

Abstract
Previous studies have shown that up-regulation of transforming growth factor β1 results in neuroprotective effects. However, the role of the transforming growth factor β1 downstream molecule, SMAD2/3, following ischemia/reperfusion remains unclear. Here, we investigated the neuroprotective effects of SMAD2/3 by analyzing the relationships between SMAD2/3 expression and cell apoptosis and inflammation in the brain of a rat model of cerebral ischemia/reperfusion. Levels of SMAD2/3 mRNA were up-regulated in the ischemic penumbra 6 hours after cerebral ischemia/reperfusion, reached a peak after 72 hours and were then decreased at 7 days. Phosphorylated SMAD2/3 protein levels at the aforementioned time points were consistent with the mRNA levels. Over-expression of SMAD3 in the brains of the ischemia/reperfusion model rats via delivery of an adeno-associated virus containing the SMAD3 gene could reduce tumor necrosis factor-α and interleukin-1β mRNA levels, down-regulate expression of the pro-apoptotic gene, capase-3, and up-regulate expression of the anti-apoptotic protein, Bcl-2. The SMAD3 protein level was negatively correlated with cell apoptosis. These findings indicate that SMAD3 exhibits neuroprotective effects on the brain after ischemia/reperfusion through anti-inflammatory and anti-apoptotic pathways.
Keywords: nerve regeneration, brain injury, neuroprotection, inflammation, apoptosis, cerebral ischemia, SMAD3, transforming growth factor β1, NSFC grant, neural regeneration
Introduction
Ischemic cerebrovascular disease is one of the most common diseases of the nervous system, accounting for 60–80% of all strokes. Its high mortality and morbidity bring about huge economic and mental burdens for the patients, families and society (Neuhaus et al., 2014; Sauser et al., 2014). Immediately following the event, nerve cells at the penumbra (Kim et al., 2014; Manning et al., 2014), the area surrounding an ischemic event, are depleted of electrical activity and are prone to hypoxic cell death (infarction). The original damage from the ischemia can be amplified by inflammation, oxidative stress and/or apoptosis (Sanchez, 2013; Baron et al., 2014; Rosso and Samson, 2014). Without effective continuous perfusion, neurological defects will progress. The effective and available therapeutic strategy to rescue nerve cells in the penumbra is recanalization through thrombolysis, which can restore blood flow at the ischemic penumbra (Dorado et al., 2014). However, clinical application of thrombolysis therapy is limited because of strict indications, a short time window, high risk, and complications. Therefore, an anti-inflammatory, anti-apoptotic, neuroprotective drug is urgently needed to provide a different avenue for clinical treatment of cerebral infarction.
Transforming growth factor-β1 (TGF-β1) is one of the most important members of the TGF-β family. It binds with membrane receptors on the surface of cells and regulates the expression of multiple genes through the SMAD signaling pathway (Konkel and Chen, 2011; Kovacic and Somanathan, 2011). The TGF-β1/SMADs signaling pathway has a variety of biological effects, and is critically involved in the physiological and pathological processes of the body, such as cell apoptosis and proliferation, stem cell differentiation and embryonic development, extracellular matrix formation, post-trauma repair and the inflammatory response (Akhurst and Hata, 2012; Araujo-Jorge et al., 2012; Beyer et al., 2013; Sakaki-Yumoto et al., 2013). TGF-β levels are upregulated in the brain after cerebral ischemia, suggesting that it may be related to nerve damage and repair after cerebral ischemia (Knuckey et al., 1996). TGF-β1 is the predominant subtype among active TGF-βs in the brain and it exerts neuroprotective effects against cerebral ischemia (Kalluri and Dempsey, 2008; Bonni and Bonni, 2012; Caraci et al., 2012; Joseph et al., 2013). In addition, the neuroprotective mechanism of TGF-β1 may include an anti-inflammatory effect, malacia healing, anti-excitatory amino acid toxicity, an anti-apoptotic effect, angiogenesis, and nerve regeneration (Pera et al., 2004).
TGF-β1 expression and its neuroprotective effect following cerebral ischemia/reperfusion have been studied in detail, but the role of the downstream signaling molecule, SMAD2/3, in neuroprotection is not fully elucidated. This study aimed to observe SMAD2 and SMAD3 expression in the brain of a cerebral ischemia/reperfusion rat model, explore the effects of SMAD2 and SMAD3 on inflammation and apoptosis after infarction, and to determine the mechanism of regulation of the TGF-β1/SMAD2/3 signaling pathway after cerebral ischemia/reperfusion.
Materials and Methods
Animals and treatments
Eighty Sprague-Dawley male rats were housed in separate specific-pathogen-free cages with free access to food and water, at 21 ± 2°C and 30–35% humidity, under a 12-hour day/night cycle. This study was approved by the Animal Ethics Committee of Jilin University in China.
To detect dynamic expression of SMAD2 and SMAD3 after cerebral ischemia/reperfusion, 10 rats served as the control group (control), 10 rats served as the sham group (sham), and cerebral ischemia/reperfusion was established in the remaining 60 rats. The ischemia/reperfusion rats were randomly divided into 6-hour, 24-hour, 72-hour and 7-day groups, with 15 rats in each group. The influence of SMADs on the expression of inflammatory factors and apoptosis was investigated in the 24 hour group.
To investigate the role of SMAD3 after cerebral ischemia/reperfusion, 15 rats were divided into three subgroups: control, negative control (vector) and SMAD3 over-expression (SMAD3), with five rats in each subgroup. In the control, SMAD3 and vector subgroups, rats were injected via the tail vein with 100-μL PBS, AAV9-SMAD3 (3 × 1013 vg/mL per kg body weight) (Cyagen Biosciences, Guangzhou, China), and AAV9-SMAD3 control AAV9-GFP (3 × 1013 vg/mL per kg body weight) (Cyagen Biosciences), respectively. Three days after injection, all rats were subjected to cerebral ischemia/reperfusion.
