

Abstract
OBJECTIVES
The management of arterial pathology in individuals with vascular Ehlers-Danlos syndrome (vEDS) remains a challenge. Here we describe the correlation between COL3A1 gene mutation type and the clinical phenotype in individuals with vEDS.
METHODS
Individuals with confirmed molecular diagnoses of vEDS were enrolled in a multi institutional natural history study. Data collected included demographics, clinical and family histories, arterial pathology (aneurysm, dissection, and rupture), operative details, and autopsy reports. Individuals were classified into two cohorts based on the type of COL3A1 mutations and their effect on the amount of normal collagen produced: MIN group had mutations that lead to minimal (10–15%) production of normal type III collagen and HI group had haploinsufficiency mutations that lead to production of half the normal type III collagen.
RESULTS
A cohort of 68 (72%) individuals from 56 families had arterial pathology (44% male, 13% HI). The HI group was older at the time of their first vascular event (mean 42 years, range 26–58 vs. 33 years, range 8–62, P = .016). The HI group had a higher incidence of aortic pathology compared to the MIN group (56% vs. 21%. P = .025). Visceral arterial pathology was seen in 43 arteries in 23 individuals in the MIN group versus only a single artery in 5 individuals in the HI group. Emergent surgical procedures were more likely to be undertaken when vEDS diagnosis was not known (81% vs. 41%, P = .005) and the majority of these procedures were open surgical repair compared to endovascular repair (81% vs. 19%, P = .019). In the elective setting, there was equal use of open and endovascular repair. Post-operative complications were highest when the diagnosis of vEDS was not known (62% vs. 14%, P < .001) and when procedures were undertaken in an emergency setting (5% vs. 55% P < .001). There was no mortality due to arterial complications in the HI cohort and 21% in the MIN cohort (P = .132).
CONCLUSIONS
Arterial pathology in vEDS individuals is related to the underlying COL3A1 mutation type. The arterial pathology in individuals with HI mutations occurs at later ages with a higher incidence of aortic disease compared with other COL3A1 mutation types. Molecular diagnosis is recommended as diagnosis confirmation, appropriate surveillance, and prophylactic interventions in an elective setting improve surgical outcomes.
Introduction
Vascular Ehlers-Danlos syndrome (vEDS) is a syndrome inherited in an autosomal dominant manner that leads to spontaneous arterial dissection or rupture. Management of these arterial complications remains a challenge.
The disorder is due to heterozygous mutations in COL3A1, which encodes the procollagen peptide for type III collagen.1 Type III collagen is especially abundant in the skin, blood vessels, and hollow organs such as the bowel and uterus. The complications of vEDS reflect the expression pattern of the gene and include rupture and dissection of primarily medium sized arteries, spontaneous rupture of the intestine or the gravid uterus, and thin, fragile skin that bruises easily and heals poorly.
Although the estimated minimum prevalence is 1 in 200,0002, vEDS diagnosis is increasing due to improvements in molecular diagnosis and recognition of mutations that lead to a milder phenotype. More than 700 COL31A mutations have been identified with 50% of affected individuals inheriting the mutation from an affected parent, and 50% due to de novo mutations. 2, 3 Two thirds of the COL3A1 mutations are caused by “missense mutations”, which are glycine substitutions in the triplets of the helical domain of collagen.4 One third of COL3A1 mutations are caused “exon skip mutations” leading to exon splicing errors causing in-frame shift in the reading frame for translation.5 Both types of mutations lead to equal production of abnormal and normal procollagen peptides, but because type III collagen is a homotrimer of three identical procollagen peptides, such mutations lead to production of 7:1 ratio of abnormal to normal collagen molecules thus there is a minimal amount (MIN) of normal collagen produced (10–15%).3, 6 The remainder of the COL3A1 mutations are nonsense mutations or frameshift mutations that lead to the creation of premature termination codons. These premature stops in translation cause rapid degeneration of the mutant mRNA by way of nonsense-mediated decay. This causes expression of a single gene thus termed haploinsufficiency (HI). The end result is production of half the amount of normal type III procollagen. HI mutations are associated with reduced penetrance and delayed onset of arterial pathology compared to MIN mutations.7 Our aim was to evaluate the current surgical management of vEDS associated arterial pathology at tertiary referral centers and to correlate presentation and outcome with the underlying type of mutation.
Methods
Approval by the Institutional Review Board (IRB) was obtained to enroll individuals with vEDS referred to the University Texas at Houston Medical School (UTH), Johns Hopkins Hospital (JHH), and the National Institute on Aging (NIA), (NCT00270686, approved by MedStar IRB #2003-086). The cohort for this study comprised a subgroup of enrolled cases with confirmed molecular diagnosis of vEDS with arterial pathology. Proper consents were obtained from the participants including written consents for clinical photography and use in medical education. All identifiable photographs in this manuscript are from deceased individuals who provided consents for use of their photos at the time of enrollment. Data collected were initially retrospective and ongoing longitudinal follow up occurred from the date of enrollment. Data were obtained from study visits, medical records, radiology images, correspondence with referring physicians, and telephone interviews. Data abstracted included demographics, age at diagnosis, family history, reasons for diagnosis, and clinical diagnostic criteria as defined by the Revised Nosology of Villefranche were collected.2 Arterial pathology was defined as arterial dissection, aneurysm, and rupture. Data collected included initial age at arterial presentation, surveillance and diagnostic imaging if performed (CT, MRI, and duplex ultrasound), management, morbidity, and mortality.
