

Abstract
Culturing human Pluripotent Stem Cells (hPSC)s in chemically defined medium and feeder-free condition can facilitate metabolome and proteome analysis of culturing cells and medium, and reduce regulatory concerns for clinical application of cells. And in addition, if hPSC are passaged and cryopreserved in single cells it also facilitates quality control of cells at single cell level. Here we report a robust single cell freezing and thawing method of hPSCs cultured in chemically-defined medium TeSRTM-E8TM and on cost-effective recombinant human Vitronectin-N (rhVTN-N)-coated dish. Cells are dissociated into single cells with recombinant TrypLETM Select and 0.5 mM EDTA/PBS (3:1 solution) in the presence of Rock inhibitor and cryopreserved with chemically defined CryoStemTM. Approximately 60% of cells were viable after dissociation. AggrewellTM 400 was used to form cell clumps of 500 cells after thaw in the presence of Rock inhibitor and cells were cultured for two days with TeSR-E8. Cells clumps were then seeded on rhVTN-N-coated dish and cultured with TeSR-E8 for two days prior to the first passage after thawing. Number of viable cells at the first passage increased around 10 times of that just before freezing. This robust single cell freezing method for hPSCs cultured in chemically defined medium will facilitate quality control of cultured cells at single cell level before cryopreservation and consequently assure the quality of cells in frozen vials for further manipulation after thawing.
Keywords: Human pluripotent stem cells (hPSCs), slow-freezing, Feeder-free, chemically-defined, single cell
Introduction
Cell therapy using human pluripotent stem cell (hPSC)-derived cells, such as human embryonic stem cell (hESC)-derived retinal pigment epithelium (RPE) or induced pluripotent stem cell (iPSC)-derived RPEs [1-3], have been conducted or scheduled by several groups [4-6]. Therefore, methods for culturing hPSCs have been developed to meet regulatory demands ensuring the safety and quality of cellular products. For example, hPSCs can be maintained with animal component-free, chemically-defined, and xeno-free medium [7-10] under feeder-free conditions e.g., using laminin-521, laminin-E8, pronectin, recombinant human vitronectin-N (rhVTN-N) [10-14]. Further, study for metabolome of cultured cells can be facilitated by culturing cells with chemically defined medium. In this context, a robust cryopreservation method for hPSCs cultured under these conditions needed to be developed to catch up with current technology of hPSCs cell culture.
Two types of cryopreservation method are currently used for hPSCs, namely vitrification and slow-freezing [15,16]. The vitrification method requires skill in rapid freezing with liquid nitrogen and is not suitable for the cryopreservation of large numbers of hPSCs [17]. In contrast, slow-freezing methods, which involve re-suspending hPSCs in cryopreservation medium followed by gradual freezing in a deep freezer, do not require special skills and allow for the cryopreservation of large numbers of hPSCs in a single batch. However, low recovery rates have been an issue. We recently reported a robust and easy cryopreservation method for hPSCs cultured on feeder cells (MEF, SNL76/7) using reagents already available in clinical settings [18]. And next we tried to develop a robust single cells cryopreservation method for hPSCs cultured in chemically defined TeSRTM-E8TM medium and rhVTN-N conditions to facilitate quality control of cells at single cell level in cryopreservation process.
In this report, we examine four different cryopreservation mediums/cocktails in combination with seven different cell dissociation conditions and discuss the mechanism of action of these agents.
Materials and methods
All experiments using human cell lines and animals were reviewed and approved by the committee for non-clinical research and committee for animal experiment of the FBRI.
Cell culture
The hESC lines KhES-1 (Riken BRC), H9 and hiPSC line PFX # 9 were used in a series of freeze/thaw experiments. These cell lines were cultured with TeSRTM-E8TM medium (Stemcell Technologies, cat # 05840) on rhVTN-N (Gibco, cat # A14700) coated-6-well tissue culture dish (BD, cat # 353046) at 37°C, 5% CO2 in an incubator (Panasonic, MCO-19AIC). These cells were dissociated with dissociation buffer (Gentle Cell Dissociation Reagent, GCDR, Stemcell Technologies, cat # 27174) at room temperature (15-25°C) for passage. Dissociated cells in small cell clumps were collected and washed once with 1 mL of PBS(-), and one fourth of the cells were seeded on rhVTN-N-coated dishes and cultured with 2 mL of TeSRTM-E8TM medium per well of a 6-well dish for four days until the next passage. Media were changed every day. Murine feeder cells SNL 76/7 (European Collection of Cell Cultures: ECACC) and human mononuclear cells (MNCs) from cord blood for research use (Riken BRC) were used for examining the expression of Neu5Gc using the FACS AriaTM II cell sorter (BD). MNCs were thawed and then resuspended in PBS(-) in the presence of human serum albumin (HSA) for detection of Neu5Gc with an anti-Neu5Gc antibody kit (BioLegend, cat # 146901).
Dissociation and cryopreservation procedure
Four different cell dissociation reagents were used to make single cell suspensions from cell colonies: I) AccutaseTM (STEMCELL Technologies, cat # 07920); II) Pronase (KYOKUTO, cat # 2810); III) TrypLETM Select (GIBCO, cat # 12563-011); and IV) EDTA (Nacalai Tesque, 15111-45)/PBS(-) (GIBCO, 14190-250). Before dissociation, TeSRTM-E8TM medium was replaced with fresh medium containing 5 µM Rock inhibitor (Y-27632, Nacalai Tesque, cat # 08945-84) and incubated for 2 hours. hPSCs cultured in two wells of 6-well culture dish were dissociated for each of the four conditions, collected with a cell scraper (IWAKI, 9000-220) and pipetted to further dissociate them into single cells. Cells were pelleted by centrifugation (120 G, 3 min) and resuspended in 1 mL PBS(-) for scoring viable cell number. Approximately 6 × 105 viable hPSCs from two wells of 6-well dish in single cell suspension were harvested and resuspended with 1 mL of one of the four cryopreservation mediums: I) CryoStemTM (Biological Industries, cat # 05-710-1E); II) CP-1 (KYOKUTO, cat # 551-27200-0); III) CP-1 with HSA (25% human serum albumin, Mitsubishi Tanabe Pharma, cat # 19553) and IV) TC protector (DS Pharma Biomedical Co., Ltd., cat # TCP-001). Cells in cryopreservation medium were transferred to cryovials (ASAHI TECHNO, 2792-002). Cryovials were placed into a freezing container (NALGENETM Cryo 1°C Freezing Container (Nalgene, cat # 5100-0001) and frozen in a -80°C freezer (Panasonic, MDF-U32V) overnight. Cells were then transferred to and stored in a -150°C freezer (Panasonic, MDF-1155AT) for at least one week before performing thawing experiments.
