

Abstract
Previous reports revealed that a significant decrease of miR-34a in oral cancer. But the role of miR-34a in oral cancer needs further research. In the present study, we will investigate the effect of miR-34a on oral cancer cell phenotypes. First, it was verified that miR-34a expression was lower in oral cancer tissues compared with their normal controls, so did the oral cancer cells. Next, it was showed that miR-34a overexpression in oral cancer cells could inhibit cell proliferation, G1 phase arrest, metastasis and epithelial mesenchymal transition. It was predicted that interleukin-6-receptor (IL6R) was a potential target gene of miR-34a by bioinformatics analysis and identified by luciferase assay. It was further showed that miR-34a inhibited oral cancer progression via IL6R. Collectively, our findings suggested that miR-34a may function as a tumor suppressor in oral cancer by targeting IL6R.
Keywords: miR-34a, oral cancer, IL6R
Introduction
Oral cancer is one of the most common type of cancer worldwide. Oral cancers are mostly squamous cell carcinomas in the oral cavity [1]. Advances in technology of diagnosis and treatment modalities, but the prognosis of oral cancer remains poor. There are numerous molecules reported to be involved in oral cancer progression [2,3]. However, the mechanism is still unclear. In order to develop therapeutic approaches that targeting those molecules, the identification of molecular targets for oral cancer is urgently required.
MicroRNAs (miRNAs) are noncoding mRNA sequences containing around 22-29 nucleotides that act as important regulators of gene expression [4]. miRNAs can silence their target genes by specifically binding and cleaving mRNAs or inhibiting their translation. Appro-ximately half of all human miRNAs can function as tumor-suppressor or oncogenic miRNAs, depending on their targets [5-7]. Oral squamous cell carcinoma (OSCC), the most frequently occurring malignant head and neck tumor, generally exhibits poor prognosis and metastases are the main cause of death. The discovery of reliable prognostic indicators of tumor progression could greatly improve clinical practice. MicroRNAs are involved in the regulation of basic cellular processes such as cell proliferation, differentiation, and apoptosis. Previous reports showed that miR-27a, miR-21, miRNA-155, miR-499a, miRNA-99a miRNA-491-5p, miRNA-9 and miR-483-3p play roles in oral cancer progression [8-15]. Since miRNAs have been shown to be abnormally expressed in different tumors their importance as potential cancer prognostic indicators is increasing. miRNA profile showed that miR-34a is one of the down-regulated miRNA in oral cancer [16,17].
miR-34a is a known tumor suppressor in many types of cancer including lung cancer, breast cancer, prostate cancer, liver cancer and others. But the role of miR-34a in oral cancer is still unknown. In this study, we will investigate the potential involvement of miR-34a in oral cancer. We examined the expression of miR-34a in human oral cancer cells and tissues and tested its effects on cell growth, cell-cycle distribution, colony formation, migration and invasion. We also investigated a potential role of miR-34a on tumorigenesis in a murine oral cancer model. Finally, we explored the underlying mechanism of miR-34a functions in oral cancer. By bioinformatics, we predicted IL6R was a potential target gene of miR-34a. We identified that miR-34a was a tumor suppressor negatively regulated IL6R signal pathway in progression of oral cancer.
Material and methods
Clinical specimens
Primary oral cancer biopsy specimens and normal biopsies were obtained from the Affiliated Hospital of Xuzhou Medical College (China). Both tumor and normal tissues were histologically confirmed by H&E (hematoxylin and eosin) staining. Informed consent was obtained from each patient, and the research protocols were approved by the Ethics Committee of the Hospital.
Cell culture
Human oral cancer cells OC3, OCM-E1, Tca8113, SCC-25 and DOK were originally obtained from the American Type Culture Collection (ATCC) were cultured in DMEM supplemented with 10% fetal bovine serum, 100 U/mL of penicillin and 100 μg/mL of streptomycin. Cells were cultured at 37°C in a humidified atmosphere of 5% CO2. Human oral keratinocytes (HOK) were purchased from ScienCell Research Laboratories and cultured in oral keratinocyte medium (OKM, ScienCell Research Laboratories, Carlsbad, CA, USA).
Antibodies
IL6R, phos-JAK2, JAK2, phos-STAT3, STAT3, E-cadherin and Snail1 primary antibodies were purchased from cell signaling (Boston, MA, USA). α-SMA primary antibody was ordered from Sigma (Saint Louis, Missouri, USA). GAPDH was purchased from Santa Cruz (Dallas, Texas, USA).
