

Abstract
Background: Beta-site Amyloid precursor protein Cleaving Enzyme 1 (BACE1) is conceived as a potential target for therapies against Alzheimer disease (AD) which is characterized by the accumulation of plaques formed of short β-amyloid (APPβ) peptides. Recently, such microRNAs, as miR-29a, miR-29b-1 have been shown to correlate with abnormally high levels of BACE1 and APPβ in sporadic AD. Methods: In order to confirm whether miR-29c correlates with the BACE1 upregulation in sporadic AD, we firstly evaluated the expression of miR-29c and BACE1, the APPβ accumulation in sporadic AD brain tissues and analyzed the correlation of miR-29c with BACE1. Then we determined the regulation of miR-29c in human heuroblastoma SH-SY5Y cells on the BACE1 expression and APPβ accumulation. And finally we determined the targeting to 3’ UTR of BACE1 by miR-29c by a luciferase reporter. Results: It was demonstrated that miR-29c was downregulated in sporadic AD brains, in an association with an upregulation of BACE1 in both mRNA and protein level of BACE1, and also an elevated APPβ accumulation. And the manipulated high level of miR-29c with miR-29c mimics transfection significantly reduced the protein level of BACE1 and APPβ accumulation in human neuroblastoma SH-SY5Y cells. Further luciferase reporter assay demonstrated that miR-29c targets the 3’ UTR of BACE1 and downregulated the BACE1 in HEK293 cells. Conclusion: Present study indicated that miR-29c was downregulated in sporadic AD brains, and it targeted the 3’ UTR of BACE1, reduced the BACE1 expression, and downregulated the APPβ accumulation in vitro.
Keywords: miR-29c, BACE1, APPβ accumulation, Alzheimer’s disease
Introduction
Alzheimer disease (AD) is an aging-related neurodegenerative disorder world widely. To date, the cause and progression of both familial and sporadic (late-onset) AD have not been fully elucidated. However, it is well confirmed that the progressive disease is characterized by the accumulation of plaques formed of short β-amyloid (Aβ) peptides [1-4], which are obtained upon proteolytic cleavage of the β-amyloid precursor protein (APP) [5], by Beta-site Amyloid precursor protein Cleaving Enzyme 1 (BACE1) [6-8]. BACE1 is conceived as a potential and attractive target for therapies against AD. Because BACE1-null mice do not demonstrate any developmental problems or aberrant behavioral phenotypes [9,10], and such agents as Sildenafil [11], GNE-892 [12], L655, 240 [13]. Moreover, targeting of BACE1 mRNA by small interfering RNAs has proven to be effective in down-regulating BACE1 protein levels and activity in cultured primary cortical neurons [14] as well as in a mouse model of AD [15]. It implies the effectiveness of endogenous RNA silencing machinery on BACE1 mRNA, which is based on microRNAs (miRNAs).
miRNAs are a group of 18-24 nt non-coding RNAs, which are known to initially repress mRNA translation or promote mRNA degradation through specific binding mainly to the 3’-untranslated region (UTR) of targeted mRNA [16]. The regulatory role of miRNAs has been recognized in a wide array of eukaryotic species [17,18], and there are about 60% of genes [19,20] to be targeted by more than 1000 miRNAs [21]. And approximately 70% of presently identified miRNAs are found to express in the brain, where they regulate diverse neuronal and glial functions [22]. Actually, most aberrantly-expressed brain-enriched miRNAs induce development of such neurodegenerative diseases, as Parkinson’s disease (PD) and AD [23-26]. Accumulating evidence indicates that deregulated miRNA expression plays a key role in AD pathogenesis. Since the pioneering identification of upregulated miR-9 and miR-128 in the hippocampus of AD brains [27], More kinds of miRNAs, such as elevated miR-146a, which targets complement factor H (CFH) and IL-1 receptor-associated kinase-1 (IRAK1) [28], reduced miR-107, which targets BACE1 [29], reduced miR-103a, which targets actin-binding protein cofilin [30], and decreased miR-29a, which targets neuronnavigator 3 (NAV3) [31], have been recognized to correlate with AD. Therefore, deregulated miRNAs might play important roles in the pathogenesis of AD.