Thawing procedure
Cryovials were warmed in a water bath at 37°C until the icy masses disappeared, and cell suspensions were transferred to 15 mL centrifuge tubes (BD, cat # 352096) and diluted by addition of 4 mL TeSR-E8 medium containing 5 M Y-27632. Cells were pelleted by centrifugation (120 G, 3 min) and resuspended in 1 mL of fresh TeSRTM-E8TM medium containing 5 M Y-27632 and seeded in AggreWellTM 400 plates (STEM CELL Technologies, cat # 27845) by pipetting in order to distribute the cells evenly throughout the wells. Culture dish were centrifuged at 100 G for 3 min in a swinging bucket rotor to pellet cells; approximately 500 cells were pelleted in each V-bottom microwell (1200 microwells/one AggreWellTM 400). For preparation of AggreWell 400 plates prior to cell seeding, 1 mL TeSR-E8 medium containing 5 M Y-27632 was added to AggreWellTM 400 plates and air bubbles were expelled by pipetting and shaking. The plates were then centrifuged at 2000 G (or at the maximum speed of the rotor) for 5 min in a swinging bucket rotor. Cell clumps were then collected from all wells after two days of incubation at 37°C, 5% CO2, transferred to three rhVTN-N-coated 6-wells dish, and cultured at 37°C, 5% CO2 for another two days with TeSR-E8 medium lacking Y-27632.
Media were changed every day. Numbers of cells and colonies in the three wells of the 6-well dish were scored four days after thawing. To compare the survival of hPSCs thawed via cell clump formation with those thawed without clump formation (single cells), hPSCs in single cell suspension were seeded onto rhVTN-N-coated wells of 24-well, flat-bottom dish (BD, cat # 353047) at cell densities of 6 × 104, 6 × 103, and 6 × 102 cells/well, cultured for four days, and scored for viable cell number.
Alkaline phosphatase (ALP) staining and colony counts
The activity of ALP was visualized after fixation with 4% (w/v) paraformaldehyde (PFA; Nacalai Tesque, cat # 09154-85) in PBS(-) (GIBCO, cat # 14190-144) using an alkaline phosphatase kit (Vector, cat # SK-5300) per the manufacturer’s instructions.
Immunohistochemistry (IHC)
hPSC colonies were fixed with 4% PFA overnight at 4°C, then washed with PBS(-) for 5 min, three times. When staining for POU5F1 and NANOG, 0.2% Triton-X (WAKO, cat # 162-24755) diluted in PBS(-) was added to the cells, followed by incubation at room temperature for 15 min for permeabilization. The fixed cells were washed with 0.1% FBS in PBS(-). Expression of POU5F1, NANOG, SSEA-4, TRA 1-60, and TRA 1-81 in cultured cells was detected with the respective antibody and visualized with a secondary antibody labeled with Alexa Fluor 488 (InvitrogenTM) or Alexa Fluor 594 (InvitrogenTM), and cells were counterstained with DAPI (InvitrogenTM). An Olympus IX71 fluorescence microscope was used for fluorescent observation. All antibodies are listed in Table S1. Image of positively stained colonies in whole dish aria was captured with image analysis software (ImageJ 1.45 s, National Institutes of Health, Bethesda, MD, USA) and percentage of area positively stained with respective antibody against that with DAPI (total cell colony) was calculated (Figure 3B).
Figure 3.
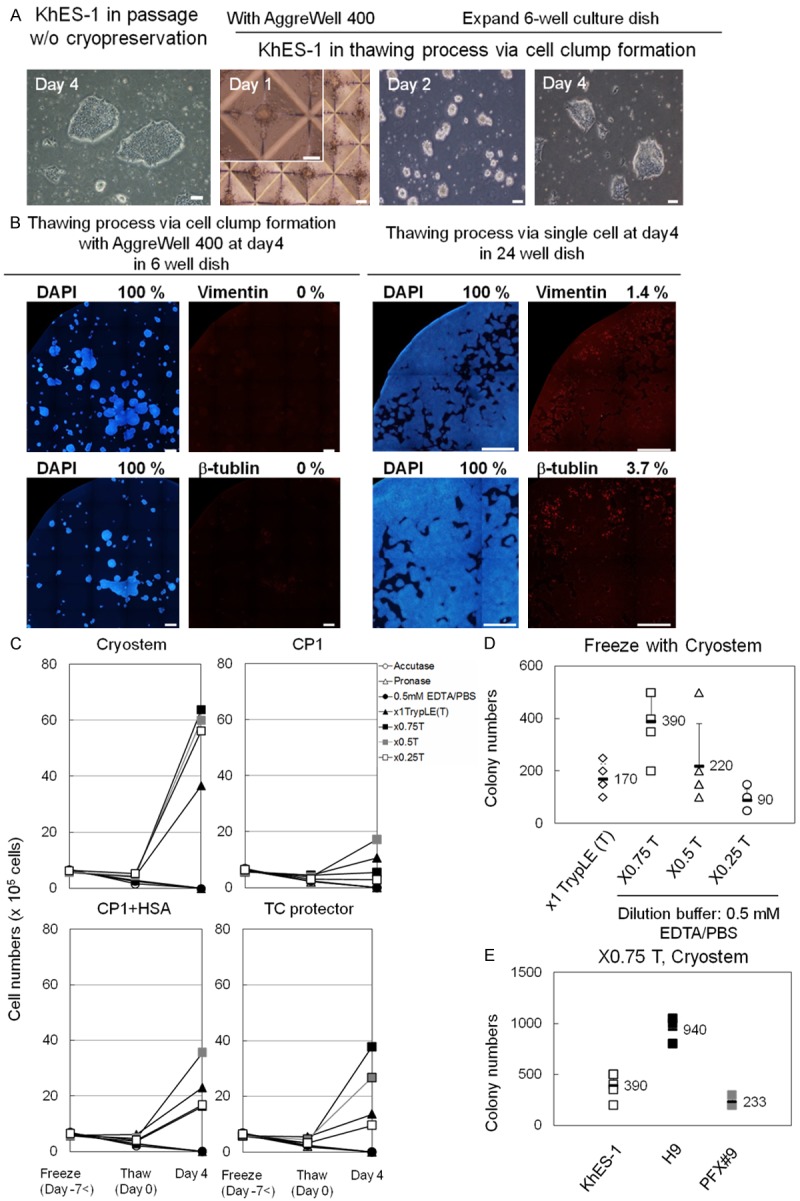