Establishment of cerebral ischemia/reperfusion
Rats were anesthetized with subcutaneous injection of ketamine (100 mg/kg), xylazine (2.5 mg/kg) and acepromazine (2.5 mg/kg; Jinan Wanxingda Chemical Co., Jinan, China), and fixed in the supine position on the operation table. A medial incision was made in the neck skin after disinfection, then the left common carotid, external carotid and internal carotid arteries were exposed. A small incision was made using ophthalmic scissors between the common carotid artery and the external carotid artery, and a 0.26-mm thread was inserted until the middle cerebral artery. After the blood flow of the left middle cerebral artery was blocked for 2 hours, the thread was removed and the external carotid artery was ligated and the wounds sutured. After recovery, rats were reared in a 20–25°C cage, with free access to water and food.
TTC staining
After sacrifice, rat brains were perfused with saline, dissected and stored at −20°C for 20 minutes. Brains were cut at the midpoint of the line between the forebrain and the optic chiasm, at the suprachiasmatic site, at the infundibular stem, and between the infundibular stem and the posterior lobe. Slices 2-mm thick were cut. Slices were stained in 2% TTC solution (JianCheng Bioengineering Institute, Nanjing, China) at 37°C for 30 minutes. The infarct volume was calculated as previously reported (Goldlust et al., 1996).
Real-time quantitative PCR
Total RNA from ischemic brain specimens was extracted with Trizol (Invitrogen, CA, USA). cDNA was synthesized from 2 μg of total RNA using a Superscript III Reverse Transcriptase kit (Invitrogen) following the manufacturer's instructions. The synthesized cDNA was used for real-time quantitative PCR in accordance with the instructions of a SYBR Green PCR kit (TransGen Biotech, Beijing, China). In brief, 1 μL specific primers (0.5 μL upstream primer and 0.5 μL downstream primer), 12.5 μL 2 × SYBR Green qPCR Master Mix, 2.5 μL diluted cDNA and 9 μL nuclease-free PCR-grade water were mixed. The mixture was denatured at 95°C for 5 minutes, followed by 40 cycles of 95°C for 30 seconds, 60°C for 1 minute, and 72°C for 30 seconds. GAPDH served as an internal reference and target mRNA concentration was expressed as 2-ΔΔCt values.
Specific primer sequences were as follows: SMAD2 upstream primer: 5′-CCA GGT CTC TTG ATG GTC GT-3′; SMAD2 downstream primer 5′-GGC GGC AGT TCT GTT AGA AT-3′; SMAD3 upstream primer: 5′-ACA AGG TCC TCA CCC AGA TG-3′; SMAD3 downstream primer: 5′-TGG CGA TAC ACC ACC TGT TA-3′; TNF-α upstream primer: 5′-CTC CAG CTG GAA GAC TCC TCC CAG-3′; TNF-α downstream primer: 5′-CCC GAC TAC GTG CTC CTC ACC-3′; IL-1β upstream primer: 5′-GAC CTG CTT CTT TGA GGC TGA C-3′; IL-1β downstream primer: 5′-TTC ATC TCG AAG CCT GCA GTG-3′; GAPDH upstream primer: 5′-GGC AAG TTC AAT GGC ACA GT-3′; GAPDH downstream primer: 5′-TGG TGA AGA CGC CAG TAG ACT C-3′.
Western blot analysis
Ischemic brain sections were homogenized in Tissue Lysate Buffer (Beyotime, Shanghai, China), and phases left to separate on ice for 30 minutes. The protein phase was then centrifuged at 8,000 × g for 20 minutes, and the supernatant removed. Protein concentration was measured with a BCA kit (Beyotime, Shanghai, China). For each sample, 15 μg total protein was mixed with 4 μL 5 × loading buffer and denatured by boiling for 5 minutes. Subsequently, specimens were separated by electrophoresis on 10% SDS-PAGE gels (Beyotime, Shanghai, China) at 80 V for 40 minutes and at 110 V for 90 minutes, and then transferred to polyvinylidene difluoride membranes (Beyotime, Shanghai, China) at 200 mA for 60 minutes. After blocking for 2 hours, membranes were washed with western solution (Beyotime) and incubated with goat anti-rat polyclonal antibody (SMAD2/3 sc-6032, pSMAD2/3 sc-11769, Bcl-2 sc-16323, caspase-3 sc-1225, cleavage casp3 sc-22139, GAPDH sc-48167) 1:100 (Santa Cruz Biotechnology, Santa Cruz, CA, USA) at 4°C overnight, and with horseradish peroxidase conjugated rabbit anti-goat polyclonal antibody (sc-2768) 1:1,000 (Santa Cruz Biotechnology) for 2 hours. Antibody binding was visualized with the ECL method, and results are expressed as the gray value ratio of target protein to GAPDH.
TUNEL staining for apoptotic cells
Paraffin sections of brain tissue were immersed and dewaxed with xylene (2 × 5 minutes), and rinsed with dehydrated alcohol (2 × 3 minutes), 95% ethanol (2 × 3 minutes), 75% ethanol (2 × 3 minutes), 0.85% sodium chloride in PBS for 5 minutes. Subsequently, sections were hydrolyzed with proteinase K solution (20 μg/mL) for 15 minutes. Brain tissue sections were placed in 2% H2O2 at room temperature for 5 minutes, then rinsed with PBS and blocked with TdT enzyme buffer for 5 minutes at room temperature. Sections were photographed under a fluorescence microscope (Olympus, Japan). Five fields of view at 100× magnification were selected from each section, to count TUNEL-positive cells.
