatment of NAC led to a decrease of HMGB1 translocation and secretion while an increase with a depletion of SOD1 expression. These results suggested that ROS are essential for enhancing autophagy and HMGB1 translocation in the differentiation of APL cells.
Moreover, HMGB1-mediated APL cell differentiation operated potentially through p62/PML-RARα-dependent pathway. The current study revealed a direct link between autophagy and ATRA-induced degradation of PML-RARα in APL cell differentiation [12]. During autophagy, ubiquitin-binding protein p62/SQSTM1 was degraded [24]. Numerous studies have shown that the degradation of PML-RARα was degraded by the polyubiquitin/proteasome system through PML-RARα bound to P62 during ATRA-induced autophagy [5,12]. Based upon our current and previous evidences, HMGB1 was proven to act as a positive regulator of autophagy [16,17]. In the present study, the interaction between p62 and PML-RARα was regulated by HMGB1-mediated ATRA-induced autophagy so that the degradation of PML-RARα oncoprotein was affected. An inhibition of p62 impaired the degradation of PML-RARα during cell differentiation. It suggested that HMGB1-mediated autophagy played an important role in regulating the differentiation of APL cells by p62.
In summary, over-expressed ROS functioned as a positive regulator of cell differentiation in ATRA-treated NB4 cells. HMGB1 was essential for ROS-mediated cell differentiation. In ATRA-treated cell model, up-regulated HMGB1 expression promoted ATRA-induced autophagy and enhanced the degradation of PML-RARα oncoprotein. The ROS production with ATRA treatment enhanced autophagy and HMGB1 translocation in the differentiation of APL cells. The HMGB1-regulated interaction between p62 and PML-RARα affected the degradation of PML-RARα and cell differentiation. Therefore up-regulated HMGB1 expression may be used for augmenting APL cell differentiation and strengthening current anti-leukemic therapeutics.
Acknowledgements
This work was supported by grants from the National Natural Sciences Foundation of China (Grant No. 81270616, 31171328 and 81400138) and the Natural Science Foundation of Hunan Province of China (Grant No. 2015JJ6110).
Disclosure of conflict of interest
None.
References
- 1.Wang ZY, Chen Z. Acute promyelocytic leukemia: from highly fatal to highly curable. Blood. 2008;111:2505–2515. doi: 10.1182/blood-2007-07-102798. [DOI] [PubMed] [Google Scholar]
- 2.Degos L. The history of acute promyelocytic leukaemia. Br J Haematol. 2003;122:539–553. doi: 10.1046/j.1365-2141.2003.04460.x. [DOI] [PubMed] [Google Scholar]
- 3.Mistry AR, Pedersen EW, Solomon E, Grimwade D. The molecular pathogenesis of acute promyelocytic leukaemia: implications for the clinical management of the disease. Blood Reviews. 2003;17:71–97. doi: 10.1016/s0268-960x(02)00075-9. [DOI] [PubMed] [Google Scholar]
- 4.Nasr R, Lallemand-Breitenbach V, Zhu J, Guillemin MC, de The H. Therapy-induced PML/RARA Proteolysis and Acute Promyelocytic Leukemia Cure. Clin Cancer Res. 2009;15:6321–6326. doi: 10.1158/1078-0432.CCR-09-0209. [DOI] [PubMed] [Google Scholar]
- 5.Nasr R, Guillemin MC, Ferhi O, Soilihi H, Peres L, Berthier C, Rousselot P, Robledo-Sarmiento M, Lallemand-Breitenbach V, Gourmel B, Vitoux D, Pandolfi PP, Rochette-Egly C, Zhu J, De The H. Eradication of acute promyelocytic leukemia-initiating cells through PML-RARA degradation (vol 14, pg 1333, 2008) Nat Med. 2009;15:117–117. doi: 10.1038/nm.1891. [DOI] [PubMed] [Google Scholar]
- 6.Klionsky DJ, Emr SD. Cell biology - Autophagy as a regulated pathway of cellular degradation. Science. 2000;290:1717–1721. doi: 10.1126/science.290.5497.1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Levine B, Kroemer G. Autophagy in the pathogenesis of disease. Cell. 2008;132:27–42. doi: 10.1016/j.cell.2007.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Livesey KM, Tang DL, Zeh HJ, Lotze MT. Autophagy inhibition in combination cancer treatment. Curr Opin Investig Drugs. 2009;10:1269–1279. [PubMed] [Google Scholar]