Recovery rate of hPSCs after thawing from various cryopreservation methods. A. Photos from left to right: Colonies of hPSCs (KhES-1) passaged without cryopreservation. Approx. 500 KhES-1 cells in AggreWell 400 after thaw at day 1. KhES-1 cells transferred to rhVTN-N-coated dish and cultured with TeSR-E8 at day 2. Cell clumps prior to the fist passage at day 4. Scale bar = 100 μm. B. KhES-1 cells cultured for four days via cell clump formation with AggreWell 400 plates were stained with DAPI, anti-Vimentin-, anti-β-tubulin-antibody at day 4 in 6-well dish (left panels). 6 × 104 KhES-1 cells seeded on rhVNT-N coated dish just after thawing without clump formation were cultured with TeSR-E8 for four days and stained with DAPI, anti-Vimentin-, anti-β-tubulin-antibody at day 4 in 24-well dish (right panels). Scale bar = 1 mm. Percentage of positively stained area with respective antibody using Image J software is shown in the right upper corner of the photo. C. Growth of KhES-1 cells after thawing, dissociated with seven dissociation reagents and frozen with four different cryopreservation mediums. Dissociation mediums: Acc:Accutage, Try:x1 TrypLE Select, pro:Pronase, EP:EDTA/PBS(-), 0.75T: x0.75 TrypLE Select diluted by 0.5 mM EDTA/PBS(-) [TrypLE Select: EDTA/PBS(-) = 3:1], 0.5T: x0.5 TrypLE Select diluted by EDTA/PBS(-) [TrypLE Select: EDTA/PBS(-) = 1:1], 0.25T: x0.25 TrypLE Select diluted by EDTA/PBS(-) [TrypLE Select: EDTA/PBS(-) = 1:3]. Cryopreservation mediums [cryo: CryoStem, CP1: CP-1, C+A: CP-1 with HSA, TC: TC protector]. Y axis: cell numbers and X axis: days after thaw. Freeze (Day-7<) mean 7 days or more prior to thaw. Means of results from five experiments are plotted and shown in line graphs. D. Numbers of colonies of KhES-1 cells scored in three wells of 6-well dish four days after thawing, dissociated with x1, x0.75, x0.5, or x0.25 TrypLE Select and frozen with CryoStem. Results from five independent experiments are plotted. Figures in graph are mean and error bars are standard deviation. E. Numbers of colonies of three hPSC lines (ESC:KhES-1, ESC:H9, iPSC:PFX#9) scored in 6-well dish four days after thawing via cell clump with AggrewWell 400. Cells were dissociated with x0.75 T (x0.75 TrypLE Select) and cryopreserved with CryoStem.
Quantitative RT-PCR
Total RNA was extracted using the RNeasy® micro kit (QIAGEN, cat # 74004) according to the manufacturer’s instructions. One mg of total RNA was used to synthesize cDNA with the Quant Tect Reverse Transcription Kit (QIAGEN, cat # 205311). Quantitative PCR (qPCR) was performed with the SYBR® Green PCR Master Mix (Applied Biosystems: cat # 4309155) and analyzed with a StepOnePlusTM Real-Time PCR System (Applied Biosystems, ABI). The sequences of all primers for pluripotency and lineage-specific genes are listed in Table S2. Gene expression was normalized to that of GAPDH as the internal control and quantified by the ΔΔCt method.
Flow cytometric analysis
hPSCs were harvested and dissociated to single cells with GCDR. The cells were washed once 0.1% FBS in PBS(-). A total of 5 × 105 cells was incubated with the same buffer containing 1/50 volume of the designated fluorescently labeled antibody for 30 min at 4°C. The cells were analyzed with a FACS AriaTM II cell sorter (BD) after washing once with 0.1% FBS in PBS(-). Alexa Fluor 647-conjugated anti-SSEA-3, FITC-conjugated anti-SSEA-4, BrilliantViolet421-conjugated anti-TRA 1-60, and PE-conjugated anti-TRA 1-81 (all antibodies from BD) were used for flow cytometric analysis.
For detection of Neu5Gc, cells were stained anti-Neu5Gc antibody (BioLegend, cat # 146901). hPSCs were dissociated into single cells with GCDR and washed once with accessory wash buffer. A total of 5 × 105 cells was incubated with the same buffer containing 1/100 volume of the designated fluorescently-labeled antibody for 1 hour at 4°C. Cells were washed once and incubated with the same buffer containing 1/100 volume of secondary antibody (Jackson Immuno Research, cat # 703-606-155) for 1 hour at 4°C. The cells were analyzed with a FACS AriaTM II cell sorter (BD) after washing once with accessory buffer. The dead cells were stained with 7-amino-actinomycin D (7AAD, BD).All antibodies for staining pluripotent stem cells are listed in Table S1.