Oligonucleotide construction and lentiviral transduction
The oligonucleotide of mature miR-34a antagomir was chemosynthesized, amplified and cloned into GV232-Puro Vectors by Genechem Co., Ltd. (Shanghai, China). The correct sequences and insertions were confirmed by DNA sequencing. Cells were lentivirally transfected with either the GV232-Puro-miR-34a recombined vector (LV-miR-34a) or emptyGV232-Puro vector (negative control, miR-control). Oligonucleotide transfection or lentivirus construction was performed using Lipofectamine 2000 reagent (Invitrogen) according to the manufacturer’s instructions. The transduced cells with a cell density of over 40% confluency were exposed to puromycin dihydrochloride (1 mg/mL, Sigma, St. Louis, MO) for resistance selection. When all of the cells in the non-transfected control culture were killed, puromycin-resistant cell clones were picked and passaged in medium containing a half concentration of puromycin (0.5 mg/mL) in the first round of selection. Lentivirus-mediated silencing of miR-30a was verified by qRT-PCR and western blot analysis.
RNA isolation and real-time RT-PCR
Total RNA, following the manufacturer’s instructions, was isolated from the cells using Trizol reagent (Invitrogen). Briefly, the cells were lysed in TRIzol and then mixed with chloroform. The lysate was centrifuged to separate RNA, DNA and protein, total RNA recovered, precipitated with isopropanol, washed in 75% ethanol to remove impurities before dissolved in water. After that, 2 μg of RNA was taken and treated with DNase to remove contaminating DNA prior to the reverse transcription to cDNA using SYBR® PCR Kit (Takara, Japan). To measure mRNA expression, real-time RT-PCR was performed using a sequence detector (ABI-Prism, Applied Biosystems). Primers were purchased from Invitrogen. The relative expression levels were calculated by comparing Ct values of the samples with those of the reference, all data normalized to the internal control GAPDH.
MTT assay
MTT assay was employed to detect the growth of cells and the growth curve was delineated. Logarithmic phase cells were collected, and the concentration of the cell suspension was adjusted to 5000 cells per well (The edge wells of the plate are filled with aseptic PBS buffer). The cells were incubated at 37°C, 5% CO2 until cells cover the bottom of the well (a flat-bottom 96-well plate), and then the cells were cultured. 20 μl of the MTT solution was added to each well (5 mg/ml, 0.5% MTT) and the cells were continued to culture for 4 h. After the incubation, the supernatant was discarded and 150 μl dimethyl sulfoxide was added to each well, and the culture plate was shaked at low speed for 10min until crystal dissolved completely. The ELISA reader was used to measure the absorbance at 570 nm.
Colony forming assay
Cells in logarithmic growth phase were digested in 0.5% trypsin/0.04% EDTA and single cell suspension was prepared. Then, these cells were added to 6-well plates (200 cells/well) followed by incubation at 37°C in an environment with saturated humidity and 5% CO2 for 24 h. Non-adherent cells were removed. After culture for 10-14 days, lcolonies were present. These cells were seeded into 96-well plates followed by incubation at 37°C in an environment with saturated humidity and 5% CO2. The colony forming efficiency and the morphology of colonies were observed. The size of colonies was measured and cells in each colony were counted. At this time, cells were divided into early passaging group and late passaging group. In the early passaging group, cells with target number were digested in 0.5% trypsin/0.04% EDTA for preparation of single cell suspension. These cells were then seeded into 96-well plates. In the late passaging group, cells were digested in 0.5% trypsin/0.04% EDTA for preparation of single cell suspension. These cells were then seeded into 96-well plates.
Cell cycle assay
Cells were harvested by trypsinization, washed in ice-cold PBS, fixed in ice-cold 80% ethanol in PBS, centrifuged at 4°C and resuspended in chilled PBS. Bovine pancreatic RNAase (Sigma-Aldrich) was added at a final concentration of 2 μg/ml, incubated at 37°C for 30 min, then 20 μg/ml propidium iodide (Sigma-Aldrich) was added and incubated for 20 min at room temperature. In each group, 50,000 cells were analyzed by flow cytometry (FACSCalibur; BD Biosciences, San Jose, CA, USA).
Western blot analysis
Western blots were performed as we described elsewhere. In brief, cells were lysed in RIPA buffer containing 1 X protease inhibitor cocktail, and protein concentrations were determined using the Bradford assay (Bio-Rad, Philadelphia, PA). Proteins were separated by 12.5% SDS/PAGE and transferred to membranes (Millipore, Bedford, MA) at 55 V for 4 h at 4°C. After blocking in 5% nonfat dry milk in TBS, the membranes were incubated with primary antibodies at 1:1,000 dilution in TBS overnight at 4°C, washed three times with TBS-Tween 20, and then incubated with secondary antibodies conjugated with horseradish peroxidase at 1:5,000 dilution in TBS for 1 hour at room temperature. Membranes were washed again in TBS-Tween 20 for three times at room temperature. Protein bands were visualized on X-ray film using an enhanced chemiluminescence detection system.