- 9.Levine B, Yuan JY. Autophagy in cell death: an innocent convict? (vol 115, pg 2679, 2005) J Clin Invest. 2006;116:3293–3293. doi: 10.1172/JCI26390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Liu L, Yang M, Kang R, Wang Z, Zhao Y, Yu Y, Xie M, Yin X, Livesey KM, Lotze MT, Tang D, Cao L. HMGB1-induced autophagy promotes chemotherapy resistance in leukemia cells. Leukemia. 2011;25:23–31. doi: 10.1038/leu.2010.225. [DOI] [PubMed] [Google Scholar]
- 11.Isakson P, Bjoras M, Boe SO, Simonsen A. Autophagy contributes to therapy-induced degradation of the PML/RARA oncoprotein. Blood. 2010;116:2324–2331. doi: 10.1182/blood-2010-01-261040. [DOI] [PubMed] [Google Scholar]
- 12.Wang Z, Cao L, Kang R, Yang M, Liu L, Zhao Y, Yu Y, Xie M, Yin X, Livesey KM, Tang D. Autophagy regulates myeloid cell differentiation by p62/SQSTM1-mediated degradation of PML-RARalpha oncoprotein. Autophagy. 2011;7:401–411. doi: 10.4161/auto.7.4.14397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tang D, Kang R, Zeh HJ 3rd, Lotze MT. High-mobility group box 1 and cancer. Biochim Biophys Acta. 2010;1799:131–140. doi: 10.1016/j.bbagrm.2009.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Andersson U, Tracey KJ. HMGB1 Is a Therapeutic Target for Sterile Inflammation and Infection. Annu Rev Immunol. 2011;29:139–162. doi: 10.1146/annurev-immunol-030409-101323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tang DL, Kang R, Livesey KM, Cheh CW, Farkas A, Loughran P, Hoppe G, Bianchi ME, Tracey KJ, Zeh HJ, Lotze MT. Endogenous HMGB1 regulates autophagy. J Cell Biol. 2010;190:881–892. doi: 10.1083/jcb.200911078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yang LC, Yu Y, Kang R, Yang MH, Xie M, Wang Z, Tang DL, Zhao MY, Liu LY, Zhang H, Cao LZ. Up-regulated autophagy by endogenous high mobility group box-1 promotes chemoresistance in leukemia cells. Leuk Lymphoma. 2012;53:315–322. doi: 10.3109/10428194.2011.616962. [DOI] [PubMed] [Google Scholar]
- 17.Zhao MY, Yang MH, Yang LC, Yu Y, Xie M, Zhu S, Kang R, Tang DL, Jiang ZG, Yuan WZ, Wu XS, Cao LZ. HMGB1 regulates autophagy through increasing transcriptional activities of JNK and ERK in human myeloid leukemia cells. Bmb Reports. 2011;44:601–606. doi: 10.5483/bmbrep.2011.44.9.601. [DOI] [PubMed] [Google Scholar]
- 18.Drexler HG, Quentmeier H, MacLeod RA, Uphoff CC, Hu ZB. Leukemia cell lines: in vitro models for the study of acute promyelocytic leukemia. Leuk Res. 1995;19:681–691. doi: 10.1016/0145-2126(95)00036-n. [DOI] [PubMed] [Google Scholar]
- 19.Yang L, Yang M, Zhang H, Wang Z, Yu Y, Xie M, Zhao M, Liu L, Cao L. S100A8-targeting siRNA enhances arsenic trioxide-induced myeloid leukemia cell death by down-regulating autophagy. Int J Mol Med. 2012;29:65–72. doi: 10.3892/ijmm.2011.806. [DOI] [PubMed] [Google Scholar]
- 20.Mostafa SS, Miller WM, Papoutsakis ET. Oxygen tension influences the differentiation, maturation and apoptosis of human megakaryocytes. Br J Haematol. 2000;111:879–889. [PubMed] [Google Scholar]
- 21.Lesser MP. Oxidative stress in marine environments: biochemistry and physiological ecology. Annu Rev Physiol. 2006;68:253–278. doi: 10.1146/annurev.physiol.68.040104.110001. [DOI] [PubMed] [Google Scholar]
- 22.Scherz-Shouval R, Elazar Z. ROS, mitochondria and the regulation of autophagy. Trends Cell Biol. 2007;17:422–427. doi: 10.1016/j.tcb.2007.07.009. [DOI] [PubMed] [Google Scholar]
- 23.Mizushima N, Yoshimori T, Levine B. Methods in mammalian autophagy research. Cell. 2010;140:313–326. doi: 10.1016/j.cell.2010.01.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pankiv S, Clausen TH, Lamark T, Brech A, Bruun JA, Outzen H, Overvatn A, Bjorkoy G, Johansen T. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J Biol Chem. 2007;282:24131–24145. doi: 10.1074/jbc.M702824200. [DOI] [PubMed] [Google Scholar]