Molecular Diagnosis
COL3A1 mutation detection was performed on a clinical basis in an outside laboratory, or on a research basis at the National Institute on Aging (NIA). At the NIA, the coding regions and flanking sequences of the COL3A1 gene were amplified and sequenced. Primers were designed by Primer3 (http://frodo.wi.mit.edu/primer3/) and are available upon request. Mutations were identified by alignment with the reference sequence (ENSG00000168542). Data collected included the COL3A1 mutation causing vEDS and biochemical analysis of type III procollagen production by dermal fibroblasts performed by a Clinical Laboratory Improvement Amendments (CLIA) certified laboratory. Results of dermal fibroblasts studies were reviewed and the amount of type III procollagen produced verified to correspond with the underlying COL3A1 mutation. The patients were divided into two groups: MIN and HI based on the predicted effect, and in some cases proven effect, of COL3A1 mutations on type III procollagen production.
Statistical Analysis
Data were analyzed using Microsoft Excel 2010 (Microsoft, Redmond, WA) and SPSS 20.0 for Windows (SPSS, Inc., Chicago, IL). Data were presented as mean ± standard error of the mean and ranges, or as numbers and percentages. Continuous variables were compared using a two-sample t-test, assuming that variance was equal. Categorical variables were compared using chi-square testing All P values were two-sided and < .05 was considered significant.
Results
Vascular EDS related arterial pathology was documented in 68 cases (44.1% male). The mean age at diagnosis was 33.0 ± 1.6 years (range 0.3–63 years). Skin biopsies were performed in 24 (35.8%) of the cohort.
Family History
Of the 68 cases, there were reported clinical manifestations of vEDS in 44 (64.7%) cases’ family members indicating a positive family history. Of those with reported family histories, 15 (22.1%) had a family member with a confirmed molecular diagnosis of vEDS as indicated by the presence of a COL3A1 mutation.
Mutation types
There were 59 cases (86.8%) with MIN mutations. MIN mutations included missense (n=42, 61.8%), exon skip (n=12, 17.6%), and unusual mutations (n=5, 7.4%). In addition, one case had biochemical diagnosis of minimal type III procollagen production, thus included in this group. There were 9 cases with HI mutations (13.2%). HI mutations included nonsense (n=5, 55.6%), and frame shift mutations (n=4, 44.4%).
The HI group was older when the first arterial pathology was diagnosed (mean age 41.6 ± 2.9 years, range 26–58) compared to the MIN group (33.1 ± 1.5 years, range 8–62 years, P = .030). Characteristic facial appearance was common (61.2%) but displayed a wide range of variability (Figure 1). Easy bruising and the characteristic thin translucent skin with visible veins (Figure 2) was seen more commonly in the MIN group than in the HI group (Table 1). Table 1 details the demographics, clinical diagnostic criteria, and reasons for diagnosis of the vEDS cohort.
Figure 1.

Spectrum of facial features in individuals with vEDS: variability among patients and does not necessarily correlate with the severity of the underlying arterial pathology. All four individuals here died from vascular complications (A) Caucasian male (MIN mutation, c.2553+1delG) presenting with characteristic vEDS facies including proptotic eyes, long and thin nose, minimal subcutaneous facial fat, and a triangular shaped face. (B) Hispanic female (MIN mutation, c.3545G>A p.G1182E) with mildly proptotic eyes, a long thin nose and a hypotrophic forehead scar, but otherwise normal facial features. (C) Caucasian male (MIN mutation, c.2870G>T, p.G957D) with down slanting palpebral fissures, long and thin nose, thin lips and attached pinna. (D) Caucasian female (c.665G>T, p.G222V) presenting with a long thin nose, but otherwise normal facial features.
Written consent was obtained at the time of enrollment for clinical photography and use in medical education.
Figure 2.
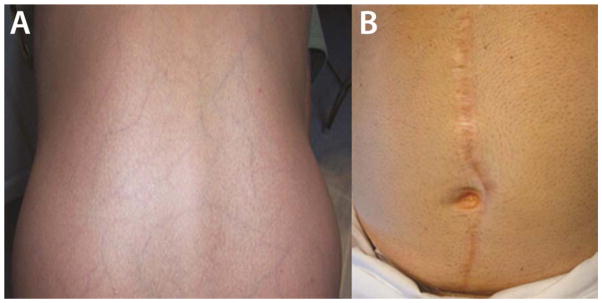
Cutaneous manifestations of vEDS. (A) Thin translucent skin with visible venous pattern on the lower back of a patient with vEDS (MIN mutation, exon skip). (B) Widened atrophic scar in patient following open abdominal aortic aneurysm repair (HI mutation)
Table 1.