Migration assay
For transwell migration assays, 1×105 pancreatic cancer cells were plated in the top chamber onto the noncoated membrane (24-well insert; pore size, 8 μm; Corning Costar) and allowed to migrate toward serum-containing medium in the lower chamber. Cells were fixed after 24 hours of incubation with methanol and stained with 0.1% crystal violet (2 mg/ml, Sigma-Aldrich). The number of cells invading through the membrane was counted under a light microscope (three random fields per well).
Invasion assay
For invasion assay, 1×105 oral cancer cells were plated in the up-chamber with Matrigel coated Membrane (24-well insert; pore size, 8 μm; Corning Costar). Each well was coated freshly with Matrigel (60 μg; BD Bioscience) before the invasion assay. Cells were plated in medium without serum or growth factors, and medium supplemented with serum was used as a chemoattractant in the lower chamber. The cells were incubated for 48 hours and cells that did invade through the pores were removed by a cotton swab. Cells on the lower surface of the membrane were fixed with methanol and stained with crystal violet. The number of cells invading through the membrane was counted under a light microscope (40×, three random fields per well).
Statistical analysis
The mean and SD were calculated for each experimental group. Differences between groups were analyzed by one or two way ANOVA, followed by Bonferoni’s multiple comparison tests using PRISM statistical analysis software (GrafPad Software, Inc., San Diego, CA). Significant differences among groups were calculated at P < 0.05.
Results
Decreased expression of miR-34a in human OSCC
To investigate the possible role of miR-34a in OSCC, we first examined the expression of miR-34a in 22 tumor samples and their compared normal tissues. As shown in Figure 1A, the expression levels of miR-34a in tumor samples were lower than those in normal samples. Similarly, miR-34a was lower in human oral cancer cells compared with normal cells (Figure 1B). These results provided us initial evidence that miR-34a may play a suppressing miRNA in the development of human OSCC.
Figure 1.
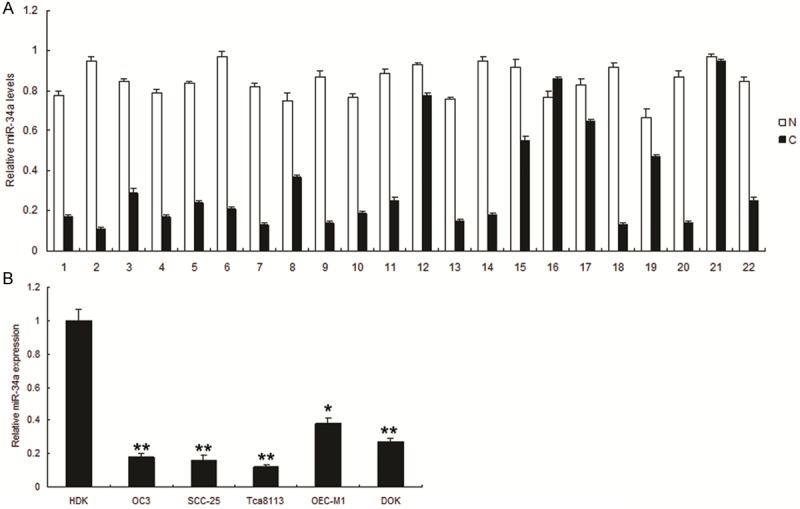
Decreased expression of miR-34a in human oral cancer tissues and cell lines. A. miR-34a was downregulated significantly in oral cancer tissues compared with normal tissues. Expression of miR-34a was normalized against an endogenous control U6. The relative expression of miR-34a was determined by normalizing the expression of miR-34a in a tumor tissue (C) to that in its corresponding non-tumor tissue (N). B. The relative expression of miR-34a in 5 OSCC cell lines and HOK cell line was evaluated using qRT-PCR analysis. Expression of miR-34a was normalized against an endogenous control U6. The relative expression of miR-34a was determined by normalizing the expression of miR-34a in OSCC cell lines to that in HOK.
miR-34a inhibits proliferation and induces G1 arrest of OSCC
To explore the effect of miR-34a on cell growth, Tca8113 and SCC-25 cells were infected with LV-miR-34a or LV-miR control respectively and miR-34a was increased significantly in the cells with LV-miR-34a (miR-34a) (Figure 2A). The results of MTT and colony formation assay displayed that miR-34a inhibited cell in Tca8113 cells (Figure 2B and 2C) and SCC-25 cells (Figure 2D and 2E). Because the miR-control had no effect on cell growth, the data was not shown the comparison of parent cells and the miR-control cells, which indicated that the effect caused by miR-34a was highly specific. To further observe miR-34a mediating growth inhibition, cells were infected with LV-miR-34a and examined cell-cycle distribution. Compared with miR-control, SCC-25 cells infected with LV-miR-34a displayed an increased percentage of cells in G1 phase and fewer cells in S phase (Figure 2F and 2G), which suggested that the growth-suppressive effect of miR-34a was partly due to a G1-phase arrest.
Figure 2.