Statistical analysis
Data were analyzed using SPSS 18.0 software (SPSS Inc, Chicago, IL, USA) and are expressed as the mean ± SD. Intergroup differences were compared using one-way analysis of variance followed by the Student-Newman-Keuls test. The test level was considered as α = 0.05. The correlation of SMAD3 expression with IL-1β levels and TUNEL-positive cells was analyzed with Spearman's rank correlation method.
Results
Over-expression of SMAD3 reduced infarct volume
Three rats from each of the control, vector and SMAD3 groups were selected for TTC staining. The infarct tissue in the control group was stained pale and involved the cortex, hippocampus and basal ganglia. The infarct volume in the vector group was the same as that in the control group, without significant difference between the two groups (P > 0.05). Compared with the control and vector groups, infarct volume was obviously reduced in the SMAD3 group (P < 0.01) (Figure 1).
Figure 1.
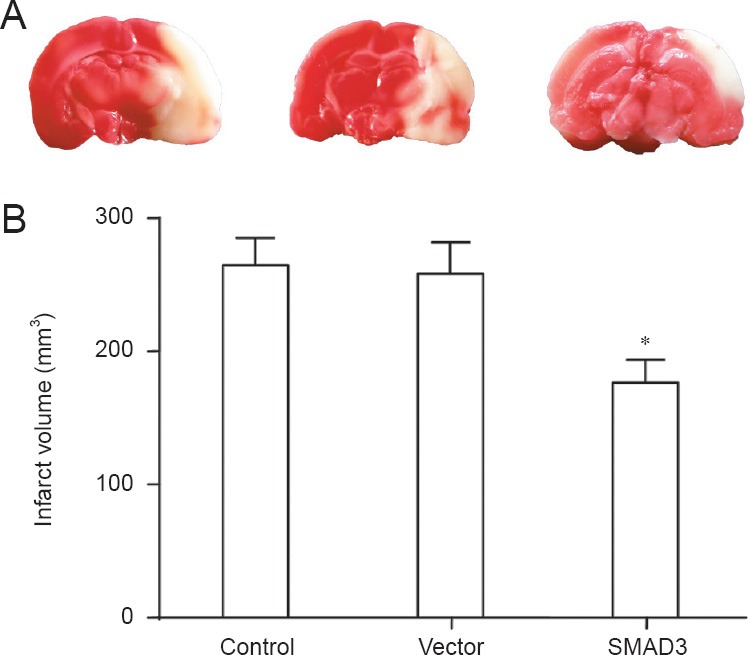
Infarct volume in ischemic brain of rats 24 hours after reperfusion (TTC staining).
In the control, SMAD3 and vector groups, rats were injected via the tail vein with 100 μL PBS, AAV9-SMAD3 (3 × 1013 vg/mL per kg body weight), and AAV9-SMAD3 control AAV9-GFP (3 × 1013 vg/mL per kg body weight), respectively. (A) Coronal brain sections of control, vector, and SMAD3 groups (left, middle, right); the pale area is the infarction. (B) Quantitative results showed that compared with the control and vector groups, infarct volume was reduced in the SMAD3 group (*P < 0.05; one-way analysis of variance followed by Student-Newman-Keul test). Data are presented as the mean ± SD from at least three independent experiments.
SMAD2/3 mRNA levels in the penumbra after cerebral ischemia/reperfusion
Real-time quantitative PCR showed a low level of SMAD2/3 mRNA in the control and sham groups. At 6 hours after cerebral ischemia/reperfusion, SMAD2/3 mRNA levels were up-regulated and were higher than those in the control and sham groups (P < 0.05). Subsequently, the level of SMAD2/3 mRNA in the ischemia/reperfusion group kept increasing with time, and the increase was still observed after 72 hours. There were significant differences between cerebral ischemia/reperfusion groups and the control group (P < 0.05; Figure 2).
Figure 2.
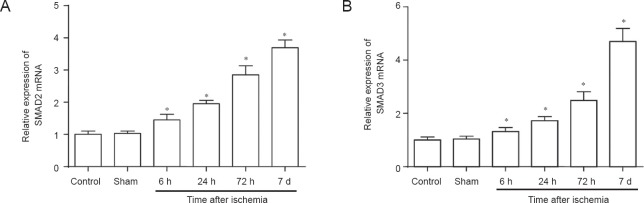
SMAD2/3 mRNA levels in the penumbra after cerebral ischemia/reperfusion in rats (real-time quantitative PCR).
(A, B) SMAD2/3 mRNA was up-regulated over time after reperfusion. *P < 0.05, vs. controls (one-way analysis of variance followed by Student- Newman-Keul test). Data are presented as the mean ± SD from at least three independent experiments. h: Hours; d: days.
SMAD2/3 and pSMAD2/3 protein in the penumbra after cerebral ischemia/reperfusion
In the model group, the level of phosphorylated SMAD2/3 protein (p-SMAD2/3) at 6 hours was higher than that in the control and sham groups. This level peaked at 72 hours, and was then decreased at 7 days. At 6, 24 and 72 hours after ischemia, SMAD2/3 protein levels in the penumbra of the model group showed no significant difference compared with the control and sham groups. The model group had a higher level of SMAD2/3 protein compared with the control and sham groups only at 7 days (Figure 3).
Figure 3.