The miR-29 family, which is processed from different precursors, includes three main mature miRNAs, known as hsa-miR-29a, hsa-miR-29b and hsa-miR-29c [32]. The miR-29a/b-1 cluster has been shown to significantly decrease in AD patients, with a correlation with abnormally high BACE1 protein. And the correlation was also found during brain development and in primary neuronal cultures [33]. In addition, it has been shown that miR-29a and miR-29b-1 can regulate BACE1 expression in vitro [33]. Therefore, it is proposed that loss of miR-29a and miR-29b-1 can contribute to increased BACE1 and APPβ levels in sporadic AD. More recently, it was reported that miR-29c, a miRNA that is highly expressed in a double transgenic mouse model of Alzheimer’s disease [34], can lower BACE1 protein in vitro and in transgenic miR-29c mice [35]. The regulation on BACE1 by miR-29c suggests that miR-29c may be also an endogenous regulator of BACE1 protein expression, and pose important role in AD.
In present study, we determined the expression of miR-29c and BACE1 in AD brain tissues, analyzed the correlation of miR-29c expression with BACE1 level. Then, we investigated the regulatory role of miR-29c on the BACE1 expression and on the followed APPβ accumulation in vitro. Present study recognized the regulatory role of miR-29c on BACE1 expression, and revealed the low level of miR-29c and the correlated BACE1 overexpression in AD brain tissues.
Materials and methods
Human brain tissue
31 AD patients and 29 control subjects were implicated in this study. The clinical variants for these subjects were shown in Table 1. Frozen tissue samples of frontal cortices from autopsied and histopathologically confirmed AD and control cases were obtained from the Tianjin Medical University General Hospital and Tianjin 4th Center Hospital (Tianjin China). Each tissue sample was homogenized to 10% (w/v) final concentration in ice-cold 1x phosphate buffer solution (PBS), supplemented with Ribonuclease inhibitor (Takara, Tokyo, Japan) and protease inhibitor cocktail (Roche Diagnostics, GmbH, Germany). And each homogenized sample was used for cellular extraction for mRNA, miRNA and protein. Prior to the operation, patients granted consent for the use of the excised brain tissue in medical or scientific research. And the use of frozen human brain tissue was in accordance with National Institutes of Health guide lines and was approved by the Institutional Review Board of the Tianjin Medical University General Hospital and Tianjin 4th Center Hospital.
Table 1.
AD and control subjects from which the brain tissue was used in this study
| Variants | AD group | Control group | P value |
|---|---|---|---|
| Average age (years) | 78.2 ± 5.45 | 79.5 ± 6.07 | 0.376 |
| Average PMI* (hours) | 2.84 ± 1.25 | 2.93 ± 1.33 | 0.142 |
| Gender | 0.821 | ||
| Male | 18 | 16 | 0.817 |
| Female | 13 | 13 |
PMI: postmortem interval.
Cell culture and treatment
Human neuroblastoma SH-SY5Y cell line was provided by the cell resource center of Chinese academy of medical sciences (Beijing, China). And cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM)/F12 (Invitrogen, Carlsbad, CA, USA) containing 10% FBS (Invitrogen, Carlsbad, CA, USA) or maintained in DMEM/F12 supplemented with 2% FBS. HEK293 cells were cultured in DMEM containing 10% FBS or maintained in DMEM supplemented with 2% FBS. miR-29c mimics or scrambled RNA (miRNA control) (Genepharma, Shanghai, China) with 25 or 50 nM were also transfected by Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA) to manipulate the miR-29c level.
RNA isolation and real-time quantitative PCR
Total cellular RNA isolation was performed with the PureLink® RNA Mini Kit (Invitrogen, Carlsbad, CA, USA), and miRNAs was isolated using mirVanaTM miRNA Isolation Kit (Ambion, Austin, TX, USA) according to manuals. Each mRNA or miRNA sample was added with Ribonuclease inhibitor (Takara, Tokyo, Japan). The expression of BACE1 in mRNA level or of miR-29c was quantified by the realtime quantitative PCR (RT-qPCR) method with Takara One Step RT-PCT kit (Takara, Tokyo, Japan). Relative quantification was determined using the ∆∆Ct method using U6 or β-actin as reference gene [36].
Western blot analysis
Approximately 2 × 105 cells were lyzed with the ProteoJET Cytoplasmic and Nuclear Protein Extraction Kit (Fermentas, Burlington, ON, Canada) and quantified using Bradford Reagent (Bio-Rad, Hercules, CA, USA), protein samples were separated by a 12% gradient SDS-PAGE gel, transferred to PVDF membrane and blocked in 5% skimmed milk. Rabbit polyclonal antibodies to BACE1, Nicastrin, APP (full), APPβ, or β-actin (Cell Signaling Technology Inc., Danvers, MA USA) were used to quantify the molecule expression, with ECL detection systems (Thermo Scientific, Rockford, IL, USA).
