

Abstract
We present a case of sclerosingrhabdomyosarcoma of the skull in a young male. This is a rare tumor that immunophenotypically shows rhabdomyoid differentiation. The lesion had a moderately aggressive course and the patient died at 19 months after diagnosis.
Keywords: Rhabdomyosarcoma, sclerosingrhabdomyosarcoma, scalp
Introduction
Sclerosing rhabdomyosarcoma (SRMS) is an extremely rare soft tissue tumor first reported by Folpe et al [1] in 2002. In contrast to alveolar and pleomorphic rhabdomyosarcomas that often arise from deep soft tissue of the extremities, SRMS is more often seen in the head or neck, similar to the localization of embryonal rhabdomyosarcoma. To date, less than 50 cases have been reported in the literature. Here we present a case involving the skull and infiltrative into the dura.
Case report
Clinical information
Beginning more than 1 year ago, a 24 year old man suffered from headaches. Recently, he also had episodes of dizziness. Physical examination revealed a 5cm hard, painless mass in the occipital region. The cranial magnetic resonance imaging (MRI) showed a mixed signal mass centered in the occiput. Gadolinium enhancement effect was inconspicuous and the diagnostic impression was of an osseous tumor (Figure 1). The tumor extended from the skull and invaded dura. The neighboring brain was also edematous. Intra-operatively, the tumor was noted to be poorly circumscribed and densely adherent to the dura. The tumor had a gray color and firm consistency without obvious necrosis, hemorrhage or calcification in the tumor. The initial clinical impression was that of an atypical or malignant meningioma.
Figure 1.
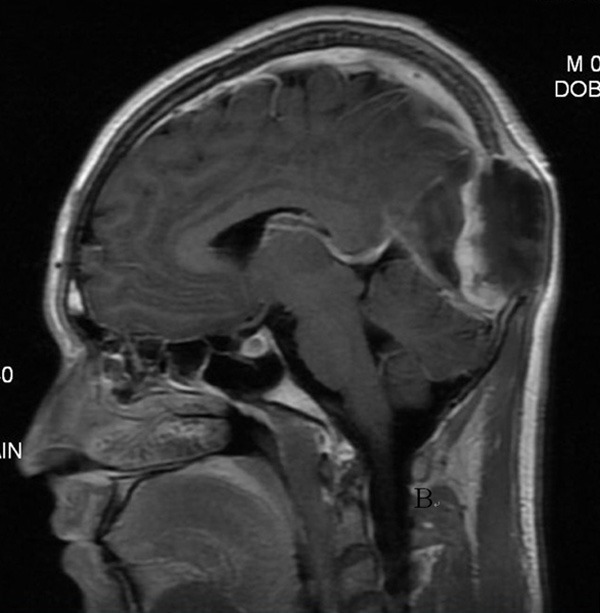
The cranial MRI: a mass in the occiput and compressed the brain tissue, with clear borderline and sclerotization.
Pathology findings
The tumor cells have round, oval or spindly vesicular nuclei with scanty cytoplasm and indistinct cell borders. The nuclei demonstrate small nucleoli and thick nuclear membranes. The tumor cells are separated by bundles of collagen fibers. Some tumor cells form vague nests while others are distributed between collagen fibers (Figure 2A, 2B). Tumoral necrosis is seen at the center of the mass. Mitoses are rare (< 1/10HPF). There are not rhabdomyomablasts such as “strap” or “spider” cells. Multinucleated giant cells are not identified. The immunohistochemical staining shows tumor cells diffusely expressing vimentin and Desmin (Figure 2C). The tumor cells are negative for CD31, CD34, D2-40, HMB-45, P63, EMA, S-100, SMA, GFAP, CK, CD99, calponin, CD10, MyoD1, myogenin, and factor VIII. The MIB-1 proliferative index is 5% on average and 10-15% focally. FISH for FUS break apart dual color probe (12p11) did not detect a FUS gene rearrangement. The final diagnosis was sclerosing rhabdomyosarcoma, primary in the skull.
Figure 2.