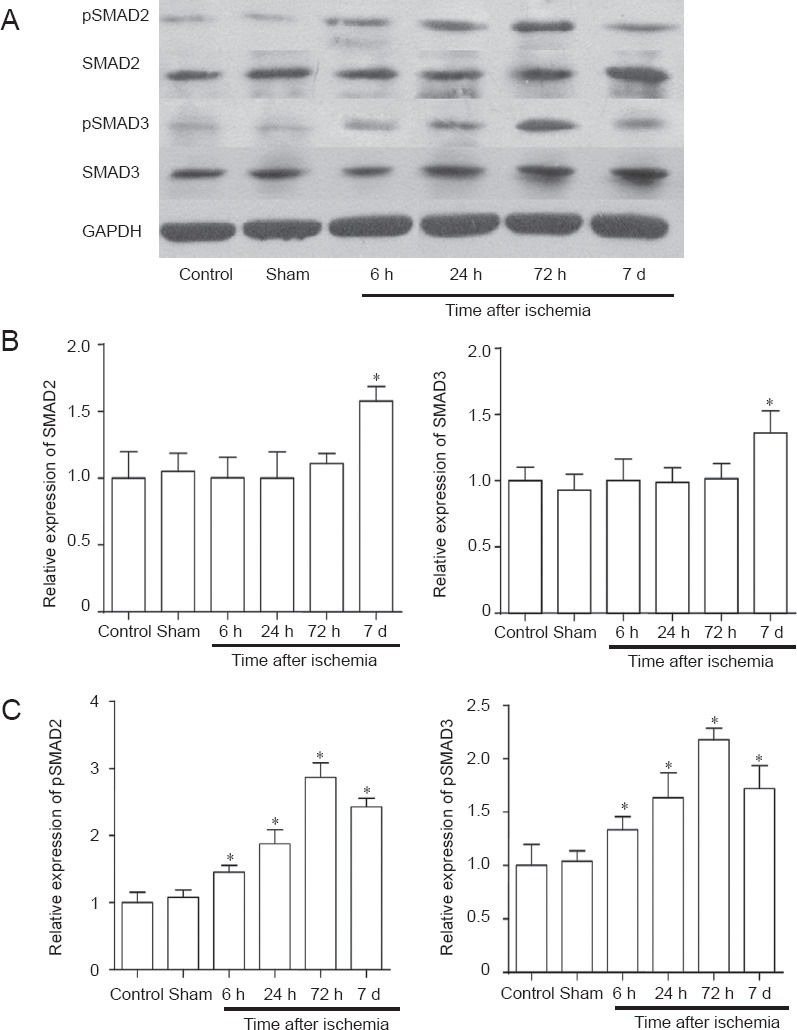
Levels of SMAD2/3 and phosphorylated-SMAD2/3 (p-SMAD2/3) in the penumbra after cerebral ischemia/reperfusion in rats.
(A) Western blot analysis of SMAD2/3 and pSMAD2/3. (B, C) Quantification of SMAD2/3 (B) and p-SMAD2/3 protein (C). (B) At 6, 24 and 72 hours (h) after ischemia, the level of SMAD2/3 protein in the ischemic penumbra showed no significant changes and was slightly up-regulated after 7 days (d). (C) p-SMAD2/3 was up-regulated at 6 h, peaked at 72 h, and was decreased after 7 d. *P < 0.05, vs. controls (one-way analysis of variance followed by Student-Newman-Keul test). Data are presented as the mean ± SD from at least three independent experiments.
Correlation between SMAD3 expression and cell apoptosis or inflammation
At 24 hours after reperfusion, Spearman correlation analysis showed that in the ischemic penumbra, SMAD3 was negatively correlated with the number of apoptotic cells (r = −0.718, P < 0.01; Figure 4A). SMAD3 was negatively correlated with the mRNA levels of IL-1β, an important pro-inflammatory cytokine (r = −0.859, P < 0.01; Figure 4B).
Figure 4.
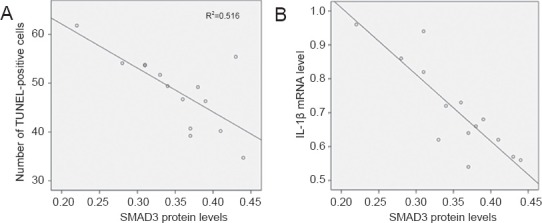
Scatter plot of the correlation between cell apoptosis and SMAD3 protein levels or interleukin-1β (IL-1β) mRNA levels in the penumbra after cerebral ischemia/reperfusion (Spearman correlation analysis).
SMAD3 was negatively correlated with the number of apoptotic cells (r = −0.718, P < 0.01) and the level of IL-1β mRNA (r = −0.859, P < 0.01).
Over-expression of SMAD3 decreased levels of TNF-α and IL-1β mRNA in the ischemic region
Twenty-four hours after reperfusion, there was no significant difference in the levels of tumor necrosis factor-α (TNF-α) or interleukin-1β (IL-1β) mRNA between control and vector groups (P > 0.05). TNF-α and IL-1β mRNA levels in the SMAD3 group were significantly lower compared with those in the control and vector groups (P < 0.05; Figure 5).
Figure 5.
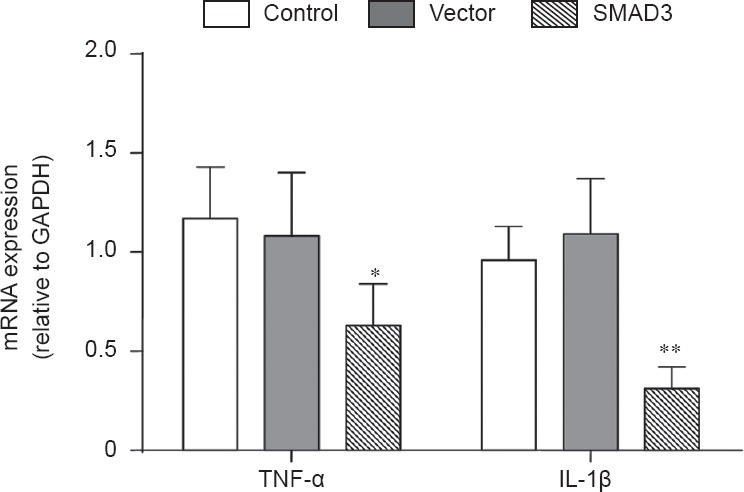
Over-expression of SMAD3 reduced the levels of tumor necrosis factor-α (TNF-α) and interleukin-1β (IL-1β) mRNA in ischemic brain of rats 24 hours after reperfusion.