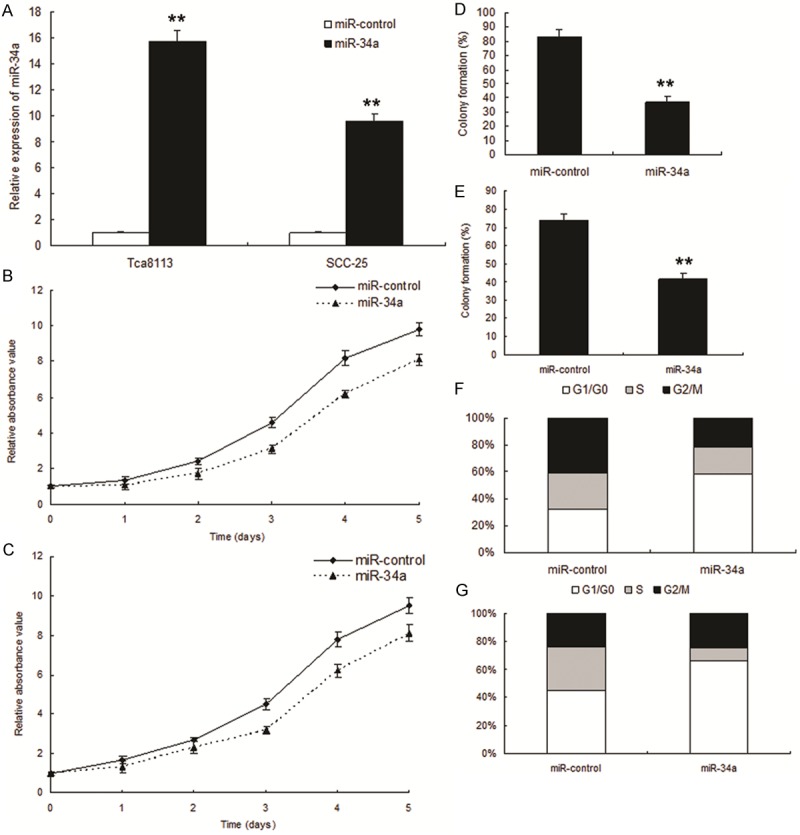
Enforced expression of miR-34a induced growth inhibition in OSCC in vitro. A. miR-34a expression in the OSCC infected with LV-miR34a and their control. B. Effect of miR-34a on cell proliferation was measured by MTT assay after miRNA infection in Tca8113 cells. C. Effect of miR-34a on cell proliferation was measured by MTT assay after miRNA infection in miR-34a SCC-25 cells. D. Effect of miR-34a on cell proliferation was measured by colony formation assay after miRNA infection in Tca8113 cells. E. Effect of miR-34a on cell proliferation was measured by colony formation assay after miRNA infection in SCC-25 cells. F. Cell-cycle distribution of Tca8113 cells infected with miRNAs for 48 hours. G. Cell-cycle distribution of SCC-25 cells infected with miRNAs for 48 hours.
miR-34a inhibits metastasis in OSCC
To investigate role of miR-34a in OSCC metastasis, Tca8113 cells were infected with LV-miR-34a, and the results showed that up-regulation of miR-34a could significantly decreased migration (Figure 3A) and invasion (Figure 3B). Metastasis associated markers were also detected by western blot and the results showed that E-cadherin increased and Snail1, α-SMA decreased in the two cell lines with LV-miR-34a (Figure 3C).
Figure 3.
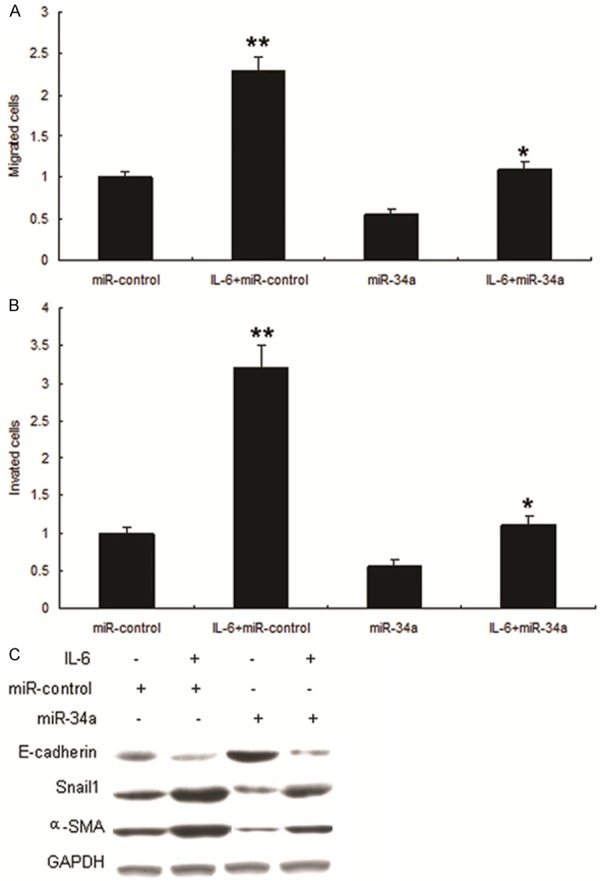
Overexpression of miR-34a suppresses metastasis of OSCC. A. miR-34a inhibited OSCC migration. Migration of Tca8113 cells with miR-34a and miR-control were assayed using transwell chamber. B. miR-34a inhibited OSCC invasion. Invasion of Tca8113 cells with miR-34a and miR control were assayed using transwell chamber. C. miR-34a suppressed EMT of Tca8113 cells. EMT markers such as E-cadherin, vimentin, Snail1 and α-SMA were assayed by Western blot.