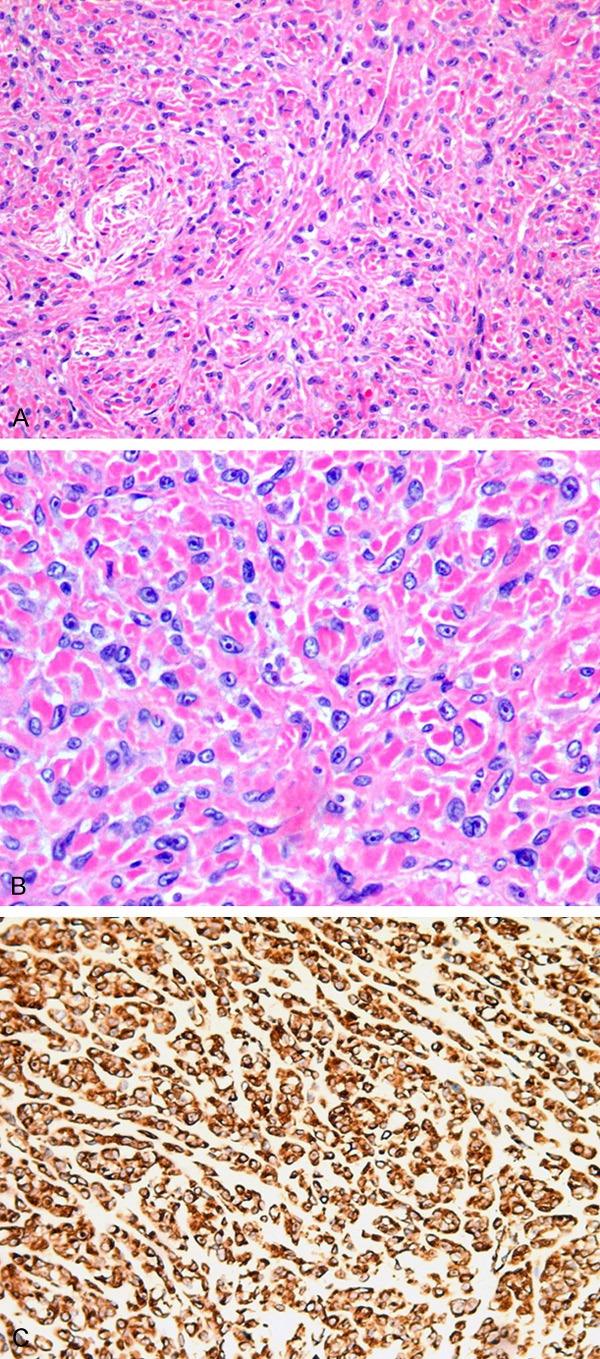
The tumor cells uniformly distributed in the hyper plastic collagen fiber (A). The tumor cells were fusiform to ovoid with vesicular nuclei, small nucleoli and mitoses were found easily (B). Desmin was strongly immunopositive in the cytoplasm (C). (A) H&E, 200 ×. (B) H&E, 400 ×. (C) IHC, 200 ×.
Prognosis and follow-up
There was no further treatment after the operation. Thirteen months after the operation, the patient complained of visual field defects and cough. A new MRI of the brain showed multiple areas of enhanced signal in the bilateral parieto-occipital lobes. In addition, CT scans showed multiple well demarcated, high density nodules in both lungs consistent with recurrence and metastasis. The patient received chemotherapy and radiotherapy, but he died 19 months after the initial surgery.
Discussion
Sclerosing rhabdomyosarcoma (SRMS) was firstly reported as a potentially novel variant of rhabdomyosarcomas by Folpe [1] in 2002. The tumor may express myogenic markers such as Desmin, Myogenin or MyoD1 and was included in the WHO soft tissue and bone tumor classification in 2013 [2]. To date, there are less than 50 cases reported in the literature [3,4]. The tumor affects males predominantly, and the ratio of male to female is about 6:1, with more than 50% cases located in head and neck [2]. SRMS has a striking dense stromal sclerosis that may be mistaken for osteoid or cartilage. Misdiagnosis as osteosarcoma, chondrosarcoma, and angiosarcoma or sclerosing epithelioidfibrosarcoma may occur.