In the control, SMAD3 and vector groups, rats were injected via the tail vein with 100 μL PBS, AAV9-SMAD3 (3 × 1013 vg/mL per kg body weight), and AAV9-SMAD3 control AAV9-GFP (3 × 1013 vg/mL per kg body weight), respectively. *P < 0.05, **P < 0.01, vs. controls (one-way analysis of variance followed by Student-Newman-Keul test). Data are presented as the mean ± SD from at least three independent experiments.
Over-expression of SMAD3 increased Bcl-2 and decreased Caspase-3 protein levels in the ischemic region
Twenty-four hours after reperfusion, the level of SMAD3 protein in the SMAD3 group (0.98 ± 0.24) was significantly higher than that in the control (0.63 ± 0.23, P < 0.05) and vector groups (0.69 ± 0.35, P < 0.05). The level of Bcl-2 protein in the SMAD3 group (0.68 ± 0.18) was increased compared with that in the control (0.39 ± 0.22, P < 0.05) and vector groups (0.37 ± 0.13, P < 0.01). Levels of Caspase-3 and cleavage Casp3 protein in the SMAD3 group were significantly lower than in the control group (P < 0.05 or P < 0.01). There was no significant difference in SMAD3, Bcl-2 or Caspase-3 protein levels between control and vector groups (P > 0.05; Figure 6).
Figure 6.
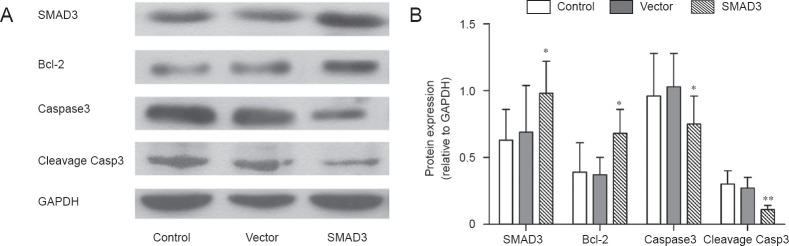
Over-expression of SMAD3 up-regulated Bcl-2 and down-regulated Caspase-3 and cleavage Casp3 proteins in ischemic brain of rats at 24 hours after reperfusion (western blot assay).
In the control, SMAD3 and vector groups, rats were injected via the tail vein with 100 μL PBS, AAV9-SMAD3 (3 × 1013 vg/mL per kg body weight), and AAV9-SMAD3 control AAV9-GFP (3 × 1013 vg/mL per kg body weight), respectively. *P < 0.05, **P < 0.01, vs. controls (one-way analysis of variance followed by Student-Newman-Keul test). Data are presented as the mean ± SD from at least three independent experiments.
Discussion
The TGF-β family consists of over 40 proteins, including activins, inhibins, Mullerian inhibitor substance, and bone morphogenetic proteins (Conway and Kaartinen, 2011). TGF-βs are important members of the TGF-β family and four TGF-β subtypes are found in mammals: TGF-β1, TGF-β2, TGF-β3 and TGF-β1β2 (Brown et al., 1985). Under physiological conditions, TGF-β1 is expressed in astrocytes and neurons. A number of studies have shown that brain TGF-β levels are up-regulated in both humans and animals with cerebral ischemia and that TGF-β1 is the predominant subtype among active TGF-βs in the brain. Although TGF-β1 has apparent neuroprotective effects against cerebral ischemia/reperfusion injury, the expression of its main downstream signaling molecules, SMAD2 and SMAD3, after cerebral ischemia/reperfusion and the role of SMAD2/3 in the neuroprotective mechanism of TGF-β1 remain unclear. In this study, we determined the dynamic changes of SMAD2/3 mRNA and protein expression in the brain after cerebral ischemia/reperfusion. The results showed that at 6 hours after cerebral ischemia/reperfusion, levels of SMAD2/3 mRNA in the ischemic penumbra were higher than those in normal brain tissue. These levels then gradually increased, peaked at 72 hours, and were then decreased after 7 days. However, at 6, 24, and 72 hours after ischemia, SMAD2/3 protein levels in the ischemic penumbra were significantly higher compared with those in normal tissue only at the time point of 7 days. Therefore, we speculate that cerebral ischemia/reperfusion contributes to induce transcriptional activation of the SMAD2/3 gene. Owing to certain post-transcriptional regulation factors, SMAD2/3 mRNA cannot be efficiently translated or translated protein is rapidly degraded, leading to different levels of the SMAD2/3 mRNA and protein.
Although the level of SMAD2/3 protein was not significantly changed in the early period following cerebral ischemia/reperfusion, we found that the level of active p-SMAD2/3 was consistent with the mRNA level, showing a gradual up-regulation. The TGF-β/SMAD2/3 signaling pathway is mediated by the phosphorylation of SMAD2/3. First, the binding of active TGF-β1 with TβRII induces phosphorylation of the TβRIGS domain. This complex then migrates toward the nucleus under the action of a small GTP enzyme, such as Rab5, and binds with SARA (Smad anchor for Receptor Activation) at the cell membrane (Lan, 2011). SARA may bind with the unactivated SMAD2/3. The binding of the activated receptor with SMAD2/3 contributes to SSXS domain phosphorylation of SMAD2/3, which decreases the affinities of TβR, SARA and SMAD2/3, leading to disintegration of the complex (Javelaud et al., 2011). Activated SMAD2/3 binds with SMAD4 to form a complex that regulates gene expression within the nucleus. Therefore, the increased p-SMAD2/3 content caused by ischemia and reperfusion may be involved in the neuroprotective effect of TGF-β.