Demographics, clinical characteristics, and reasons for diagnosis in patients with vEDS
| N (%), Mean (range) | All (N=67) | MIN (N=58) | HI (N=9) | P* |
|---|---|---|---|---|
| Male | 30 (44.1) | 23 (39.7) | 7 (77.8) | .032 |
| Caucasian | 60 (89.6) | 51 (87.9) | 9 (100.0) | |
| African American and Hispanic | 7 (10.4) | 7 (12.1) | 0 | |
| Age | 40.9 (8–70.2) | 39.9 (8–70.2) | 47.5 (26–61.6) | .116 |
| Age at diagnosis | 33 (0.3–63) | 31.5 (0.3–63) | 42.6 (26–58) | .016 |
| Age at first arterial pathology diagnosis | 34.2 (8–62) | 33.0 (8–62) | 41.6 (26–58) | .030 |
| Major and Minor criteria | ||||
| Family history of vEDS | 43 (64.2) | 35 (60.3) | 8 (88.9) | .097 |
| Easy Bruising | 51 (76.1) | 46 (79.3) | 5 (55.6) | .120 |
| Colon Perforation | 8 (11.9) | 8 (13.8) | 1 (1.7) | .235 |
| Gravid Uterine Rupture (Females) | 1 (2.7) | 1 | 0 | |
| Thin translucent skin | 38 (56.7) | 36 (62.1) | 7 (12.1) | .025 |
| Skin hyperextensibilty | 7 (10.4) | 7 (12.1) | 0 | .271 |
| Characteristic facial appearance | 41 (61.2) | 38 (65.5) | 3 (33.3) | .065 |
| Carotid-cavernous sinus fistula | 4 (6) | 4 (6.9) | 0 | .417 |
| Small joint hypermobility | 29 (43.3) | 28 (48.3) | 1 (11.1) | .036 |
| Tendon or muscle rupture | 6 (9) | 6 (10.3) | 0 | .312 |
| Early-onset varicose veins | 9 (13.4) | 9 (15.5) | 0 | .204 |
| Pneumothorax/Hemothorax | 15 (22.4) | 14 (24.1) | 1 (11.1) | .383 |
| Clubfoot | 6 (9) | 6 (10.3) | 0 | .312 |
| Other comorbidities | ||||
| Hypertension | 15 (22.4) | 11 (19) | 4 (44.4) | .088 |
| Pyloric Stenosis | 3 (4.5) | 3 (5.2) | 0 | .485 |
| Shoulder Surgery or dislocation | 4 (6) | 4 (6.9) | 0 | .417 |
| Migraine Headaches | 6 (9) | 6 (10.3) | 0 | |
| Reason for diagnosis | ||||
| Personal History of Vascular Pathology | 29 (43.3) | 22 (37.9) | 7 (77.8) | .025 |
| Family History | 21 (31.1) | 19 (32.8) | 2 (22.2) | |
| Personal History of Colon Perforation | 5 (7.5) | 5 (8.6) | 0 | |
| History of excessive bruising | 4 (6) | 4 (6.9) | 0 | |
| Diagnosed at autopsy | 2 (3) | 2 (3.4) | 0 | |
| History of excessive scarring | 1 (1.5) | 1 (1.7) | 0 | |
| History of ruptured spleen | 1 (1.5) | 1 (1.7) | 0 | |
| Strong phenotype | 1 (1.5) | 1 (1.7) | 0 | |
| Unknown reason | 2 (3) | 2 (3.4) | 0 | |
MIN: mutations that lead to minimal normal type III collagen production. HI: mutations that lead to 50% normal type III collagen production.
P value calculated using the Pearson Chi-square test for categorical data and independent samples t-test with equal variances assumed for the continuous variables.
Arterial Pathology
The distribution of arterial pathology based on genotype is illustrated in Figure 3.
Figure 3.

Arterial involvement in vEDS cases by COL3A1 mutations. Though vEDS involves medium sized arteries more commonly, there was an increased prevalence of aortic involvement in the HI cohort compared to the MIN cohort (P = .025)
MIN: mutations that lead to minimal normal type III collagen production. HI: mutations that lead to 50% normal type III collagen production. CCF: Carotid Cavernous Fistula.
Aorta involvement was seen in 16 cases. The prevalence of aortic disease was significantly higher in the HI group (55.6%) compared to the MIN group (7%, P < 0.001). Surgical management and outcomes are detailed in Table 2. Non-operative management was undertaken in one case of descending thoracic aorta aneurysm (3.5 cm) that was diagnosed in a patient with MIN mutation without complications.
Table 2.