Karyotype analysis and CGH array
G-band analysis: Cells were treated with Colcemid® Solution (GIBCO, cat # 15212-012) and CRA solution (Genial, cat # GGS-JL-003a) and harvested with 0.1% trypsin [2.5% solution (GIBCO, cat # 15090-046) diluted PBS(-)]. Cells were then suspended in hypotonic KCl solution (Nacalai Tesque, 28514-75) and incubated for 15 min at room temperature. Cells were fixed with Carnoy’s solution (acetic acid (Nacalai Tesque, cat # 00212-85): MeOH (Nacalai Tesque, cat # 21915-93), 1:3) and dropped onto glass slides. Glass slides were soaked in Coplin jars with pre-warmed (37°C) 0.005% trypsin [2.5% solution, GIBCO, diluted in Gurr’s 6.8 buffer (GIBCO, cat # 10582-013)] for a few seconds. Glass slides were transferred to ethanol (Nacalai Tesque, cat # 14713-53) for 2-3 seconds and then stained in 6.8% Giemsa Stain Solution (GIBCO, cat # 10092-013) diluted in Gurr’s 6.8 buffer. Cells in metaphase were identified at 64x magnification using an Axio Imager Z2 Upright Microscope (ZEISS) and analyzed using Ikaros Version 5.4 software (Metasystems). Interpretation of chromosome structure by G-band staining was performed by Nihon Gene Research Laboratories, Inc., (Sendai, Japan).
mFISH and mBAND analysis: hPSCs fixed on glass slides were hybridized with a 24XCyte mFISH probe kit (MetaSystems, cat # 000000-0514-056) or mBAND probe kit (MetaSystems, META mBAND-XCyte) overnight. Sections on glass slides were hybridized, and DAPI/antifade (MetaSystems, cat # 000000-0542-060) was applied per the manufacturer’s instructions for multi-color fluorescence in situ hybridization (mFISH) analysis or mBAND analysis. Metaphase cells were identified at 64x magnification using an Axio Imager. Z2 Upright Microscope (ZEISS) and analyzed using Isis Version 5.4 software (Metasystems). Array Comparative Genomic Hybridization (CGH) was performed with a CytoSureTM Genomic DNA Labeling Kit (Oxford Gene Technology, cat # 020020) and CytoSureTM ISCA 8x 60 k, v2 (Oxford Gene Technology, cat # 020040). Briefly, one g hPSC DNA before and after cryopreservation was prepared with the CytoSureTM Genomic DNA Labeling Kit (Oxford Gene Technology, cat # 020020) and labeled with the fluorescent tags Cy3- or Cy5-dUTP, respectively. The labeled samples, along with human Cot-1 DNA (InvitrogenTM, 15279-011), were added together and hybridized on the array slides. The slides were scanned on a GenePix® 4400A Microarray Scanner (Molecular Devices, LLC). Data were quantified with the GenePix® Pro 7.0 (Molecular Devices, LLC) and analyzed with CytoSureTM Interpret Software (Oxford Gene Technology).
Results
Safe and robust freezing/thawing methods for hPSCs in single cell suspension were explored by evaluating the combination of three distinct processing steps. Namely, Step 1: selection of optimum dissociation reagents that can generate single cell suspension with minimal cell damage. Step 2: selection of a cryopreservation medium that produces a high recovery rate after thawing of cells. Step 3: selection of thawing methods that can prevent differentiation and minimize apoptosis of thawed hPSCs. A schema for the freezeing/thawing is shown in Figure 1. The recovery rate after thawing was calculated by scoring the number of cells harvested four days after thawing against cell number before cryopreservation. The best combination was determined to give the highest recovery rate. Approx. 1 × 106 hPSCs cultured on rhVTN-N-coated 6-well dish inTeSR-E8 medium were dissociated into single cells with Accutase, Pronase, x1 TrypLE Select, x0.75 TrypLE Select (mix x1 TrypLE Select and 0.5 mM EDTA/PBS(-) at 3:1 ratio), x0.5 TrypLE Select (at 1:1), x0.25 TrypLE Select (at 1:3), or 0.5 mM EDTA/PBS(-) respectively in the presence of 5 M Rock inhibitor (Y-27632) and viability of cells after dissociation was evaluated (Figure 1, Step 1). Cells in single cell suspension were frozen with CryoStem, CP-1, CP-1 with human serum albumin (HSA), or TC protector (Figure 1, Step 2). All possible combination of dissociation reagents and cryopreservation mediums were tested. In thawing step, Aggrewell 400 was used to form cell clumps of 500 cells after thawing in the presence of 5 M Rock inhibitor and cells were cultured for two days with TeSR-E8 medium. Cells clumps were then seeded on rhVTN-N-coated 6-well dish and cultured with TeSR-E8 for two days prior to the first passage after thawing (Figure 1, Step 3A). Viable cells were scored at the first passage to determine the best combination of dissociation reagent and freezing medium. Finally, to examine how cell clump formation after thawing could contribute to the prevention of differentiation and minimize apoptosis of cells, cells were seeded on rhVTN-N-coated 24-well flat bottom dish in single cells at various cell concentrations (6 × 104, 6 × 103, or 6 × 102/well) and cultured with TeSR-E8 for four days (Figure 1, Step 3B). The best combination of dissociation buffer and freezing medium determined by Step 1, 2, 3A and 3B was used for Step 3 experiment.
Figure 1.
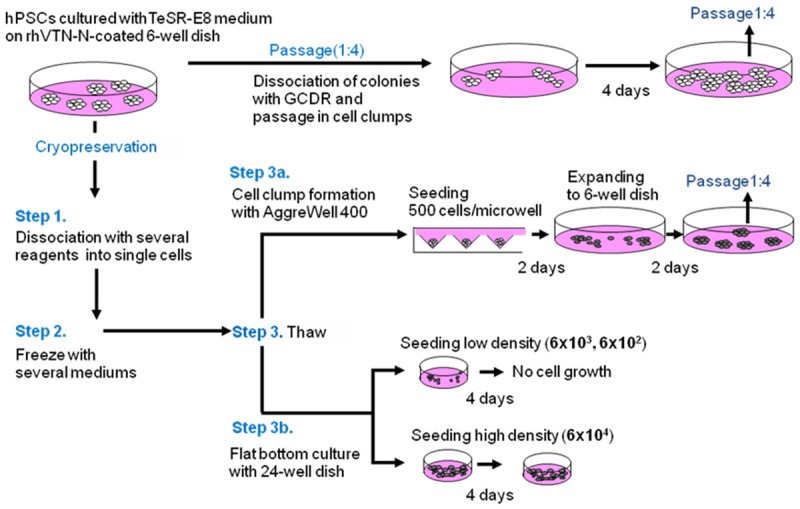
Schema for freezing/thawing of hPSCs. Passage: One fourth of cell clumps after dissociation with GCDR were seeded onto rhVitronectin (rhVTN)-N-coated dishes and cultured with TeSR-E8 medium for four days before the next passage. Cells were dissociated into single cells with several dissociation reagents (Step 1) and frozen with several cryopreservation mediums (Step 2). All possible combinations of dissociation reagent and cryopreservation medium were tested. Cells were thawed (Step 3) either via cell clumping with AggreWell 400 plates for two days followed by seeding and culturing in TeSR-E8 medium on rhVTN-N-coated dishes for two days (Step 3A) or seeded on rhVTN-N-coated 24-well flat bottom dish at various cell concentrations (6 × 104, 6 × 103, 6 × 102 cells/well) as non-cell clump formation (single cell suspension) controls (Step 3B).