Luciferase reporting assay
The 3’ UTR of BACE1 and the CMV promoter were amplified from human chromosomal DNA and pcDNA3.1 (+) and cloned into the pGL3-luciferase basic vector (Promega, Madison, WI, USA). Sequences of primers and cloning strategy are available on request. For the luciferase assays, 50 nM of miR-29c mimics or scrambled RNA were co-transfected with the reporter vector and the Renilla control vector (Promega, Madison, WI, USA) into the HEK293 cells by Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA). 24 h post transfection, the measurements were performed using the Dual luciferase re-porter assay kit (Promega, Madison, WI, USA). Or the HEK293 cells post the transfection for 24 h was lyzed for western blot analysis.
Statistical evaluation
For the analysis of miR-29c and U6, BACE1 and β-actin in mRNA or in protein level, APP (full), APPβ and Nicastrin expression in protein level, between two groups, statistical evaluations are presented as mean ± SE, and data were analyzed using the Student’s t test. The nonparametric Pearson’s correlation test was performed to evaluate the correlation of miR-29c with the BACE1 mRNA level. A statistical significance was considered when P < 0.05.
Results
Reduced miR-29c in AD brains correlates with high BACE1 mRNA
31 AD patients and 29 control subjects were implicated in this study. There was no significant difference in the average age (78.2 ± 5.45 vs. 79.5 ± 6.07), average PMI value (2.84 ± 1.25 vs. 2.93 ± 1.33) and gender distribution (P = 0.821) between the AD and control groups (Table 1). We chose miR-29c, which was recently confirmed to regulate BACE1 [35], for investigation in AD brain tissues. RT-qPCR was performed to evaluate miR-29c expression level in 31 sporadic AD brain tissues and 29 brain specimens from subjects other than AD. Results demonstrated in Figure 1A that the miR-29c level in AD brains was 0.548 ± 0.058, significantly less than 1.000 ± 0.093 in control group (P < 0.001, paired t test). To further examine in AD brain samples the level of BACE1, which catalyzes the cleavage of the β-amyloid precursor protein (APP) [5], we quantified by RT-qPCR the BACE1 mRNA level in those samples. As shown in Figure 1B, compared to the mean level (1.002 ± 0.082) in control group, there was a significantly high level (1.761 ± 0.147) of BACE mRNA in AD brains samples (P < 0.001, paired t test). Then we determined the correlation of the downregulated miR-29c with the upregulated BACE1 mRNA level by Pearson’s correlation test. It was shown that the miR-29c level was negatively correlated with the BACE1 mRNA level in AD brain tissues. (R2 = 0.2362, P < 0.0056) (Figure 1C). Interestingly, there was either a negative correlation of miR-29c level with the BACE1 mRNA level in control brain specimens (R2 = 0.2276, P < 0.0067; Figure 1D), implying a widely regulation on BACE1 by miR-29c.
Figure 1.
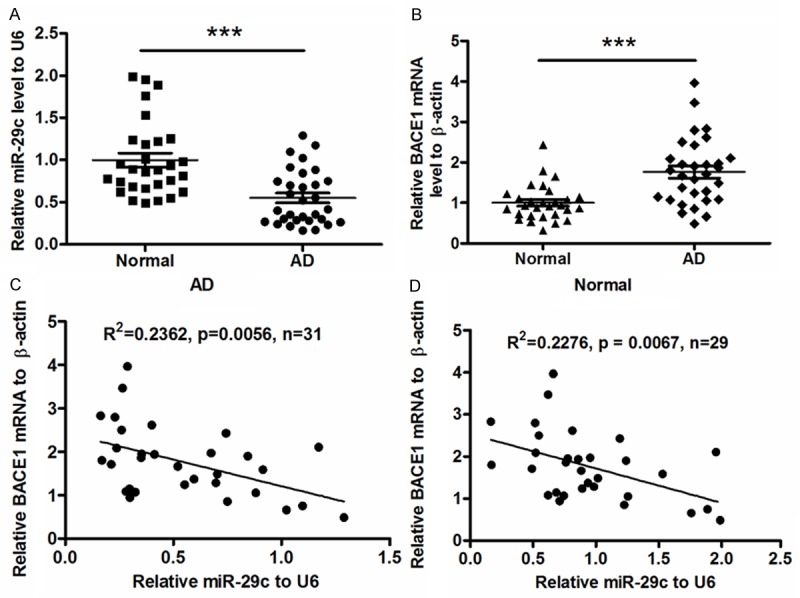
Downregualted miR-29c negatively correlates with the BACE1 mRNA upregulation in sporadic AD brain tissues. (A) miR-29c level was quantified by qRT-PCR in sporadic AD patients or control subjects, with high BACE1 mRNA (B) in AD patients, miR-29c or BACE mRNA was shown as a relative expression to the control group. (C and D) Correlation of miR-29c expression with BACE mRNA level in sporadic AD patients (C) or in control subjects (D). Statistical significance was considered when P < 0.05. ***P < 0.001.