SMAD2 and SMAD3 are highly homologous and both can be simultaneously activated by TGF-β in many tissues and cells, although accumulating evidence shows that SMAD2 and SMAD3 have some different functions. However, it is unclear if SMAD2 and SMAD3 have different responses to nerve damage after cerebral ischemia/reperfusion. Therefore, we analyzed the correlation between SMAD2/3 protein and cell apoptosis or inflammation following cerebral ischemia/reperfusion. The correlation analysis results showed that, SMAD2 was not correlated with apoptosis or inflammation, while SMAD3 had a negative correlation with the inflammatory response and apoptosis. We speculate that, SMAD3 may have neuroprotective effects and that the effect is mediated by a major downstream signaling molecule.
Gene vectors are important tools for studies of pathogenesis and gene therapy, and have been widely applied in the research of nervous system diseases (Park et al., 2012; Taniyama et al., 2012; Schlenk et al., 2013). Gene expression vectors can be divided into viral vectors and non-viral vectors (Humbert and Halary, 2012). Viral vectors have a high transfection efficiency, produce stable expression and low immunogenicity, and can effectively infect and integrate into non-dividing cells such as neurons; therefore, viral vectors are especially useful in nervous system research (Hester et al., 2009; Betley and Sternson, 2011; Low and Aebischer, 2012). The present study aimed to observe the effect of SMAD3 over-expression in neurons after cerebral ischemia/reperfusion injury; therefore, we used an adeno-associated virus carrying the SMAD gene. Western blot analysis showed that, SMAD3 levels in the brains of rats after intracerebral injection of Ad-RSV-SMAD3 were significantly higher compared with the empty viral vector and control groups.
The present study showed that SMAD3 levels were negatively correlated with apoptosis and inflammation in the ischemic region after cerebral ischemia/reperfusion, suggesting that SMAD3 may mediate neuroprotective effects of TGF-β. Furthermore, SMAD3 over-expression reduced the infarct volume. This evidence indicated that SMAD3 ameliorated cell apoptosis and brain injury after cerebral ischemia/reperfusion. Previous studies showed that the TGF-β/SMAD2/3 signaling pathway was responsible for the apoptosis and growth of a variety of cells (Heldin et al., 2009). Conversely, TGF-β/SMAD2/3 signaling protects neurons from stress-induced apoptosis, and SMAD3 plays a crucial role in the development, growth and differentiation of nerve cells (Docagne et al., 2002; Garcia-Campmany and Marti, 2007; Katsuno et al., 2011; Estaras et al., 2012). However, the protective effect of SMAD3 on neurons is not fully understood. In this rat model of cerebral ischemia/reperfusion, SMAD3 over-expression caused the up-regulation of Bcl2 and the down-regulation of Caspase-3. Apoptosis is the result of a series of highly regulated Caspase cascades, and Caspase-3 is the most critical apoptotic protease in the downstream cascade. Bcl-2 can induce a neuroprotective effect and Bcl-2 over-expression contributes to inhibit the apoptosis of nerve cells caused by a variety of factors. Previous studies demonstrated that, after removal of trophic factors, TGF-β exerts a neuroprotective effect through the up-regulation of Bcl2. Therefore, we speculate that SMAD3 inhibits neuronal apoptosis after cerebral ischemia/reperfusion by increasing Bcl-2 and decreasing Caspase-3 expression.
Like apoptosis, inflammation is another indicator of ischemia/reperfusion injury. SMAD3 is closely involved with inflammation. The TGF-β pathway contributed to reduce inflammation and β amyloid deposition in an Alzheimer's disease model, through inhibition of NF-κB and MAPK (Flores and von Bernhardi, 2012). Moreover, SMAD3 mediated a TGF-β1 inhibition effect on microglial activation, thus reducing inflammation in the central nervous system (Le et al., 2004; Huang et al., 2010). To date, no study has addressed the correlation between SMAD3 and inflammation after cerebral ischemia/reperfusion. The present study demonstrated that over-expression of SMAD3 triggered the down-regulation of TNF-α and IL-1β after ischemia/reperfusion. This evidence indicated that SMAD3 can not only play an anti-apoptotic role after cerebral ischemia and reperfusion, but that it also has the potential to inhibit inflammation and reduce acute brain injury. However, further studies are needed to understand the mechanism associated with the down-regulation of TNF-α and IL-1β, as well as interactions with other inflammatory signaling pathways after injury.
In summary, our study showed that levels of SMAD2 and SMAD3 mRNAs, as well as levels of pSMAD2 and pSMAD3, were up-regulated in the penumbra after cerebral ischemia. Furthermore, apoptosis and neuro-inflammation in ischemic penumbra were negatively correlated with levels of SMAD3 and could be ameliorated by SMAD3 over-expression in a rat model of cerebral ischemia/reperfusion. Our findings indicate that, SMAD3 has anti-apoptotic and anti-inflammatory effects after cerebral ischemia, and is thus a potential drug target in the clinical treatment of cerebral infarction.
Footnotes
Funding: This work was supported by the National Natural Science Foundation of China, No. 81460193.
Conflicts of interest: None declared.