x0.75 TrypLE Select dissociates hPSCs into single cells effectively
Among dissociation reagents tested [Accutase, Pronase, x1 TrypLE Select, and 0.5 mM EDTA/PBS(-)], x1 TrypLE Select dissociated hESCs (KhES-1) on rhVTN-N effectively in a relatively short period of time (5 min) with over 60% viability (Figures 2A, S1A). It is not able that enzymatic reagents (TrypLE Select, Pronase, and Accutase) detached KhES-1 cells from the rims of colonies (Figure S1A), while the chelating agent EDTA/PBS(-) dissociated cells from the centers of the colonies (Figure S1B). These observations prompted us to test the dissociation potential of a cocktail composed of the enzymatic reagent TrypLE Select and the chelating agent 0.5 mM EDTA/PBS(-), at ratios of 3:1, 1:1, and 1:3. Among those tested, x0.75 TypLE Select dissociated hPSCs efficiently with the highest vitality (Figure 2B). A time course of the dissociation process with x0.75 TrypLE Select/PBS(-) is shown in Figure 2C.
Figure 2.
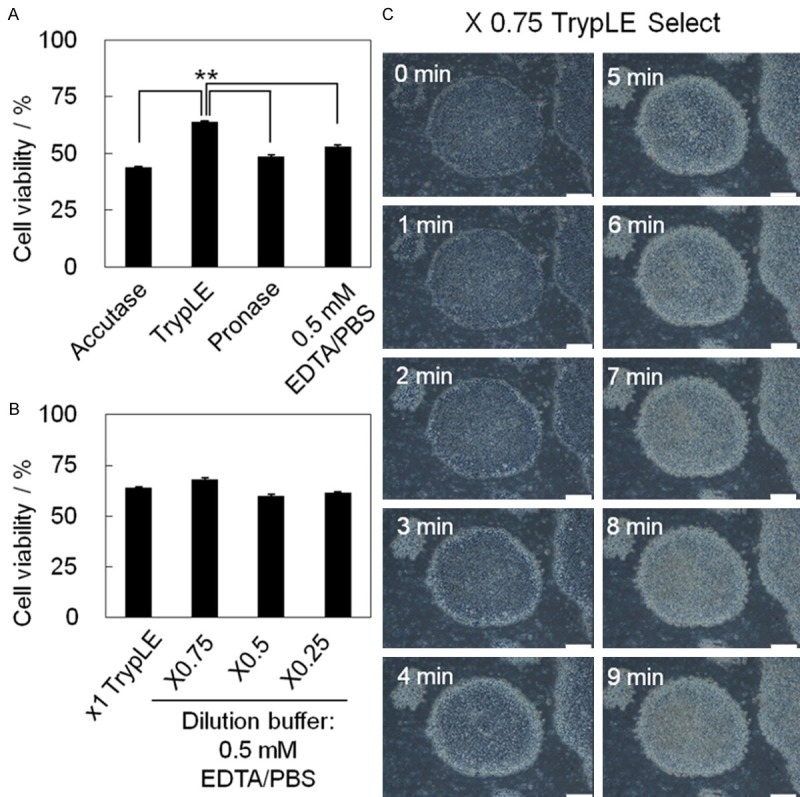
Selection of dissociation buffers for single cell suspension. A. Approximately 1 × 106 hPSCs (KhES-1) cultured in TesR-E8 medium on rhVTN-N-coated dish were dissociated into single cells with the following dissociation reagents: Accutase, x1 TrypLE Select, Pronase, and 0.5 mM EDTA/PBS(-). The percentage of cells that were viable after dissociation is shown as a bar (means) with error bars (SD: Standard Deviation) (n = 5). **p = 0.0112 < 0.05. B. The percentage of cells that were viable after treatment with a cocktail consisting of TrypLE Select and 0.5 mM EDTA/PBS(-) at ratios of 1:0 (x1 TrypLE Select), 0.75:0.25 (x0.75), 0.5:0.5 (x0.5), and 0.25:0.75 (x0.25) is shown in bar graphs with error bars (n = 5). C. Time-lapse photographs of dissociation of KhES-1 cells on rhVTN-N-coated dishes treated with x0.75 TrypLE Select. Scale bar = 200 μm.
Dissociation with x0.75 TrypLE Select followed by cryopreservation with CryoStem showed the best recovery rate for hPSCs after thawing
The numbers of KhES-1 cells that were dissociated with each of the seven dissociation reagents (Accutase, x1 TrypLE Select, Pronase, EDTA/PBS(-), x0.75 TrypLE Select, x0.5 TrypLE Select, and x0.25 TrypLE Select) in the presence of 5 mM Rock inhibitor, resuspended with one of the four different cryopreservation mediums (CryoStem, CP-1, CP-1 with HSA, and TC protector) and freezed with slow freezing method. Then, cells were thawed quickly with 37°C water bath and transferred to AggreWell 400 plates having 1200 V-bottom microwells to form cell clump of approx. 500 cells in well in the presence of 5 mM Rock inhibitor. Cell were cultured with TeSR-E8 for two days and then seeded on rhVTN-N-coated dish. Viable cells were scored two days after seeding on rhVTN-N-coated dish for the first passage and the optimum combination of dissociation reagent and freezing medium was determined that gives the highest ratio of viable cells after thawing against that before freezing. The cell clumps in each well of the AggreWell 400 plates at day 1, 2 and 4 were shown in Figure 3A. With the aid of the AggreWell 400 plates, hPSCs formed cell colonies with relatively uniform size on rhVTN-N-coated flat 6-well dish with no detection of differentiation markers such as vimentin (mesoderm), β-tubulin (ectoderm) at four days after thawing (Figure 3B). Numbers of KhES-1 cells harvested four days after thawing via cell clump formation from each combination of dissociation reagent [Accutage, Pronase, 0.5 mM EDTA/PBS, x1 TrypLE Select (x1T), x0.75, x0.5 and x0.25] and cryopreservation medium (CryoStem, CP-1, CP-1 with HAS and TC protector) are shown in Figure 3C. We found that viable cells were increased approx. 10 times when ES cell line (KhES-1) were dissociated with x0.75 TrypLE Select and cryopreserved with CryoStem. Numbers of colonies of KhES-1 cells scored in three wells of rhVTN-N-coated 6-well dish four days after thawing that were dissociated with x1, x0.75, x0.5 or x0.25 TrypLE Select and frozen with CryoStem are shown in Figure 3D. Numbers of colonies of hES cell line H9 [19], KhES-1 [20] or iPS cell line PFX # 9 [13] scored in three wells of 6-well dish four days after thawing that were dissociated with x0.75 TrypLE Select and frozen with CryoStem are shown in Figure 3E. In thawing process, formation of cell clump consisting approx. 500 cells with AggreWell 400 is essential to maintain undifferentiated state of hPSC, as vimentin (mesoderm) and β-tubulin (ectoderm) were detected in cell colonies when 6 × 104 KhES-1 cells were seeded directly on rhVTN-N-coated dish after thaw (Figure 3B). Expression of vimentin and β-tubulin in differentiated cells in hPSC colony was shown in Figure S2. No cell colonies were observed when 6 × 103 cells or fewer cells were seeded on rhVTN-N-coated dish after thaw and cultured with TeSR-E8 for four days.