Surgical management and outcomes of aortic pathology in patients with vEDS
| Aortic Pathology Age/Sex | Mutation | Operation | Outcome |
|---|---|---|---|
| Thoracic Aorta | |||
| Type A aortic dissection 40M | HI |
|
Graft infection |
| Type B aortic dissection with extension into iliacs 35M | HI | Endovascular Stent Graft | Renal Infarcts due to embolization, alive at age 42 |
| Chronic type B dissection with 6 cm DTA aneurysm 19M | MIN |
|
Enlargement of intercostals patch 2 years later Alive at age 25 |
|
| |||
| Abdominal Aorta | |||
| AAA (40M) | HI |
|
Iliac limb thrombosis, IH Abdominal wound dehiscence. (Figure 2B). Alive at age 52 |
| AAA saccular (4 cm), bilateral CIAA with dissection (58M) | HI | Aortobiiliac repair, jump graft to RIIA | Alive at age 61 |
| AAA, dissection, rupture (51M) | HI | Aortobiiliac repair. “Buttery” Aorta | Alive at age 57 |
| Abdominal aortic dissection (trauma related) to R CFA with RLE ischemia (24M) | HI | Fem-Fem bypass | Abdominal aorta is at 3 cm. Alive at age 32 |
| AAA (4.6 cm) (41M) | HI | Interposition graft | Alive at age 47 |
| AAA, bilateral CIAA (21F) | MIN | Aortobiiliac repair, fragile tissue | Abdominal wound dehiscence. Died 10 years later |
| AAA ( 4.5 cm), bilateral CIAA due to aortic dissection(trauma related) (27M) | MIN | Aortobiiliac repair | Alive at age 27 |
ATA: Ascending Thoracic Aorta. DTA: Descending Thoracic Aorta. AAA: Abdominal Aortic Aneurysm. M: Male. F: Female. MIN: mutations that lead to minimal normal type III collagen production. HI: mutations that lead to 50% normal type III collagen production.
Brachiocephalic Arteries Pathology
Details of the brachiocephalic arteries involved and management are shown in Table 3. Multi-brachiocephalic arterial involvement was seen in 9 cases, all with MIN mutations.
Table 3.
Presentation, management, and complications of brachiocephalic arteries pathology in patients with vEDS.
| Arterial Pathology | N | Mutation | Age range | Presentation | Management | Complications/Comments | |
|---|---|---|---|---|---|---|---|
| Carotid Artery | Dissection | 5 | MIN (4), HI (1) | 26–51 | Surveillance (2), Symptomatic (1), Trauma (2) | Non-operative | None |
| Stenosis | 2 | MIN (1), HI (1) | 36–41 | Surveillance | Non-operative | FMD appearance (HI) | |
| Ectasia | 3 | MIN (2), HI (1) | 36–63 | Surveillance | Non-operative | None | |
| Aneurysm | 7 | MIN | 22–56 | Surveillance (6), Symptomatic (1) | Non-operative (6), Coiling & stenting (1) | TIA post coiling (case in Figure 5) | |
| CCF | 5 | MIN | 25–46 | Symptomatic | Endovascular | One required repeat embolization 2 years later | |
|
| |||||||
| Vertebral Artery | Dissection | 6 | MIN (5), HI (1) | 22–52 | Surveillance (3), Symptomatic (3) | Non-operative | PCA infarct (age 52), Stenosis on F/U (n=2) |
| Ectasia | 1 | MIN | 36 | Surveillance | Non-operative | None | |
| Aneurysm | 1 | MIN | 29 | Surveillance | Non-operative | 0.5 cm at V2 | |
|
| |||||||
| Subclavian Artery | Dissection | 2 | MIN | 41 | Surveillance | Non-operative | Bilateral in same case |
| Ectasia | 1 | MIN | 52 | Surveillance | Non-operative | None | |
| Aneurysm | 3 | MIN | 26–34 | Surveillance | Non-operative (3), | None | |
| Aneurysm | 1 | MIN | 26 | Symptomatic | R SCA ligation, L axillary to R SCA bypass | Bypass Thrombosis, reintervention, alive at age 27 | |
| Aneurysm | 1 | MIN | 18–32 | Rupture (R SCA) | Endovascular stenting R CCA/ R SCA | No complications (case in Figure 7) | |
| Aneurysm | 1 | MIN | 32 | Rupture (L SCA) | Carotid Subclavian bypass | Post op infarcts, prolonged vent dependence, tracheostomy | |
CCF: Carotid-Cavernous fistula, CCA: Common Carotid Artery. SCA: Subclavian artery, Op: Operative, F/U: Follow-up, R: right, L: Left, MIN: mutations that lead to minimal normal type III collagen production. HI: mutations that lead to 50% normal type III collagen production.
Visceral Arteries
Multi-visceral arterial involvement was seen in 23 patients (43 arteries), all in the MIN group. In the HI group, there were five cases (with visceral arterial involvement, all single artery pathology.
Celiac artery pathology occurred only in individuals with MIN mutation (n=5): three aneurysms (1.5–2 cm in diameter), and two cases of dissection that progressed to aneurysms (1.6–1.8 cm) during one year of follow up. All the cases were managed non-operatively.
Hepatic artery pathology occurred in 10 cases (MIN n=7, 12.1% vs. HI n=3, 33.3%, P = .096). Management was non-operative in aneurysms smaller than 2 cm in diameter (n=6). Two cases had coil embolization: right hepatic artery 2.2 cm in diameter and an intra-parenchymal hepatic artery rupture. One case remains under observation due to high surgical risk for a stable 3 cm proper hepatic artery aneurysm (34 year old female, c.1772G>A, p.Gly591Asp, MIN type mutation).
Splenic artery pathology occurred in 18 cases (MIN n=17, 29.3% vs. HI=1, 11.1%, P = .252). Management was non-operative in isolated dissections or aneurysms smaller than 1.7 cm in diameter (n=6). Coil embolization was performed successfully in 4 cases of aneurysms larger than 2 cm in diameter (one with a contained rupture). Ruptured splenic artery of unknown size with hemodynamic compromise occurred in two female patients requiring operative exploration and splenectomy (62 year old, c.3319G>C, p.Gly1107Arg, and 28 year old, c.3499G>C, p.Gly1000Arg, both MIN type mutations, both diagnosed after the rupture event).