- 25.Cecconi F, Levine B. The role of autophagy in mammalian development: Cell makeover rather than cell death. Developmental Cell. 2008;15:344–357. doi: 10.1016/j.devcel.2008.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Scherz-Shouval R, Elazar Z. Regulation of autophagy by ROS: physiology and pathology. Trends Biochem Sci. 2011;36:30–38. doi: 10.1016/j.tibs.2010.07.007. [DOI] [PubMed] [Google Scholar]
- 27.Hoppe G, Talcott KE, Bhattacharya SK, Crabb JW, Sears JE. Molecular basis for the redox control of nuclear transport of the structural chromatin protein Hmgb1. Exp Cell Res. 2006;312:3526–3538. doi: 10.1016/j.yexcr.2006.07.020. [DOI] [PubMed] [Google Scholar]
- 28.Owusu-Ansah E, Banerjee U. Reactive oxygen species prime Drosophila haematopoietic progenitors for differentiation. Nature. 2009;461:537–U109. doi: 10.1038/nature08313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Vafa O, Wade M, Kern S, Beeche M, Pandita TK, Hampton GM, Wahl GM. c-Myc can induce DNA damage, increase reactive oxygen species, and mitigate p53 function: A mechanism for oncogene-induced genetic instability. Molecular Cell. 2002;9:1031–1044. doi: 10.1016/s1097-2765(02)00520-8. [DOI] [PubMed] [Google Scholar]
- 30.Behrend L, Henderson G, Zwacka RM. Reactive oxygen species in oncogenic transformation. Biochem Soc Trans. 2003;31:1441–1444. doi: 10.1042/bst0311441. [DOI] [PubMed] [Google Scholar]
- 31.Yang BX, Duan YJ, Dong CY, Zhang F, Gao WF, Cui XY, Lin YM, Ma XT. Novel Functions for mda-7/IL-24 and IL-24 delE5: Regulation of Differentiation of Acute Myeloid Leukemic Cells. Mol Cancer Ther. 2011;10:615–625. doi: 10.1158/1535-7163.MCT-10-0863. [DOI] [PubMed] [Google Scholar]
- 32.Muller S, Ronfani L, Bianchi ME. Regulated expression and subcellular localization of HMGB1, a chromatin protein with a cytokine function. J Intern Med. 2004;255:332–343. doi: 10.1111/j.1365-2796.2003.01296.x. [DOI] [PubMed] [Google Scholar]
- 33.Prasad S, Thakur MK. Distribution of high mobility group proteins in different tissues of rats during aging. Biochem Int. 1990;20:687–695. [PubMed] [Google Scholar]
- 34.Cabart P, Kalousek I, Jandova D, Hrkal Z. Differential expression of nuclear HMG1, HMG2 proteins and H1(zero) histone in various blood cells. Cell Biochem Funct. 1995;13:125–133. doi: 10.1002/cbf.290130209. [DOI] [PubMed] [Google Scholar]
- 35.Seyedin SM, Pehrson JR, Cole RD. Loss of chromosomal high mobility group proteins HMG1 and HMG2 when mouse neuroblastoma and Friend erythroleukemia cells become committed to differentiation. Proc Natl Acad Sci U S A. 1981;78:5988–5992. doi: 10.1073/pnas.78.10.5988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dumitriu IE, Baruah P, Bianchi ME, Manfredi AA, Rovere-Querini P. Requirement of HMGB1 and RAGE for the maturation of human plasmacytoid dendritic cells. Eur J Immunol. 2005;35:2184–2190. doi: 10.1002/eji.200526066. [DOI] [PubMed] [Google Scholar]
- 37.Jia L, Clear A, Liu FT, Matthews J, Uddin N, McCarthy A, Hoxha E, Durance C, Iqbal S, Gribben JG. Extracellular HMGB1 promotes differentiation of nurse-like cells in chronic lymphocytic leukemia. Blood. 2014;123:1709–1719. doi: 10.1182/blood-2013-10-529610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chen Y, Azad MB, Gibson SB. Superoxide is the major reactive oxygen species regulating autophagy. Cell Death Differ. 2009;16:1040–1052. doi: 10.1038/cdd.2009.49. [DOI] [PubMed] [Google Scholar]
- 39.Azad MB, Chen YQ, Gibson SB. Regulation of Autophagy by Reactive Oxygen Species (ROS): Implications for Cancer Progression and Treatment. Antioxid Redox Signal. 2009;11:777–790. doi: 10.1089/ars.2008.2270. [DOI] [PubMed] [Google Scholar]
- 40.Kang R, Livesey KM, Zeh HJ, Lotze MT, Tang DL. HMGB1 as an autophagy sensor in oxidative stress. Autophagy. 2011;7:904–906. doi: 10.4161/auto.7.8.15704. [DOI] [PubMed] [Google Scholar]