Neu5Gc is expressed on animal cells and human cells cultured with animal component [20]. Expression of Neu5Gc on KhES-1 after thaw that were maintained with TeSR-E8 on rhVTN-N-coated dish, dissociated with x0.75T TrypLE Select and cryopreserved with CryoStem was examined by flow cytometry after staining with anti-Neu5Gc antibody. Neu5Gc was detected on mouse feeder cell SNL (positive control), but neither on human mononuclear cells (MNCs) cultured with human serum albumin (HSA) contained PBS(-) (negative control), nor on thawed KhES-1 by flow cytometry (Figure S3).
Maintenance of the self-renewal potential of thawed hPSCs
Expression of pluripotency-related molecules (POU5F1, SSEA-4, TRA 1-60, NANOG and TRA 1-81) in hPSCs after thawing was examined by IHC (Figures 4A, S4A). Expression of the pluripotency markers POU5F1, NANOG, SOX2, KLF4, MYC, hTERT, NODAL, LEFTY2, REX were examined by qRT-PCR (Figures 4B, S4B). Expression of SSEA-4, SSEA-3, and TRA 1-60 on the surfaces of hPSCs was examined by flow cytometry (Figures 4C, S4C). Growth of hPSCs after thawing and without cryopreservation is shown in Figure 4D and suggests that the growth potential of cryopreserved hPSCs recovered after three passages.
Figure 4.
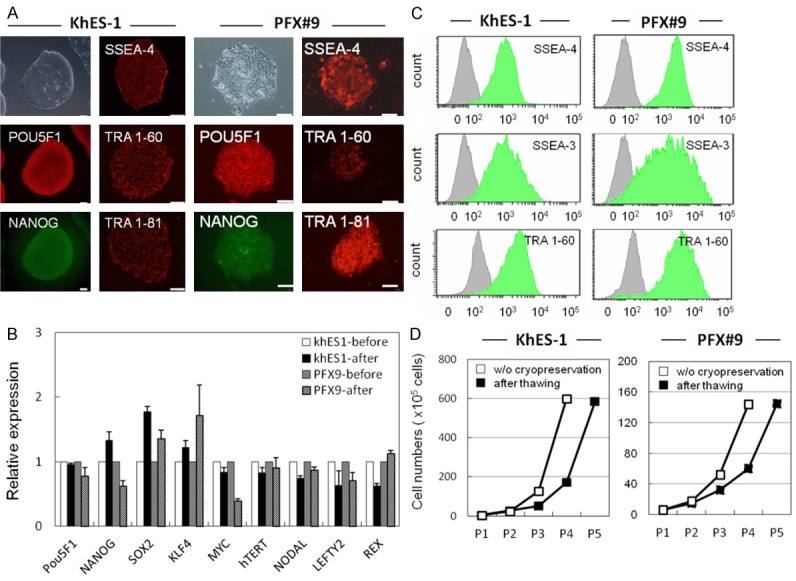
Self-renewal potential of thawed hPSCs. A. IHC of pluripotency-related molecules (POU5F1, SSEA-4, TRA 1-60, NANOG and TRA 1-81) in hPSCs (KhES-1 and PFX#9) at five passages after thawing. These molecules were detected by specific antibodies and visualized with secondary Alexa Fluor 488 (green) or 594 (red) -labeled antibodies. Phase contrast image of KhES-1 and PFX#9 cells is shown in upper left. Scale bar = 100 μm. B. Pluripotency-related genes (POU5F1, NANOG, SOX2, KLF4, cMYC, hTERT, NODAL, LEFTY, and REX1) in KhES-1 and PFX#9 cells before and six passages after thawing were examined by qRT-PCR. Gene expression was compared by the ΔΔCt method. C. Flow cytometric analysis of pluripotency-related surface markers (SSEA-3, SSEA-4, and TRA 1-60) in KhES-1 and PFX#9 cells at five passages after thawing. D. Growth of KhES-1 and PFX#9 cells after thawing and passaging without cryopreservation. X axis represents cell numbers, Y axis represents passage number.
These results suggested that a combination of rhVTN-N and TeSR-E8 medium is a potent cell culture system for effective amplification of hPSC after thawing.
Gene stability of hPSCs after thawing
Karyotype analysis of hPSCs after thawing was conducted by G-banding, mFISH, and mBAND analysis (Figure 5A-C), and chromosomal changes in hPSCs before and after thawing were analyzed by CGH array (Figure 5D). We evaluated normal karyotypes and no regional genomic aberrations in these cells after the freeze/thaw process.
Figure 5.