Superior mesenteric artery (SMA) pathology occurred only in individuals with MIN mutation. One case presented with rupture not amenable to repair thus necessitating ligation and right hemicolectomy (20 year old male, c.2482G>T, p. Gly828Trp). One case of SMA dissection was managed non-operatively, and another SMA dissection was managed by placement of a 6×18mm bare metal stent (39 year old female, c.2356G>A, p.Gly786Arg). Initial attempt at access via the brachial artery for intended embolization was complicated by brachial artery dissection, necessitating transfer to a tertiary center (JHH). The case was then performed via a femoral artery access with manual compression for 5F sheath removal and no adverse events.
Renal artery pathology occurred in 17 cases (MIN n=16, 27.6% vs. HI n=1, 11.1%, P = .291). This involved small aneurysms and dissections (n=14) leading to renal infarcts (n=8), all managed non-operatively. In one case the renal artery had fibromuscular dysplasia type beaded appearance in a patient who had undiagnosed vEDS (41 years old female, c.766delA, p. Ile256Fx, HI type mutation). The patient underwent an attempted open repair complicated by nephrectomy and subsequent incisional hernia. Coil embolization was performed successfully in one case of an intra-parenchymal renal artery aneurysm and on subsequent follow up was noted to have a dissection treated with non-operative management. Ruptured renal artery with hemodynamic compromise occurred in one case (17 year old female, IVS43+2T>G, MIN type mutation). The patient died intraoperatively due to hemorrhagic shock.
Iliac Arteries
Iliac arteries aneurysms and dissections independent of abdominal aortic involvement occurred in 22 cases (MIN n=20, 34.5% vs. HI n=2, 22.2%, P = .466). Successful non-operative management was utilized in cases of dissection (n=7) aneurysms smaller than 2.5 cm in diameter (n=3). Details of iliac arteries operative management are shown in Table 4. There were two cases of death due to presumed iliac artery rupture: One case with known maximum iliac artery size of 1.4 cm (28 year old female, c.3201+2T>C, MIN type mutation), and the second case had known iliac artery dissection (29 year old female, c.665G>T, p.Gly222Val, MIN type mutation). Both presented with pain and had cardiac arrest during diagnostic workup.
Table 4.
Iliac artery interventions and complications in cases of vEDS
| Pathology | Mutation | Age | Management | Complications |
|---|---|---|---|---|
| AVF/ symptomatic | MIN | 28 | Elective Endovascular repair | Alive at age 34 |
| AVF/ rupture | MIN | 28 | Emergent open repair | Intraoperative death |
| Dissection | MIN | 55 | Elective prophylactic iliofemoral bypass | Alive at age 59 |
| Dissection | MIN | 43 | Elective prophylactic iliofemoral bypass | Alive at age 48 |
| Aneurysm/dissection | HI | 27 | Elective prophylactic aortobiiliac repair | Alive at age 26 (case in figure 6) |
| Dissection/ rupture | MIN | 42 | Emergent open repair | Wound infection, alive at age 50 |
| Aneurysm/ rupture | MIN | 22 | Emergent open repair | Intraoperative death |
AVF: Arteriovenous fistula. MIN: mutations that lead to minimal normal type III collagen production. HI: mutations that lead to 50% normal type III collagen production.
Impact of preoperative diagnosis of vEDS on operative outcomes
Fifty procedures (29 emergent, 21 elective) were performed in 35 patients (9 HI, 25.7%) and involved the brachiocephalic arteries (n=9), coronary arteries (n=3), aorta (n=12), iliac arteries (n=8), visceral arteries (n=14), tibial arteries (n=2), inferior epigastric artery (n=1), and ruptured chordae tendineae (n=1). Emergent procedures were performed in 62.2% and 46.2% of the MIN and HI groups respectively. Post-operative complications were not different between the MIN and HI groups (MIN=13, 35.1%, vs. HI=4, 30.7%, P = .775)
Diagnosis of vEDS was known in 42% cases prior to surgical intervention. In these cases, arterial repairs were approached in an elective manner with an equal mix of open and endovascular repair, 48.4% and 51.7% respectively, as necessitated by the underlying arterial pathology (Figure 4). In the cases requiring emergency interventions, surgical technique was altered to accommodate the fragility of tissues (Figure 5).
Figure 4.

A 6 cm carotid artery aneurysm which developed in a 34 year old woman acutely in the post-partum period. (A, B) 3-D reconstruction and representative axial image of a computed tomography scan demonstrating the large right internal carotid artery aneurysm. Her arterial pathology led to vEDS diagnosis (MIN mutation, c.1772G>A, p.Gly591Asp) and preoperative multidisciplinary planning accordingly. She was successfully treated with stent-assisted coil embolization of the ICA aneurysm (C). Images courtesy of Johns Hopkins/NIH
Figure 5.