To reconfirm the negative correlation of miR-29c with BACE1, we analyzed the BACE1 and APPβ accumulation in protein level in the sporadic AD brain samples. As shown by the western blot assay results in Figure 2A, BACE1 was significantly higher in AD group than in control group, the percentage of BACE1 to β-actin was 25.6 ± 8.32 in AD group, whereas was 19.4 ± 6.79 in control group, with a p value less than 0.01 (0.0026). And the difference in APPβ accumulation in protein level between both groups was also significant, the percentage of APPβ/APP (full length) was higher in AD group than in control group (P = 0.00087). However, we did not find a difference in the APP (full length) or Nicastrin between two groups.
Figure 2.
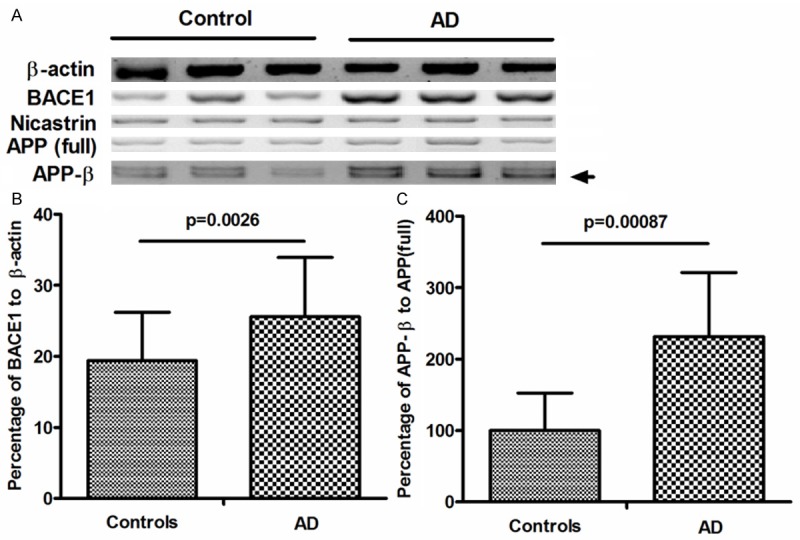
Upregulated BACE1 and APPβ in protein level in sporadic AD brain tissues. A: Representative western blot of BACE1, Nicastrin, APP (full-length), APPβ in AD and matched control subjects. β-actin was used as normalization control. B: Quantifications of BACE1 in AD and control groups, which are shown as percentage to β-actin. C: quantifications of APPβ in AD and control groups, which are shown as percentage to APP (full-length). The relative BACE1 value was averaged for 23 of the 31 AD brain samples or 18 of 29 control brain samples; and the relative APPβ value was averaged for 25 of the 31 AD brain samples or 20 of 29 control brain samples. Statistical significance was considered when P < 0.05.