IL6R is a new target gene of miR-34a in OSCC
We then investigated the mechanisms by which miR-34a inhibit OSCC progression. Bioinfor-matic analysis showed that IL6R was directly suppressed by miR-34a (Figure 4A). As shown in Figure 4B, the luciferase activity of IL6R in Tca8113 cells was much lower than in control cells. The luciferase activity of mutation was rescued in the cells. We next examined whether miR-34a could regulate endogenous IL6R expression in the cells. Compared with control, endogenous IL6R mRNA levels (Figure 4C) were down-regulated when cells were transfected with miR-34a. IL6R protein increased in the cells with anti-miR-34a (Figure 4D).
Figure 4.
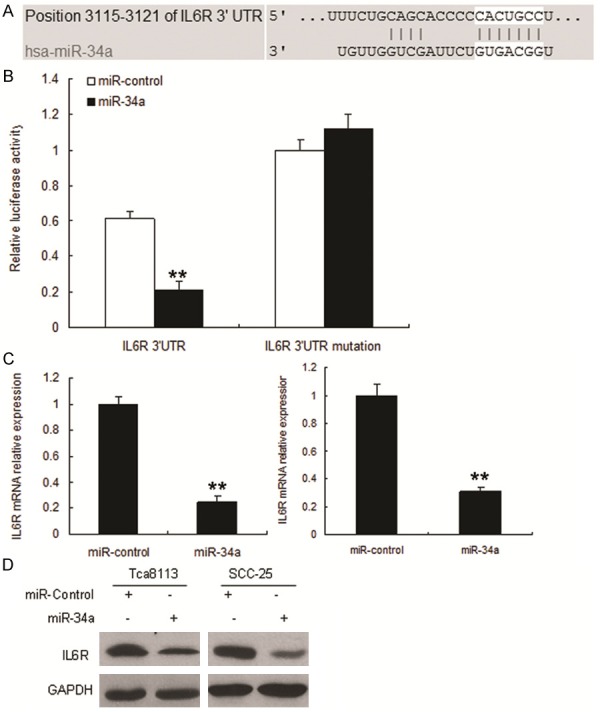
Restoration of miR-34a down-regulates IL6R expression. A. The 3’-UTR of the IL6R gene contains binding sites for miR-34a according to bioinformatic analysis. B. miR-34a suppressed the expression of a luciferase reporter gene harbouring the 3’-UTR of IL6R. The pGL4 plasmid was modified by adding the human 3’-UTR or the 3’-UTR with mutations in regions complementary to miR-34a seed regions behind the firefly luciferase gene. Cells were transiently co-transfected with miR-34a or control together with the indicated luciferase constructs, and luciferase activity was analyzed 48h later. Data are presented as relative firefly luciferase activity normalized to Renilla luciferase activity from the same construct. The data presented are shown as means ± S.D. collected from three independent experiments. C. miR-34a restoration down-regulated IL6R in OSCC cells. Tca8113 (left panel) and SCC-25 (right panel) cells were transfected with miR-34a or miR-control for 48 hours, then collected for Real-time PCR. D. miR-34a restoration down-regulated IL6R in OSCC cells. Cells were transfected with miR-34a or miR- control for 48 hours, and then collected for Western blot analysis.
miR-34a inhibits oral cancer metastasis by targeting IL6R and its signal pathway
We next tested whether miR-34a inhibits metastasis of Tca8113 cells with over-expression of IL6R. The result showed that miR-34a inhibited the proliferation and migration with IL6R overexpression in Tca8113 cells respectively (Figure 5A and 5B). Since IL-6/STAT3 signaling pathway is an important pathway involved in cancer development, we then detected several proteins and downstream proteins in the pathway. STAT3 was decreased by miR-34a over-expression (Figure 5C). In order to investigate miR-34a mediating growth inhibition in vivo, glioma nude mice were set up using Tca8113 -miR-34a and Tca8113-miR control, the results showed that miR-34a suppressed oral cancer growth (Figure 5D).
Figure 5.