Endovascular repair of a right subclavian artery rupture presenting with sudden onset of left shoulder pain radiating to the neck in an 18 year old man with known vEDS (MIN mutation, (c.3847C>T). (A) Arteriogram demonstrating the rupture site and arteriovenous fistula to right internal jugular vein. (B) Knowledge of his diagnosis allowed for the surgical treatment to be tailored to his diagnosis with 7 mm Self expanding covered stents in the common carotid and subclavian arteries without complications. Images courtesy of Dr. Ali Azizzadeh, MD (UT Houston).
The undiagnosed vEDS cases were significantly more likely to present in an emergent fashion (80.9% vs. 41.3%, P = .005) and undergo open surgical repair (81% vs. 48.3%, P = .019). Postoperative complications were substantially increased in these cases (P < 0.001, Table 5). All intraoperative deaths (n=3) occurred in cases with MIN type mutation who presented in an emergent fashion requiring management without the benefit of preoperative vEDS diagnosis.
Table 5.
Fifty Operative procedures classified according to preoperative knowledge of vEDS diagnosis
| N=50 (%) | Diagnosis Known N=29 |
Diagnosis not known N=21 |
P* |
|---|---|---|---|
| Family History | 13 (44.8) | 9 (42.9) | .890 |
| Haploinsufficiency Mutation | 6 (20.7) | 7 (33.3) | .314 |
| Open Surgical Repair | 14 (48.3) | 17 (81.0) | .019 |
| Endovascular Procedures | 15 (51.7) | 4 (19.0) | .019 |
| Emergent Repairs | 12 (41.3) | 17 (80.9) | .005 |
| Intraoperative Death | 0 | 3 (14.2) | .036 |
| Post-operative complications | 4 (13.7) | 13 (61.9) | <.001 |
P value calculated using the Pearson Chi-square test for categorical data
Mortality
Mean follow up from the time of diagnosis was 8 ± 0.82 years (range 1–26 years). There were 12 deaths all occurring in the MIN group (21%) with mean age of death was 31.4 ± 2.6 years (range 17–45 years). Causes of death were the following: intraoperative deaths during attempts to repair ruptured iliac (n=2) and renal artery (n=1), stroke post carotid cavernous fistula embolization (n=1), mesenteric arterial thrombosis and necrosis after admission for small bowel obstruction (n=1) (Figure 1A), central line insertion complication (n =1) (Figure 1B), rapid aneurysmal degeneration of iliac and gluteal arteries complicated by retroperitoneum and spontaneous hemothorax post colostomy take down (n=1) (Figure 1C), presumed iliac artery rupture (n=2) (Figure 1D), and unexpected death of unknown causes (n=3).
Discussion
Management of vascular pathology associated with vEDS remains a clinical challenge. This report highlights the importance of establishing the molecular diagnosis in cases of vEDS. Although clinical findings can offer a suggestion of the diagnosis, molecular testing should be pursued when the diagnosis is suspected.8 Confirmation of the underlying molecular diagnosis and classifying the mutation based on the impact on type III collagen production are important elements in the counseling and management of vEDS patients. Furthermore, it differentiates the vEDS from other syndromes such as Loeys-Dietz syndrome (LDS) which has similar age at presentation and features that overlap with vEDS patients.9 Molecular testing allows for critical genotype-phenotype correlations to be identified, such as the fact that patients with HI mutations present at an older age, have milder arterial disease than MIN mutations7, and have a high prevalence of aortic disease.
Our data support that elective repair for arterial complications can be performed with acceptable morbidity. Given the small cohort size it is difficult to set absolute guidelines beyond what is currently recommended for patients without vEDS. We propose elective interventions in cases in which rapid change such as growth or dissection occurs in a previously stable aneurysm and we individualize the recommendations to the patient based on the risk/benefit profile for each patient. Knowledge of the molecular diagnosis allows preoperative counseling and modification of the surgical techniques which are critical to successful outcomes. Patients with an established preoperative diagnosis treated in an elective setting have significantly improved outcomes when compared to cases repaired in an emergent fashion and when the diagnosis is not known. We have shown that open repairs of the aorta are well tolerated in the elective setting, and endovascular repairs of the medium sized arteries are also well tolerated.10 The preoperative diagnosis allows the surgeon to prepare for perioperative and intraoperative management. In the perioperative setting, communication with the anesthesiologists is a key element in management as it allows for careful intubation, use of ultrasound guided vascular access, and stringent blood pressure control. Indeed one case survived an abdominal aortic aneurysm repair, only to die at a separate hospitalization ten years later due to a central line complication. Intraoperatively, the surgeon should be prepared to deal with the fragile tissues. Clamping techniques are modified, this includes a more proximal “fall-back position” to avoid placing vascular clamps on the same location, avoiding Rommel tourniquet use as they tend to create circumferential adventitial hematoma, and external pneumatic tourniquets are used to control limbs proximally. In the case of aortic repairs, buttressing the sutures lines with circumferential individual felt pledgets to promote hemostasis has been described. 11
In the endovascular setting for arterial embolization in medium sized arteries the surgeon can apply these same principles of access vessel management with open exposure for large diameter sheaths, minimal sheath exchanges, and circumferential individual pledget reinforcement for closure.10 During the post-operative period, there should be specific attention to blood pressure control. We have used a stepwise increase in allowed-systolic pressure (SBP) in aortic patients with SBP maintained at <90mmHg for the first 24–48 hours postoperatively, gradually increasing to normotension on postoperative days 3–5. Since treatment with beta-adrenergic blocking agents has been shown to decreased vascular events in vEDS patients,12 these agents should be considered for blood pressure control. Other postoperative considerations include adequate pain management and diligent monitoring and immediate treatment of constipation since it may lead to bowel rupture.