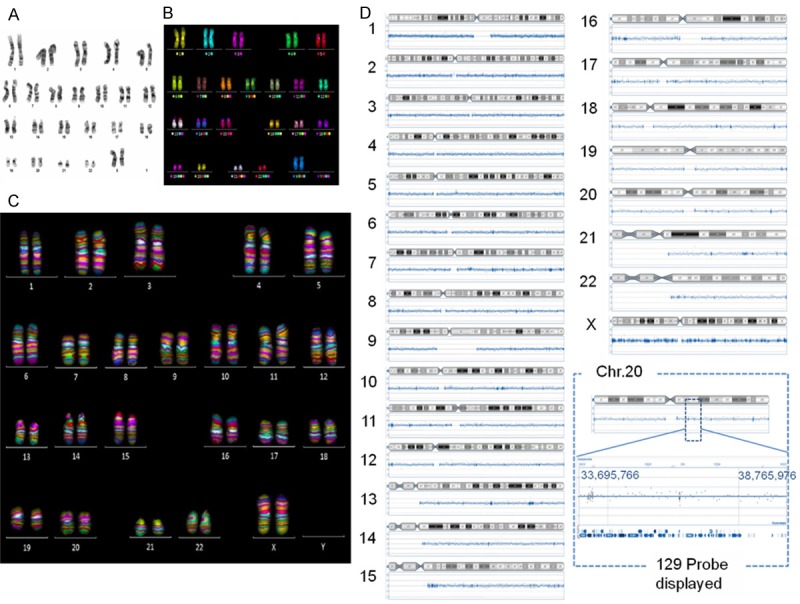
Karyotype analysis of hPSCs after thawing. A. G-band, B. mFISH, C. mBAND of KhES-1 cells six passages after thawing. D. CGH array of KhES-1 cells before/after (passage 6) cryopreservation.
Differentiation potential of thawed hPSCs
The differentiation potential of KhES-1 after thawing was also assessed by checking the potential for embryoid bodies (EBs) formation and following the differentiation of the three germ layers (Figure S5A-H). A teratoma formation assay was conducted by transplanting KhES-1 cells into the testicular capsules of NSG mice. We found teratomas consisting of three germ layers in mice by 12 weeks (Figure S5I-N).
Discussion
Culturing hPSCs in chemically defined medium without BSA or HSA and feeder-free condition [10,14] promises a great advantage for metabolome and proteome analysis of cells and culture medium with less back ground of unanalyzed protein fraction and less concern for a regulatory issue. Single cell passage and single cell cryopreservation will facilitate quality control of cells by scoring viable cells or checking expression of surface molecules by a flow cytometry during passage and before cryopreservation. In this report, we showed that hPSCs cultured with chemically-defined TeSR-E8 medium on rhVTN-N-coated dishes can be effectively cryopreserved and recovered after thaw maintaining robust growth rate and undifferentiated state.
We presumed that the loss of cell-cell contact of hPSCs during dissociation into single cells may confer unbalanced stresses in cytoskelton structure [21], in addition to the frost damage during freezing and thawing manipulation. But chemically defined medium may not provide enough physical and chemical protective action during dissociation, freezing and thawing to the cells compared with albumin contained medium, therefore cell clumps formation with AggreWell just after thawing would be effective to protect cells from cell death and differentiation.
In this report, we used rhVNT-N for coating of culture dish, as it is a cost-effective recombinant protein and suitable for large-scale culture and cell cryopreservation, compared with laminin-521 [11,14] or laminin-E8 [12]. As for the dissociation reagents, a cocktail of TrypLE Select, a recombinant enzyme that digests integrin, collagen and laminin, and EDTA, that disrupts cadherin-mediated cell-cell interactions by chelating Ca2+, would minimize the time needed for dissociation by multi-actions and consequently minimize cell damage in hPSCs during dissociation process. Freezing medium, Cryostem contains methylcellulose and dimethyl sulfoxide (DMSO), but no human or animal serum-derived component, which can reduce safety concern in a clinical application. But the exact mechanism how these compositions demonstrated a superior cryopreservation potential over other reagents needed to be addressed in further study in the combination of culture medium, culture dish coating material, dissociation reagents and post thaw culture method.
In conclusion, our methodology is suitable for a large-scale cryopreservation of hPSCs that are cultured in chemically defined medium on feeder-free conditions. It also enables quality control of hPSCs at the single cell level before cryopreservation and makes post thawing cell manipulation plan manageable by knowing exact number of viable hPSCs.
Acknowledgements
This work was partly supported by Japanese Governmental Research Grant; JST “Safety test for iPSC-derived cell products” 2011-15, Health and Labour Sciences Research Grants; MHLW “Research on Regenerative Medicine for Clinical Application” 2013-2015 and Grant-in Aid for Scientific Research (KAKENHI) JSPS, 20142018.
Disclosure of conflict of interest
None.