miR-29c mimics transfection reduces the protein level of BACE1 and APPβ in human neuroblastoma SH-SY5Y cells
To further confirm the regulation on BACE1 and APPβ accumulation by miR-29c in vitro, we manipulated the miR-29c level by miR-29c mimics transfection, and then examined the level of BACE1 and APPβ in SH-SY5Y cells. Firstly, 25 or 50 nM miR-29c mimics or scrambled miRNA oligonucleotide was transfected into SH-SY5Y cells, and the miR-29c level was examined using RT-qPCR. Figure 3A demonstrated that the 25 or 50 nM miR-29c mimics transfection significantly promoted the miR-29c level, compared to the scrambled miRNA transfection (P < 0.001 respectively, paired t test). Then the BACE1 mRNA was examined in the miR-29c mimics- or scrambled miRNA-transfected SH-SY5Y cells. And the miR-29c mimics transfection with either 25 or 50 nM, significantly reduced the BACE1 mRNA level (either P < 0.05). Then BACE1 and APPβ in protein level were analyzed by western blot assay. And it was indicated in Figure 3C that the BACE1 reduced in SH-SY5Y cells by the miR-29c mimics transfection (both 25 and 50 nM), rather than the scrambled miRNA transfection (both 25 and 50 nM). And what’s more, the APPβ also decreased post the miR-29c mimics transfection (both 25 and 50 nM), though the APP (full length) was not regulated by the miR-29c mimics transfection. Therefore, the miR-29c mimics transfection significantly downregulated the BACE1 and APPβ in SH-SY5Y cells.
Figure 3.
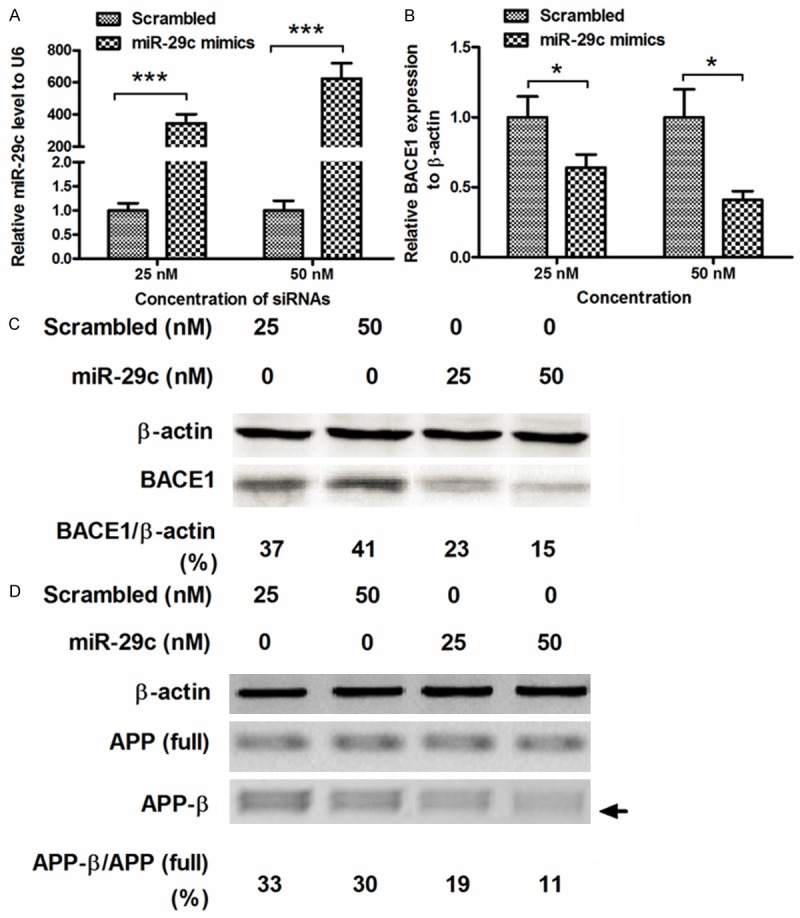
miR-29c mimics transfection inhibits BACE1 expression and APPβ accumulation in SH-SY5Y cells. A: Manipulated high level of miR-29c level in SH-SY5Y cells, by miR-29c mimics transfection, scrambled oligonucleotide as control. miR-29c level was examined by RT-qPCR, with U6 as internal control. B: miR-29c mimics transfection reduced the BACE1 expression in mRNA level, as was quantified by RT-qPCR; C: Western blot assay indicated a reduced BACE1 expression in protein level; D: Western blot analysis of accumulated APPβ in SH-SY5Y cells which were transfected with miR-29c mimics or scrambled oligonucleotide. All results were averaged from triplicate independent experiments. *P < 0.05, ***P < 0.001.