Copyedited by Allen J, Raye W, Li CH, Song LP, Zhao M
References
- Akhurst RJ, Hata A. Targeting the TGFbeta signalling pathway in disease. Nat Rev Drug Discov. 2012;11:790–811. doi: 10.1038/nrd3810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Araujo-Jorge TC, Waghabi MC, Bailly S, Feige JJ. The TGF-beta pathway as an emerging target for Chagas disease therapy. Clin Pharmacol Ther. 2012;92:613–621. doi: 10.1038/clpt.2012.102. [DOI] [PubMed] [Google Scholar]
- Baron JC, Yamauchi H, Fujioka M, Endres M. Selective neuronal loss in ischemic stroke and cerebrovascular disease. J Cereb Blood Flow Metab. 2014;34:2–18. doi: 10.1038/jcbfm.2013.188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Betley JN, Sternson SM. Adeno-associated viral vectors for mapping, monitoring, and manipulating neural circuits. Hum Gene Ther. 2011;22:669–677. doi: 10.1089/hum.2010.204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beyer TA, Narimatsu M, Weiss A, David L, Wrana JL. The TGFbeta superfamily in stem cell biology and early mammalian embryonic development. Biochim Biophys Acta. 2013;1830:2268–2279. doi: 10.1016/j.bbagen.2012.08.025. [DOI] [PubMed] [Google Scholar]
- Bonni S, Bonni A. SnoN signaling in proliferating cells and postmitotic neurons. FEBS Lett. 2012;586:1977–1983. doi: 10.1016/j.febslet.2012.02.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown JP, Twardzik DR, Marquardt H, Todaro GJ. Vaccinia virus encodes a polypeptide homologous to epidermal growth factor and transforming growth factor. Nature. 1985;313:491–492. doi: 10.1038/313491a0. [DOI] [PubMed] [Google Scholar]
- Caraci F, Spampinato S, Sortino MA, Bosco P, Battaglia G, Bruno V, Drago F, Nicoletti F, Copani A. Dysfunction of TGF-beta1 signaling in Alzheimer's disease: perspectives for neuroprotection. Cell Tissue Res. 2012;347:291–301. doi: 10.1007/s00441-011-1230-6. [DOI] [PubMed] [Google Scholar]
- Conway SJ, Kaartinen V. TGFbeta superfamily signaling in the neural crest lineage. Cell Adh Migr. 2011;5:232–236. doi: 10.4161/cam.5.3.15498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Docagne F, Nicole O, Gabriel C, Fernandez-Monreal M, Lesne S, Ali C, Plawinski L, Carmeliet P, MacKenzie ET, Buisson A, Vivien D. Smad3-dependent induction of plasminogen activator inhibitor-1 in astrocytes mediates neuroprotective activity of transforming growth factor-beta 1 against NMDA-induced necrosis. Mol Cell Neurosci. 2002;21:634–644. doi: 10.1006/mcne.2002.1206. [DOI] [PubMed] [Google Scholar]
- Dorado L, Millan M, Davalos A. Reperfusion therapies for acute ischemic stroke: an update. Curr Cardiol Rev. 2014;10:327–335. doi: 10.2174/1573403X10666140320144637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Estaras C, Akizu N, Garcia A, Beltran S, de la Cruz X, Martinez-Balbas MA. Genome-wide analysis reveals that Smad3 and JMJD3 HDM co-activate the neural developmental program. Development. 2012;139:2681–2691. doi: 10.1242/dev.078345. [DOI] [PubMed] [Google Scholar]
- Flores B, von Bernhardi R. Transforming growth factor beta1 modulates amyloid beta-induced glial activation through the Smad3-dependent induction of MAPK phosphatase-1. J Alzheimers Dis. 2012;32:417–429. doi: 10.3233/JAD-2012-120721. [DOI] [PubMed] [Google Scholar]
- Garcia-Campmany L, Marti E. The TGFbeta intracellular effector Smad3 regulates neuronal differentiation and cell fate specification in the developing spinal cord. Development. 2007;134:65–75. doi: 10.1242/dev.02702. [DOI] [PubMed] [Google Scholar]
- Goldlust EJ, Paczynski RP, He YY, Hsu CY, Goldberg MP. Automated measurement of infarct size with scanned images of triphenyltetrazolium chloride-stained rat brains. Stroke. 1996;27:1657–1662. doi: 10.1161/01.str.27.9.1657. [DOI] [PubMed] [Google Scholar]
- Heldin CH, Landstrom M, Moustakas A. Mechanism of TGF-beta signaling to growth arrest, apoptosis, and epithelial-mesenchymal transition. Curr Opin Cell Biol. 2009;21:166–176. doi: 10.1016/j.ceb.2009.01.021. [DOI] [PubMed] [Google Scholar]
- Hester ME, Foust KD, Kaspar RW, Kaspar BK. AAV as a gene transfer vector for the treatment of neurological disorders: novel treatment thoughts for ALS. Curr Gene Ther. 2009;9:428–433. doi: 10.2174/156652309789753383. [DOI] [PubMed] [Google Scholar]
- Huang WC, Yen FC, Shie FS, Pan CM, Shiao YJ, Yang CN, Huang FL, Sung YJ, Tsay HJ. TGF-beta1 blockade of microglial chemotaxis toward Abeta aggregates involves SMAD signaling and down-regulation of CCL5. J Neuroinflammation. 2010;7:28. doi: 10.1186/1742-2094-7-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humbert JM, Halary F. Viral and non-viral methods to genetically modify dendritic cells. Curr Gene Ther. 2012;12:127–136. doi: 10.2174/156652312800099580. [DOI] [PubMed] [Google Scholar]