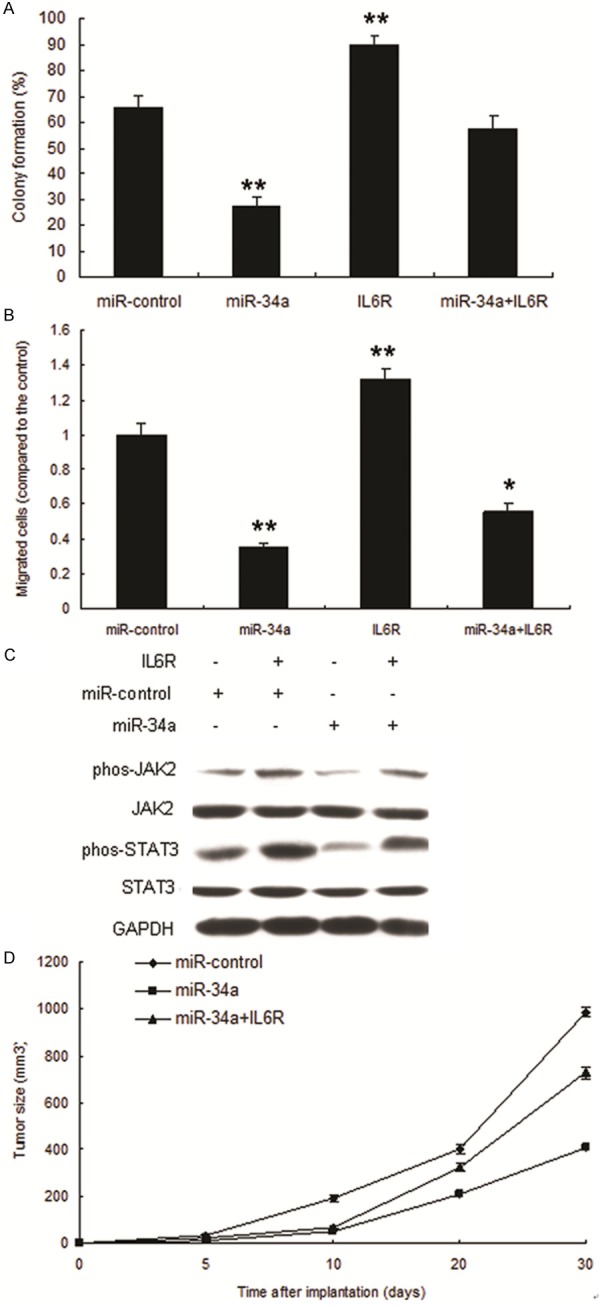
miR-34a inhibits progression of OSCC targeting IL6R. A. miR-34a inhibited proliferation of Tca8113 cells with IL6R overexpression. B. miR-34a inhibited migration of Tca8113 cells with IL6R overexpression. C. miR-34a inhibited IL6R signal pathway in Tca8113 cells. D. Tumor growth on nude mice set up using cells LV-miR-34a or LV-miR control.
Discussion
Previous studies used miRNA microarray and qRT-PCR analyses to characterize the miRNA expression profiles in oral cancer cell lines and clinical samples of different stages [8,9]. Reports showed that miR-27a, miR-21, miRNA-155, miR-499a, miRNA-99a miRNA-491-5p, miRNA-9 and miR-483-3p play roles in oral cancer progression [10-17], which suggested the dysregulation of miRNAs in the initiation and progression of oral cancers. Our data showed that miR-34a is frequently downregulated in OSCC cell lines and tissues.
The biological roles of miR-34a in oral tumorigenesis remain unclear. In Tca8113 and SCC-25 cells, ectopic miR-34a expression has significant effects on cell growth (Figure 2) and decreases cell migration, invasion (Figure 3). This suggested that the downregulation of miR-34a might play a critical role in invasion and metastasis of OSCC and supported evidence that ectopic miR-34a expression inhibits cell migration, invasion and lung colonization in OSCC cells. However, miR-34a doesn’t influence EMT of Tca8113 and SCC-25 oral cancer cells. These observations suggested that the diverse biological functions of miR-34a may be cell type- and context-dependent.
With bioinformatic prediction and we identified that IL6R is the direct target of miR-34a. In our study, the expression of miR-34a correlated inversely with IL6R (Figure 4). Ectopic expression of miR-34a decreased the levels of IL6R protein, suggesting that IL6R is a specific target of miR-34a in OSCC cells. However, it remains possible that miR-34a might functionally target other genes in oral cancer cells or in different biological systems. It is, therefore, possible that other molecules or signaling pathways influenced by miR-34a could be involved in OSCC pathogenesis, and that some of them have yet to be identified. IL6R is overexpressed in several malignancies and plays a crucial role in promoting cell proliferation and metastasis [18]. Our results demonstrated that miR-34a expression decreases IL6R protein levels by directly targeting the 3’ UTR of IL6R mRNA. Previous studies have identified that IL6R is also a specific target of miR-23a [19], miR-124 [20], miR-125b [21] and miR-107, miR-449a [22]. A recent study showed that the administration of tumor suppressive miRNA to mice could effectively suppress tumorigenesis in liver cancer without causing toxicity [23]. This indicated the possibility of using miRNA to target IL6R in cancer therapy. Although previous studies identified reduced miR-34a expression in several types of tumors [24,25], the detailed mechanisms underlying its repression remain unclear. As previously reported, epigenetic mechanisms, including DNA methylation and histone modification, chromosome deficiency, and transcriptional regulation can influence the downregulation of miRNA [26].