3’ UTR of BACE1 is targeted by miR-29c in HEK293 cells
To investigate whether miR-29c targeted the 3’ UTR of BACE1 and exerted its regulatory role, we analyzed by miRTarbase the alignment of miR-29c with the 3’ UTR of BACE1. It was shown that there were three targeted sites in the 3’ UTR of BACE1, which well paired with miR-29c. Then we constructed a luciferase reporter containing the 3’ UTR of human BACE1, with CMV promoter (Figure 4A), the inserted 3’ UTR of human BACE1 mainly included a miR-29c paired sequence (Figure 4B). As shown in Figure 4C, 25 nM miR-29c mimics, rather than scrambled miRNA, significantly reduced the luciferease activity of the reporter plasmid (P < 0.05) in HEK293 cells. And the 50 nM transfection group reconfirmed the luciferase activity reduction (P < 0.05). In addition, to reconfirm the regulation by miR-29c in HEK293 cells, we also analyzed the influence of miR-29c transfection on the BACE1 expression in protein level in HEK293 cells, and the western blot assay confirmed the reduction of BACE1 in such kind of cells. Taken together, miR-29c was indicated to target the 3’ UTR of BACE1 and reduced the expression of it.
Figure 4.
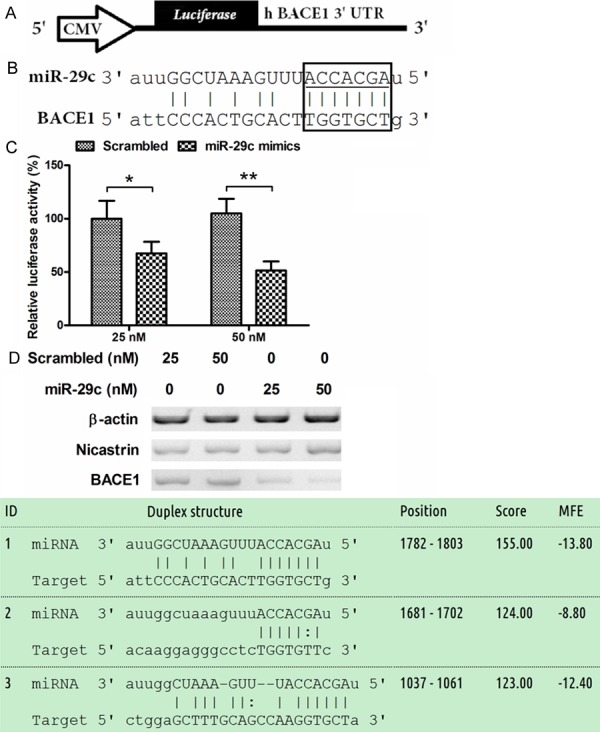
miR-29c targets the 3’ UTR of BACE1 gene and blocks the BACE1 expression. A: Schematic representation of a luciferase reporter with the BACE1 3’ UTR. CMV: Cytomegalovirus promoter; B: Pairment of miR-29c with the 3’ UTR of BACE1, The sequence and putative binding sites of the miR-29c was shown in the box. The miR-29c seed sequence is highlighted with an underscore. C: Relative luciferase of the reporter with a 3’ UTR of BACE1, with renilla luciferase as internal control, in HEK293 cells, post miR-29c mimics or scrambled oligonucleotide transfection. Error bars represent standard deviations derived from three independent experiments. Statistical significance was considered when P < 0.05 or less.
Discussion
miR-29c has been recognized to mediate initiation of gastric carcinogenesis by directly targeting ITGB1 [37], to induce cell cycle arrest in esophageal squamous cell carcinoma by modulating cyclin E expression [38], and even to promote the growth of bladder cancer cells [39]. Besides the role in tumorigenesis, the microRNA is also indicated to play a role in renal interstitial fibrosis. miR-29c is downregulated in renal interstitial fibrosis in humans and rats and restored by HIF-α activation [40], and it was suggested to be a novel, noninvasive marker for renal fibrosis [41]. And more recently, it was reported that miR-29c can lower BACE1 protein in vitro and in transgenic miR-29c mice [35]. The regulation on BACE1 by miR-29c suggests miR-29c to be an endogenous BACE1 regulator, and to pose important role in AD.