The SRMS cells are spindled with ovoid to elongate nuclei, vesicular chromatin, small nucleoli, and scant lightly eosinophilic cytoplasm. However, many cells in this case show epithelioid cytoplasm with obvious though not large nucleoli and rare mitoses, raising the question of sclerosing epithelioid fibrosarcoma (SEF). SEF is an uncommon soft tissue sarcoma with conspicuous sclerosis. Recent studies have shown an overlap in the histology of SEF and low grade fibromyxoid sarcoma, with a FUS aberration characteristic of fibromyxoid sarcoma though sometimes also present in SEF [5,6]. There was no FUS gene aberration located on chromosome 16p detected by FISH in this case. Immunohistochemically however, desmin positivity is noted in the cytoplasm or nucleolus of tumor cells of this case indicating myogenic differentiation. SEF’s immunohistochemical phenotype includes expression of EMA or S-100 but not desmin [7]. In addition to desmin, SRMS can express MyoD1 and myogenin. In this case, the tumor did not express MyoD1 and myogenin. All in all, this case is most consistent with a SRMS.
The prognosis in adults SRMS is significantly worse than the pediatric population. The rate of recurrence and metastasis is about 40-50%. The prognosis of tumors located in the head and neck are poorer than those located in the extremities. The inability to completely resect tumor in the head and neck region due to the complex local anatomy may contribute to the difference in prognosis. There is not a consensus in SRMS treatment. Lacking significant pleomorphism and showing scant mitotic activity and a low Ki-67 index in the tumor, the patient was spared adjunctive radiation and chemotherapy. However, the tumor had an aggressive biology and course. The patient developed multiple metastases in the brain and lungs 13 months after the operation. Some authors have suggested radical resection with adjunctive radiation and chemotherapy. Our case supports the premise that adjunctive treatment such as radiotherapy or chemotherapy is indicated especially for incomplete resection.
Disclosure of conflict of interest
None.
References
- 1.Folpe AL, McKenney JK, Bridge JA, Weiss SW. Sclerosingrhabdomyosarcoma in adults: report of four cases of a hyalinizing, matrix-rich variant of rhabdomyosarcoma that may be confused with osteosarcoma, chondrosarcoma, or angiosarcoma. Am J Surg Pathol. 2002;26:1175–1183. doi: 10.1097/00000478-200209000-00008. [DOI] [PubMed] [Google Scholar]
- 2.Nascimento AF, Barr FG. Spindle cell/sclerosing rhabdomyosarcoma. In: Fletcher CDM, Bridge JA, Hogendoorn PCW, et al., editors. WHO classification of tumors of soft tissue and bone. 4th edition. Lyon: IARC; 2013. pp. 134–135. [Google Scholar]
- 3.Wang J, Tu X, Sheng W. Sclerosingrhabdomyosarcoma: a clinicopathologic and immunohistochemical study of five cases. Am J Clin Pathol. 2008;129:410–415. doi: 10.1309/EP3FRRY6GMP555QC. [DOI] [PubMed] [Google Scholar]
- 4.Robinson JC, Richardson MS, Neville BW, Day TA, Chi AC. Sclerosingrhabdomyosarcoma: report of a case arising in the head and neck of an adult and review of the literature. Head Neck Pathol. 2013;7:193–202. doi: 10.1007/s12105-012-0398-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yoon N, Kwon JW, Seo SW, Ahn G, Choi YL. Sclerosing epithelioidfibrosarcoma: cytogenetic analysis of FUS rearrangement. Pathol Int. 2012;62:65–68. doi: 10.1111/j.1440-1827.2011.02752.x. [DOI] [PubMed] [Google Scholar]
- 6.Wang WL, Evans HL, Meis JM, Liegl-Atzwanger B, Bovee JV, Goldblum JR, Billings SD, Rubin BP, López-Terrada D, Lazar AJ. FUS rearrangement is rare in pure sclerosing epithelioid fibrosarcoma. Mod Pathol. 2012;25:846–853. doi: 10.1038/modpathol.2011.214. [DOI] [PubMed] [Google Scholar]
- 7.Kindblom LG, Mertens F, Coindre JM, et al. Sclerosing epithelioid fibrosarcoma. In: Fletcher CDM, Bridge JA, Hogendoorn PCW, et al., editors. WHO classification of tumors of soft tissue and bone. 4th edition. Lyon: IARC; 2013. pp. 97–98. [Google Scholar]