In summary, our present study demonstrated that the deregulated expression of miR-34a was associated with poor prognosis and aggressive phenotype of oral cancer. This study also implied that miR-34a played an important role in the regulation of OSCC malignant behavior including cell proliferation and invasion by directly targeting IL6R. In general, these results indicated that miR-34a might be applied as a potential prognostic biomarker and inhibitor in OSCC.
Acknowledgements
The study was supported by the National Natural Science Foundation of China (No. 31060167 and 81160242).
Disclosure of conflict of interest
None.
References
- 1.Warnakulasuriya S. Global epidemiology of oral and oropharyngeal cancer. Oral Oncol. 2009;45:309–316. doi: 10.1016/j.oraloncology.2008.06.002. [DOI] [PubMed] [Google Scholar]
- 2.Walker DM, Boey G, McDonald LA. The pathology of oral cancer. Pathology. 2003;35:376–383. doi: 10.1080/00310290310001602558. [DOI] [PubMed] [Google Scholar]
- 3.Jerjes W, Upile T, Petrie A, Riskalla A, Hamdoon Z, Vourvachis M, Karavidas K, Jay A, Sandison A, Thomas GJ, Kalavrezos N, Hopper C. Clinicopathological parameters, recurrence, locoregional and distant metastasis in 115 T1-T2 oral squamous cell carcinoma patients. Head Neck Oncol. 2010;2:9. doi: 10.1186/1758-3284-2-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dalmay T. MicroRNAs and cancer. J Intern Med. 2008;263:366–375. doi: 10.1111/j.1365-2796.2008.01926.x. [DOI] [PubMed] [Google Scholar]
- 5.Calin GA, Croce CM. MicroRNA signatures in human cancers. Nat Rev Cancer. 2006;6:857–866. doi: 10.1038/nrc1997. [DOI] [PubMed] [Google Scholar]
- 6.Esquela-Kerscher A, Slack FJ. Oncomirs-microRNAs with a role in cancer. Nat Rev Cancer. 2006;6:259–269. doi: 10.1038/nrc1840. [DOI] [PubMed] [Google Scholar]
- 7.Negrini M, Ferracin M, Sabbioni S, Croce CM. MicroRNAs in human cancer: from research to therapy. J Cell Sci. 2007;120:1833–1840. doi: 10.1242/jcs.03450. [DOI] [PubMed] [Google Scholar]
- 8.Kariya A, Furusawa Y, Yunoki T, Kondo T, Tabuchi Y. A microRNA-27a mimic sensitizes human oral squamous cell carcinoma HSC-4 cells to hyperthermia through downregulation of Hsp110 and Hsp90. Int J Mol Med. 2014;34:334–40. doi: 10.3892/ijmm.2014.1758. [DOI] [PubMed] [Google Scholar]
- 9.Hedbäck N, Jensen DH, Specht L, Fiehn AM, Therkildsen MH, Friis-Hansen L, Dabelsteen E, von Buchwald C. MiR-21 expression in the tumor stroma of oral squamous cell carcinoma: an independent biomarker of disease free survival. PLoS One. 2014;9:e95193. doi: 10.1371/journal.pone.0095193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shi LJ, Zhang CY, Zhou ZT, Ma JY, Liu Y, Bao ZX, Jiang WW. MicroRNA-155 in oral squamous cell carcinoma: Overexpression, localization, and prognostic potential. Head Neck. 2014 doi: 10.1002/hed.23700. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 11.Hou YY, Lee JH, Chen HC, Yang CM, Huang SJ, Liou HH, Chi CC, Tsai KW, Ger LP. The association between miR-499a polymorphism and oral squamous cell carcinoma progression. Oral Dis. 2015;21:195–206. doi: 10.1111/odi.12241. [DOI] [PubMed] [Google Scholar]
- 12.Yen YC, Shiah SG, Chu HC, Hsu YM, Hsiao JR, Chang JY, Hung WC, Liao CT, Cheng AJ, Lu YC, Chen YW. Reciprocal regulation of microRNA-99a and insulin-like growth factor I receptor signaling in oral squamous cell carcinoma cells. Mol Cancer. 2014;13:6. doi: 10.1186/1476-4598-13-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Huang WC, Chan SH, Jang TH, Chang JW, Ko YC, Yen TC, Chiang SL, Chiang WF, Shieh TY, Liao CT, Juang JL, Wang HC, Cheng AJ, Lu YC, Wang LH. miRNA-491-5p and GIT1 serve as modulators and biomarkers for oral squamous cell carcinoma invasion and metastasis. Cancer Res. 2014;74:751–64. doi: 10.1158/0008-5472.CAN-13-1297. [DOI] [PubMed] [Google Scholar]