With the understanding that diagnosed vEDS patients can do well with elective surgical repair of arterial pathology, it raises the question as to the extent these patients should have ongoing surveillance for vascular diseases, a topic that is controversial.8 We recommend surveillance with carotid artery ultrasound, and noninvasive imaging of the thoracic and abdominal aorta, visceral arteries, and iliac arteries by using magnetic resonance angiography (MRA) during the initial evaluation of these patients, followed by targeted computed tomography as needed based on vascular pathology identified. The young age of most patients makes routine use of computed tomography as a surveillance method problematic due to lifetime radiation exposure issues. Since rare vascular complications have occurred in patients in their teenage years, it is reasonable to obtain a baseline study before the age of 20 years. Patients with a known vascular lesion can be followed in a manner tailored to the lesion and the patient. However, further data regarding intensity of surveillance is needed. In patients without vascular manifestation, periodic arterial screening is recommended, however, frequency is not known and further data regarding intensity of surveillance is needed.2 Without ongoing surveillance, vascular complications were generally described as sudden and catastrophic, and our data support this statement. It was generally believed that gradual vascular dilatation such as seen in Marfan syndrome was not a feature of vEDS.13 With surveillance, asymptomatic dissections and aneurysms can be identified in vEDS patients and our data suggest that surgical repair of this arterial pathology can be done. Further research regarding these asymptomatic vascular anatomical events is needed to determine natural history and morphology changes that may motivate surgical or endovascular intervention.
It is imperative that the disease be properly diagnosed when encountering peripheral arterial aneurysms or dissections in a young patient. Features of vEDS such as facial characteristics, unexplained bruises, translucent or thin skin should be examined, and history should be elicited to note club foot, dislocations or pneumothorax. However, the absence of the facial appearance or suspicious clinical history does not rule out the disease, since there are a considerable number of patients without major features (Figure 1). Obtaining a family history of sudden death due to unknown causes at a young age or arterial complications is important for identifying possible cases of vEDS, and referral to a medical geneticist for counseling and diagnosis is appropriate. Establishing the diagnosis allows for genetic counseling, proper surveillance to detect the arterial pathology, and for elective repair planning by a multidisciplinary team involving vascular surgeons, cardiac surgeons, interventionists, and medical geneticists. Correct diagnosis also allows for the first degree relatives to undergo counseling and testing, thus identifying asymptomatic vEDS individuals prior to experiencing a potentially catastrophic event. The development of medical treatment options, such as the use of celiprolol,12,14 as well as growing experience with prophylactic endovascular and open procedures stand to improve the prognosis for vEDS. Although the disease is rare, the morbidity and mortality is significant and warrants the study of long term outcomes with contemporary surgical and medical interventions.
Conclusions
Establishing the molecular diagnosis of vEDS is imperative to disease management. When surgical repair is indicated, elective repairs offered by a multidisciplinary team and using surgical techniques applied to address the fragile tissues are associated with favorable outcomes. This approach is expected to improve the overall prognosis of patients with vEDS.
Acknowledgments
The authors are extremely grateful to patients involved in this study. We are deeply indebted to Kristofer M Charlton-Ouw, MD, Ali Azizzadeh, MD, Sheila M Coogan, MD, and Anthony L Estrera, MD from the Department of Cardiothoracic and Vascular Surgery, University of Texas Medical School at Houston, Houston, TX who have critically reviewed this manuscript, contributed their clinical expertise, and individual patient cases for this study. We also are grateful to Hanci Zhang BS and Jason D Lehman MD from the National Institute on Aging, Baltimore, Maryland who assisted in data collection. Without their help, this work would have not been possible. Both authors D.M.M. and N.B.M. contributed equally as senior authors to this manuscript
The following sources provided funding for these studies: John J. Roberts Aortic Research Fund (J.H.B), NIA/NIH Intramural funds (N.B.M), R01HL109942-01A1 (D.M.M.), the Richard T. Pisani Funds (D.M.M.), UL1 RR024148 (University of Texas Health Science Center at Houston), and Vivian L. Smith Foundation (D.M.M.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
Sherene Shalhub, Email: shalhub@uw.edu, Division of Vascular Surgery, Department of General Surgery, University of Washington, Seattle, WA.
James H Black, III, Email: jhblack@jhmi.edu, Division of Vascular Surgery and Endovascular Therapy, Department of Surgery, The Johns Hopkins Hospital, Baltimore, MD.
Alana C. Cecchi, Email: Alana.C.Cecchi@uth.tmc.edu, Division of Medical Genetics, Department of Internal Medicine, University of Texas Health Science Center at Houston, Houston, TX.
Zhi Xu, Email: zhi.xu@nih.gov, National Institute on Aging, Baltimore, Maryland.