- Javelaud D, Alexaki VI, Dennler S, Mohammad KS, Guise TA, Mauviel A. TGF-beta/SMAD/GLI2 signaling axis in cancer progression and metastasis. Cancer Res. 2011;71:5606–5610. doi: 10.1158/0008-5472.CAN-11-1194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joseph JV, Balasubramaniyan V, Walenkamp A, Kruyt FA. TGF-beta as a therapeutic target in high grade gliomas - promises and challenges. Biochem Pharmacol. 2013;85:478–485. doi: 10.1016/j.bcp.2012.11.005. [DOI] [PubMed] [Google Scholar]
- Kalluri HS, Dempsey RJ. Growth factors, stem cells, and stroke. Neurosurg Focus. 2008;24:E14. doi: 10.3171/FOC/2008/24/3-4/E13. [DOI] [PubMed] [Google Scholar]
- Katsuno M, Adachi H, Banno H, Suzuki K, Tanaka F, Sobue G. Transforming growth factor-beta signaling in motor neuron diseases. Curr Mol Med. 2011;11:48–56. doi: 10.2174/156652411794474356. [DOI] [PubMed] [Google Scholar]
- Kim BJ, Kang HG, Kim HJ, Ahn SH, Kim NY, Warach S, Kang DW. Magnetic resonance imaging in acute ischemic stroke treatment. J Stroke. 2014;16:131–145. doi: 10.5853/jos.2014.16.3.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knuckey NW, Finch P, Palm DE, Primiano MJ, Johanson CE, Flanders KC, Thompson NL. Differential neuronal and astrocytic expression of transforming growth factor beta isoforms in rat hippocampus following transient forebrain ischemia. Brain Res Mol Brain Res. 1996;40:1–14. doi: 10.1016/0169-328x(96)00016-2. [DOI] [PubMed] [Google Scholar]
- Konkel JE, Chen W. Balancing acts: the role of TGF-beta in the mucosal immune system. Trends Mol Med. 2011;17:668–676. doi: 10.1016/j.molmed.2011.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovacic P, Somanathan R. Cell signaling and receptors in toxicity of advanced glycation end products (AGEs): alpha-dicarbonyls, radicals, oxidative stress and antioxidants. J Recept Signal Transduct Res. 2011;31:332–339. doi: 10.3109/10799893.2011.607171. [DOI] [PubMed] [Google Scholar]
- Lan HY. Diverse roles of TGF-beta/Smads in renal fibrosis and inflammation. Int J Biol Sci. 2011;7:1056–1067. doi: 10.7150/ijbs.7.1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Y, Iribarren P, Gong W, Cui Y, Zhang X, Wang JM. TGF-beta1 disrupts endotoxin signaling in microglial cells through Smad3 and MAPK pathways. J Immunol. 2004;173:962–968. doi: 10.4049/jimmunol.173.2.962. [DOI] [PubMed] [Google Scholar]
- Low K, Aebischer P. Use of viral vectors to create animal models for Parkinson's disease. Neurobiol Dis. 2012;48:189–201. doi: 10.1016/j.nbd.2011.12.038. [DOI] [PubMed] [Google Scholar]
- Manning NW, Campbell BC, Oxley TJ, Chapot R. Acute ischemic stroke: time, penumbra, and reperfusion. Stroke. 2014;45:640–644. doi: 10.1161/STROKEAHA.113.003798. [DOI] [PubMed] [Google Scholar]
- Neuhaus AA, Rabie T, Sutherland BA, Papadakis M, Hadley G, Cai R, Buchan AM. Importance of preclinical research in the development of neuroprotective strategies for ischemic stroke. JAMA Neurol. 2014;71:634–639. doi: 10.1001/jamaneurol.2013.6299. [DOI] [PubMed] [Google Scholar]
- Park J, Singha K, Son S, Kim J, Namgung R, Yun CO, Kim WJ. A review of RGD-functionalized nonviral gene delivery vectors for cancer therapy. Cancer Gene Ther. 2012;19:741–748. doi: 10.1038/cgt.2012.64. [DOI] [PubMed] [Google Scholar]
- Pera J, Zawadzka M, Kaminska B, Szczudlik A. Influence of chemical and ischemic preconditioning on cytokine expression after focal brain ischemia. J Neurosci Res. 2004;78:132–140. doi: 10.1002/jnr.20232. [DOI] [PubMed] [Google Scholar]
- Rosso C, Samson Y. The ischemic penumbra: the location rather than the volume of recovery determines outcome. Curr Opin Neurol. 2014;27:35–41. doi: 10.1097/WCO.0000000000000047. [DOI] [PubMed] [Google Scholar]
- Sakaki-Yumoto M, Katsuno Y, Derynck R. TGF-beta family signaling in stem cells. Biochim Biophys Acta. 2013;1830:2280–2296. doi: 10.1016/j.bbagen.2012.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez EC. Mechanisms of action of hyperbaric oxygenation in stroke: a review. Crit Care Nurs Q. 2013;36:290–298. doi: 10.1097/CNQ.0b013e318294e9e3. [DOI] [PubMed] [Google Scholar]
- Sauser K, Burke JF, Reeves MJ, Barsan WG, Levine DA. A systematic review and critical appraisal of quality measures for the emergency care of acute ischemic stroke. Ann Emerg Med. 2014;64:235–244.e5. doi: 10.1016/j.annemergmed.2014.01.034. [DOI] [PubMed] [Google Scholar]
- Schlenk F, Grund S, Fischer D. Recent developments and perspectives on gene therapy using synthetic vectors. Ther Deliv. 2013;4:95–113. doi: 10.4155/tde.12.128. [DOI] [PubMed] [Google Scholar]
- Taniyama Y, Azuma J, Kunugiza Y, Iekushi K, Rakugi H, Morishita R. Therapeutic option of plasmid-DNA based gene transfer. Curr Top Med Chem. 2012;12:1630–1637. doi: 10.2174/156802612803531342. [DOI] [PubMed] [Google Scholar]