- 14.Yu T, Liu K, Wu Y, Fan J, Chen J, Li C, Yang Q, Wang Z. MicroRNA-9 inhibits the proliferation of oral squamous cell carcinoma cells by suppressing expression of CXCR4 via the Wnt/β-catenin signaling pathway. Oncogene. 2013;33:5017–27. doi: 10.1038/onc.2013.448. [DOI] [PubMed] [Google Scholar]
- 15.Bertero T, Bourget-Ponzio I, Puissant A, Loubat A, Mari B, Meneguzzi G, Auberger P, Barbry P, Ponzio G, Rezzonico R. Tumor suppressor function of miR-483-3p on squamous cell carcinomas due to its pro-apoptotic properties. Cell Cycle. 2013;12:2183–93. doi: 10.4161/cc.25330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Scapoli L, Palmieri A, Lo Muzio L, Pezzetti F, Rubini C, Girardi A, Farinella F, Mazzotta M, Carinci F. MicroRNA expression profiling of oral carcinoma identifies new markers of tumor progression. Int J Immunopathol Pharmacol. 2010;23:1229–34. doi: 10.1177/039463201002300427. [DOI] [PubMed] [Google Scholar]
- 17.Cui J, Li D, Zhang W, Shen L, Xu X. Bioinformatics analyses combined microarray identify the deregulated microRNAs in oral cancer. Oncol Lett. 2014;8:218–222. doi: 10.3892/ol.2014.2070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Weidle UH, Klostermann S, Eggle D, Krüger A. Interleukin 6/interleukin 6 receptor interaction and its role as a therapeutic target for treatment of cachexia and cancer. Cancer Genomics Proteomics. 2010;7:287–302. [PubMed] [Google Scholar]
- 19.Zhu LH, Liu T, Tang H, Tian RQ, Su C, Liu M, Li X. MicroRNA-23a promotes the growth of gastric adenocarcinoma cell line MGC803 and downregulates interleukin-6 receptor. FEBS J. 2010;277:3726–34. doi: 10.1111/j.1742-4658.2010.07773.x. [DOI] [PubMed] [Google Scholar]
- 20.Hatziapostolou M, Polytarchou C, Aggelidou E, Drakaki A, Poultsides GA, Jaeger SA, Ogata H, Karin M, Struhl K, Hadzopoulou-Cladaras M, Iliopoulos D. An HNF4α-miRNA inflammatory feedback circuit regulates hepatocellular oncogenesis. Cell. 2011;147:1233–47. doi: 10.1016/j.cell.2011.10.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jia HY, Wang YX, Yan WT, Li HY, Tian YZ, Wang SM, Zhao HL. MicroRNA-125b Functions as a Tumor Suppressor in Hepatocellular Carcinoma Cells. Int J Mol Sci. 2012;13:8762–74. doi: 10.3390/ijms13078762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sarma NJ, Tiriveedhi V, Crippin JS, Chapman WC, Mohanakumar T. Hepatitis C virus-induced changes in microRNA 107 (miRNA-107) and miRNA-449a modulate CCL2 by targeting the interleukin-6 receptor complex in hepatitis. J Virol. 2014;88:3733–43. doi: 10.1128/JVI.03060-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rokavec M, Öner MG, Li H, Jackstadt R, Jiang L, Lodygin D, Kaller M, Horst D, Ziegler PK, Schwitalla S, Slotta-Huspenina J, Bader FG, Greten FR, Hermeking H. IL-6R/STAT3/miR-34a feedback loop promotes EMT-mediated colorectal cancer invasion and metastasis. J Clin Invest. 2014;124:1853–67. doi: 10.1172/JCI73531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hermeking H. The miR-34 family in cancer and apoptosis. Cell Death Differ. 2010;17:193–9. doi: 10.1038/cdd.2009.56. [DOI] [PubMed] [Google Scholar]
- 25.Wong KY, Yu L, Chim CS. DNA methylation of tumor suppressor miRNA genes: a lesson from the miR-34 family. Epigenomics. 2011;3:83–92. doi: 10.2217/epi.10.74. [DOI] [PubMed] [Google Scholar]
- 26.Choi JD, Lee JS. Interplay between Epigenetics and Genetics in Cancer. Genomics Inform. 2013;11:164–73. doi: 10.5808/GI.2013.11.4.164. [DOI] [PMC free article] [PubMed] [Google Scholar]