Ben F Griswold, Email: griswoldb@mail.nih.gov, National Institute on Aging, Baltimore, Maryland.
Hazim J Safi, Email: Hazim.J.Safi@uth.tmc.edu, Department of Cardiothoracic and Vascular Surgery, University of Texas Medical School at Houston, Houston, TX.
Dianna M. Milewicz, Email: Dianna.M.Milewicz@uth.tmc.edu, Division of Medical Genetics, Department of Internal Medicine, University of Texas Health Science Center at Houston, Houston, TX.
Nazli B. McDonnell, Email: naznaz567@gmail.com, National Institute on Aging, Baltimore, Maryland.
Reference List
- 1.Pope FM, Narcisi P, Nicholls AC, Germaine D, Pals G, Richards AJ. COL3A1 mutations cause variable clinical phenotypes including acrogeria and vascular rupture. Br J Dermatol. 1996 Aug;135(2):163–81. [PubMed] [Google Scholar]
- 2.Pepin MG, Byers PH. GeneReviews™ [Internet] Vol. 1993 Seattle (WA): University of Washington, Seattle; 1993–1999. Sep 02, Ehlers-Danlos Syndrome Type IV. [updated 2011 May 03] [Google Scholar]
- 3.Pepin M, Schwarze U, Superti-Furga A, Byers PH. Clinical and genetic features of Ehlers-Danlos syndrome type IV, the vascular type. N Engl J Med. 2000 Mar 9;342(10):673–80. doi: 10.1056/NEJM200003093421001. [DOI] [PubMed] [Google Scholar]
- 4.Smith LT, Schwarze U, Goldstein J, Byers PH. Mutations in the COL3A1 gene result in the Ehlers-Danlos syndrome type IV and alterations in the size and distribution of the major collagen fibrils of the dermis. J Invest Dermatol. 1997 Mar;108(3):241–7. doi: 10.1111/1523-1747.ep12286441. [DOI] [PubMed] [Google Scholar]
- 5.Schwarze U, Goldstein JA, Byers PH. Splicing defects in the COL3A1 gene: marked preference for 5′ (donor) spice-site mutations in patients with exon-skipping mutations and Ehlers-Danlos syndrome type IV. Am J Hum Genet. 1997 Dec;61(6):1276–86. doi: 10.1086/301641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pyeritz RE. Ehlers-Danlos syndrome. N Engl J Med. 2000 Mar 9;342(10):730–2. doi: 10.1056/NEJM200003093421009. [DOI] [PubMed] [Google Scholar]
- 7.Leistritz DF, Pepin MG, Schwarze U, Byers PH. COL3A1 haploinsufficiency results in a variety of Ehlers-Danlos syndrome type IV with delayed onset of complications and longer life expectancy. Genet Med. 2011 Aug;13(8):717–22. doi: 10.1097/GIM.0b013e3182180c89. [DOI] [PubMed] [Google Scholar]
- 8.Oderich GS, Panneton JM, Bower TC, Lindor NM, Cherry KJ, Noel AA, et al. The spectrum, management and clinical outcome of Ehlers-Danlos syndrome type IV: a 30-year experience. J Vasc Surg. 2005 Jul;42(1):98–106. [Google Scholar]
- 9.Loeys BL, Chen J, Neptune ER, Judge DP, Podowski M, Holm T, et al. A syndrome of altered cardiovascular, craniofacial, neurocognitive and skeletal development caused by mutations in TGFBR1 or TGFBR2. Nat Genet. 2005 Mar;37(3):275–81. doi: 10.1038/ng1511. [DOI] [PubMed] [Google Scholar]
- 10.Brooke BS, Arnaoutakis G, McDonnell NB, Black JH., III Contemporary management of vascular complications associated with Ehlers-Danlos syndrome. J Vasc Surg. 2010 Jan;51(1):131–8. doi: 10.1016/j.jvs.2009.08.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lum YW, Brooke BS, Black JH., III Contemporary management of vascular Ehlers-Danlos syndrome. Curr Opin Cardiol. 2011 Nov;26(6):494–501. doi: 10.1097/HCO.0b013e32834ad55a. [DOI] [PubMed] [Google Scholar]
- 12.Ong KT, Perdu J, De BJ, Bozec E, Collignon P, Emmerich J, et al. Effect of celiprolol on prevention of cardiovascular events in vascular Ehlers-Danlos syndrome: a prospective randomised, open, blinded-endpoints trial. Lancet. 2010 Oct 30;376(9751):1476–84. doi: 10.1016/S0140-6736(10)60960-9. [DOI] [PubMed] [Google Scholar]
- 13.Purohit N, Marsland D, Roberts N, Townsend E. Haemo-pneumothorax and haemoptysis in a patient with suspected Ehlers-Danlos syndrome. Interact Cardiovasc Thorac Surg. 2009 Jul;9(1):130–1. doi: 10.1510/icvts.2009.204313. [DOI] [PubMed] [Google Scholar]
- 14.Brooke BS. Celiprolol therapy for vascular Ehlers-Danlos syndrome. Lancet. 2010 Oct 30;376(9751):1443–4. doi: 10.1016/S0140-6736(10)61155-5. [DOI] [PubMed] [Google Scholar]