In present study, we recognized the downregulation of miR-29c and upregulation of BACE1, in AD brain tissues, and there was a correlation of the downregulated miR-29c with the BACE1 upregulation, which promotes the accumulation of APPβ [42]. And further western blot analysis of BACE1 expression and APPβ accumulation in protein level in those sporadic AD brain samples reconfirmed the negative correlation of miR-29c with BACE1. We then investigated the negative regulation of miR-29c on the BACE1 expression and APPβ accumulation in SH-SY5Y cells. The manipulated upregulation of miR-29c significantly downregulated the expression of BACE1 and APPβ in both mRNA and protein levels in SH-SY5Y cells. Moreover, the miRTarbase alignment demonstrated a well pairment of miR-29c with the 3’ UTR of human BACE1, and the miR-29c targeted the 3’ UTR of human BACE1, directly downregulated the expression of BACE1. Thus, present study recognized the downregulation of miR-29c in sporadic Alzheimer’s disease, which promoted the increased BACE1 expression and APPβ accumulation in AD.
BACE1, also known as Beta-secretase 1, and memapsin-2 [43], is an enzyme that in humans is encoded by the BACE1 gene [44]. BACE1 is important in the formation of myelin sheaths in peripheral nerve cells [45]. And what’s more, BACE1 is necessary for the generation of the 40 or 42 amino acid-long amyloid-β peptides that aggregate in the brain of Alzheimer’s patients [6-8]. Levels of this enzyme have been shown to be elevated in the far more common late-onset sporadic Alzheimer’s [46]. On the other side, the inhibition of BACE1 overexpression by chemicals reduces the APPβ accumulation [47,48]. And the BACE1 level has shown to be regulated by microRNAs, such as miR-29a and miR-29b-1 in vitro [33]. Recently, miR-29c was also indicated to regulate BACE1 expression in a double transgenic mouse model of Alzheimer’s disease [34,35]. Moreover, present study recognized the regulatory role of miR-29c downregulation in sporadic Alzheimer’s disease, by promoting the increased BACE1 expression.
In summary, in present study, we confirmed that miR-29c was downregulated in sporadic Alzheimer’s disease, in association with BACE1 upregulation. And miR-29c targeted the 3’ UTR of human BACE1, directly downregulated the expression of BACE1. Therefore, attenuated miR-29c promoted the BACE1 upregulation and APPβ accumulation in AD.
Disclosure of conflict of interest
None.
References
- 1.Mattson MP. Pathways towards and away from Alzheimer’s disease. Nature. 2004;430:631–9. doi: 10.1038/nature02621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tanzi RE, Bertram L. Twenty years of the Alzheimer’s disease amyloid hypothesis: a genetic perspective. Cell. 2005;120:545–55. doi: 10.1016/j.cell.2005.02.008. [DOI] [PubMed] [Google Scholar]
- 3.Reddy PH, McWeeney S. Mapping cellular transcriptosomes in autopsied Alzheimer’s disease subjects and relevant animal models. Neurobiol Aging. 2006;27:1060–77. doi: 10.1016/j.neurobiolaging.2005.04.014. [DOI] [PubMed] [Google Scholar]
- 4.Thajeb P, Wang P, Chien CL, Harrigan R. Novel polymorphisms of the amyloid precursor protein (APP) gene in Chinese/Taiwanese patients with Alzheimer’s disease. J Clin Neurosci. 2009;16:259–63. doi: 10.1016/j.jocn.2008.04.006. [DOI] [PubMed] [Google Scholar]
- 5.Selkoe DJ, Schenk D. Alzheimer’s disease: molecular understanding predicts amyloid-based therapeutics. Annu Rev Pharmacol Toxicol. 2003;43:545–84. doi: 10.1146/annurev.pharmtox.43.100901.140248. [DOI] [PubMed] [Google Scholar]
- 6.Hussain I, Powell D, Howlett DR, Tew DG, Meek TD, Chapman C, Gloger IS, Murphy KE, Southan CD, Ryan DM Smith TS, Simmons DL, Walsh FS, Dingwall C, Christie G. Identification of a novel aspartic protease (Asp 2) as beta-secretase. Mol Cell Neurosci. 1999;14:419–27. doi: 10.1006/mcne.1999.0811. [DOI] [PubMed] [Google Scholar]