Supporting Information
References
- 1.Thomson JA, Itskovitz-Eldor J, Shapiro SS, Waknitz MA, Swiergiel JJ, Marshall VS, Jones JM. Embryonic stem cell lines derived from human blastocysts. Science. 1998;282:1145–1157. doi: 10.1126/science.282.5391.1145. [DOI] [PubMed] [Google Scholar]
- 2.Takahashi K, Tanabe K, Ohnuki M, Narita M, Ichisaka T, Tomoda K, Yamanaka S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. 2007;131:861–872. doi: 10.1016/j.cell.2007.11.019. [DOI] [PubMed] [Google Scholar]
- 3.Schwartz SD, Hubschman JP, Heilwell G, Franco-Cardenas V, Pan CK, Ostrick RM, Mickunas E, Gay R, Klimanskaya I, Lanza R. Embryonic stem cell trials for macular degeneration: a preliminary report. Lancet. 2012;379:713–20. doi: 10.1016/S0140-6736(12)60028-2. [DOI] [PubMed] [Google Scholar]
- 4.Kanemura H, Go MJ, Shikamura M, Nishishita N, Sakai N, Kamao H, Mandai M, Morinaga C, Takahashi M, Kawamata S. Tumorigenicity studies of induced pluripotent stem cell (iPSC)-derived retinal pigment epithelium (RPE) for the treatment of age-related macular degeneration. PLoS One. 2014;9:e85336. doi: 10.1371/journal.pone.0085336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kamao H, Mandai M, Okamoto S, Sakai N, Suga A, Sugita S, Kiryu J, Takahashi M. Characterization of human induced pluripotent stem cell-derived retinal pigment epithelium cell sheets aiming for clinical application. Stem Cell Reports. 2014;2:205–18. doi: 10.1016/j.stemcr.2013.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kuroda T, Yasuda S, Kusakawa S, Hirata N, Kanda Y, Suzuki K, Takahashi M, Nishikawa S, Kawamata S, Sato Y. Highly sensitive in vitro methods for detection of residual undifferentiated cells in retinal pigment epithelial cells derived from human iPS cells. PLoS One. 2013;7:e37342. doi: 10.1371/journal.pone.0037342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Beers J, Gulbranson DR, George N, Siniscalchi LI, Jones J, Thomson JA, Chen G. Passaging and colony expansion of human pluripotent stem cells by enzyme-free dissociation in chemically defined culture conditions. Nat Protoc. 2012;7:2029–40. doi: 10.1038/nprot.2012.130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Meng G, Liu S, Rancourt DE. Synergistic effect of medium, matrix, and exogenous factors on the adhesion and growth of human pluripotent stem cells under defined, xeno-free conditions. Stem Cells Dev. 2012;21:2036–48. doi: 10.1089/scd.2011.0489. [DOI] [PubMed] [Google Scholar]
- 9.Nakagawa M, Taniguchi Y, Senda S, Takizawa N, Ichisaka T, Asano K, Morizane A, Doi D, Takahashi J, Nishizawa M, Yoshida Y, Toyoda T, Osafune K, Sekiguchi K, Yamanaka S. A novel efficient feeder-free culture system for the derivation of human induced pluripotent stem cells. Sci Rep. 2014;4:3594. doi: 10.1038/srep03594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chen G, Gulbranson DR, Hou Z, Bolin JM, Ruotti V, Probasco MD, Smuga-Otto K, Howden SE, Diol NR, Propson NE, Wagner R, Lee GO, Antosiewicz-Bourget J, Teng JM, Thomson JA. Chemically defined conditions for human iPSC derivation and culture. Nat Methods. 2011;8:424–429. doi: 10.1038/nmeth.1593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rodin S, Antonsson L, Niaudet C, Simonson OE, Salmela E, Hansson EM, Domogatskaya A, Xiao Z, Damdimopoulou P, Sheikhi M, Inzunza J, Nilsson AS, Baker D, Kuiper R, Sun Y, Blennow E, Nordenskjöld M, Grinnemo KH, Kere J, Betsholtz C, Hovatta O, Tryggvason K. Clonal culturing of human embryonic stem cells on laminin-521/E-cadherin matrix in defined and xeno-free environment. Nat Commun. 2014;5:3195. doi: 10.1038/ncomms4195. [DOI] [PubMed] [Google Scholar]
- 12.Miyazaki T, Futaki S, Suemori H, Taniguchi Y, Yamada M. Laminin E8 fragments support efficient adhesion and expansion of dissociated human pluripotent stem cells. Nat Commun. 2012;3:1236. doi: 10.1038/ncomms2231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nishishita N, Shikamura M, Takenaka C, Takada N, Fusaki N, Kawamata S. Generation of virus-free induced pluripotent stem cell clones on a synthetic matrix via a single cell subcloning in the naïve state. PLoS One. 2012;7:e38389. doi: 10.1371/journal.pone.0038389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tano K, Yasuda S, Kuroda T, Saito H, Umezawa A, Sato Y. A novel in vitro method for detecting undifferentiated human pluripotent stem cells as impurities in cell therapy products using a highly efficient culture system. PLoS One. 2014;9:e110496. doi: 10.1371/journal.pone.0110496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Reubinoff BE, Pera MF, Fong CY, Trouson AO, Bongso A. Effective cryopreservation of human embryonic stem cells by open pulled straw vitrification method. Hum Reprod. 2001;16:2187–2194. doi: 10.1093/humrep/16.10.2187. [DOI] [PubMed] [Google Scholar]
- 16.Holm F, Ström S, Inzunza J, Baker D, Strömberg AM, Rozell B, Feki A, Bergström R, Hovatta O. An effective serum- and xeno-free chemically defined freezing procedure for human embryonic and induced pluripotent stem cells. Hum Reprod. 2010;25:1271–1279. doi: 10.1093/humrep/deq040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.T’Joen V, Grande LD, Declercq H, Cornelissen M. An efficient, economical slow-freezing method for large-scale human embryonic stem cell banking. Stem Cells Dev. 2012;21:721–8. doi: 10.1089/scd.2011.0192. [DOI] [PubMed] [Google Scholar]
- 18.Imaizumi K, Nishishita N, Muramatsu M, Yamamoto T, Takenaka C, Kawamata S, Kobayashi K, Nishikawa S, Akuta T. A simple and highly effective method for slow-freezing human pluripotent stem cells using dimethyl sulfoxide, hydroxyethyl starch and ethylene glycol. PLoS One. 2014;9:e88696. doi: 10.1371/journal.pone.0088696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Levenstein ME, Ludwig TE, Xu RH, Llanas RA, VanDenHeuvel-Kramer K, Manning D, Thomson JA. Basic fibroblast growth factor support of human embryonic stem cell self-renewal. Stem Cells. 2006;24:568–74. doi: 10.1634/stemcells.2005-0247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Suemori H, Yasuchika K, Hasegawa K, Fujioka T, Tsuneyoshi N, Nakatsuji N. Efficient establishment of human embryonic stem cell lines and long-term maintenance with stable karyotype by enzymatic bulk passage. Biochem Biophys Res Commun. 2006;345:926–32. doi: 10.1016/j.bbrc.2006.04.135. [DOI] [PubMed] [Google Scholar]
- 21.Hayashi Y, Chan T, Warashina M, Fukuda M, Ariizumi T, Okabayashi K, Takayama N, Otsu M, Eto K, Furue MK, Michiue T, Ohnuma K, Nakauchi H, Asashima M. Reduction of N-glycolylneuraminic acid in human induced pluripotent stem cells generated or cultured under feeder- and serum-free defined conditions. PLoS One. 2010;5:e14099. doi: 10.1371/journal.pone.0014099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen G, Hou Z, Gulbranson DR, Thomson JA. Actin-myosin contractility is responsible for the reduced viability of dissociated human embryonic stem cells. Cell Stem Cell. 2010;7:240–248. doi: 10.1016/j.stem.2010.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.