- 7.Sinha S, Anderson JP, Barbour R, Basi GS, Caccavello R, Davis D, Doan M, Dovey HF, Frigon N, Hong J Jacobson-Croak K, Jewett N, Keim P, Knops J, Lieberburg I, Power M, Tan H, Tatsuno G, Tung J, Schenk D, Seubert P, Suomensaari SM, Wang S, Walker D, Zhao J, McConlogue L, John V. Purification and cloning of amyloid precursor protein beta-secretase from human brain. Nature. 1999;402:537–40. doi: 10.1038/990114. [DOI] [PubMed] [Google Scholar]
- 8.Yan R, Bienkowski MJ, Shuck ME, Miao H, Tory MC, Pauley AM, Brashier JR, Stratman NC, Mathews WR, Buhl AE Carter DB, Tomasselli AG, Parodi LA, Heinrikson RL, Gurney ME. Membrane-anchored aspartyl protease with Alzheimer’s disease beta-secretase activity. Nature. 1999;402:533–7. doi: 10.1038/990107. [DOI] [PubMed] [Google Scholar]
- 9.Roberds SL, Anderson J, Basi G, Bienkowski MJ, Branstetter DG, Chen KS, Freedman SB, Frigon NL, Games D, Hu K Johnson-Wood K, Kappenman KE, Kawabe TT, Kola I, Kuehn R, Lee M, Liu W, Motter R, Nichols NF, Power M, Robertson DW, Schenk D, Schoor M, Shopp GM, Shuck ME, Sinha S, Svensson KA, Tatsuno G, Tintrup H, Wijsman J, Wright S, McConlogue L. BACE knockout mice are healthy despite lacking the primary beta-secretase activity in brain: implications for Alzheimer’s disease therapeutics. Hum Mol Genet. 2001;10:1317–24. doi: 10.1093/hmg/10.12.1317. [DOI] [PubMed] [Google Scholar]
- 10.Luo Y, Bolon B, Kahn S, Bennett BD, Babu-Khan S, Denis P, Fan W, Kha H, Zhang J, Gong Y Martin L, Louis JC, Yan Q, Richards WG, Citron M, Vassar R. Mice deficient in BACE1, the Alzheimer’s beta-secretase, have normal phenotype and abolished beta-amyloid generation. Nat Neurosci. 2001;4:231–2. doi: 10.1038/85059. [DOI] [PubMed] [Google Scholar]
- 11.Orejana L, Barros-Minones L, Jordan J, Cedazo-Minguez A, Tordera RM, Aguirre N, Puerta E. Sildenafil Decreases BACE1 and Cathepsin B Levels and Reduces APP Amyloidogenic Processing in the SAMP8 Mouse. J Gerontol A Biol Sci Med Sci. 2014 doi: 10.1093/gerona/glu106. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 12.Takahashi R, Ma S, Deese A, Yue Q, Kim-Kang H, Yi Y, Siu M, Hunt KW, Kallan NC, Hop CE, Liu X, Khojasteh SC. Elucidating the mechanism of cytochrome P450-mediated pyrimidine ring conversion to pyrazole metabolites with the BACE1 inhibitor GNE-892 in rats. Drug Metab Dispos. 2014;42:890–8. doi: 10.1124/dmd.114.057141. [DOI] [PubMed] [Google Scholar]
- 13.Lu Q, Chen WY, Zhu ZY, Chen J, Xu YC, Kaewpet M, Rukachaisirikul V, Chen LL, Shen X. L655, 240, acting as a competitive BACE1 inhibitor, efficiently decreases beta-amyloid peptide production in HEK293-APPswe cells. Acta Pharmacol Sin. 2012;33:1459–68. doi: 10.1038/aps.2012.74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kao SC, Krichevsky AM, Kosik KS, Tsai LH. BACE1 suppression by RNA interference in primary cortical neurons. J Biol Chem. 2004;279:1942–9. doi: 10.1074/jbc.M309219200. [DOI] [PubMed] [Google Scholar]
- 15.Singer O, Marr RA, Rockenstein E, Crews L, Coufal NG, Gage FH, Verma IM, Masliah E. Targeting BACE1 with siRNAs ameliorates Alzheimer disease neuropathology in a transgenic model. Nat Neurosci. 2005;8:1343–9. doi: 10.1038/nn1531. [DOI] [PubMed] [Google Scholar]
- 16.Ouellet DL, Perron MP, Gobeil LA, Plante P, Provost P. MicroRNAs in gene regulation: when the smallest governs it all. J Biomed Biotechnol. 2006;2006:69616. doi: 10.1155/JBB/2006/69616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tanzer A, Stadler PF. Molecular evolution of a microRNA cluster. J Mol Biol. 2004;339:327–35. doi: 10.1016/j.jmb.2004.03.065. [DOI] [PubMed] [Google Scholar]
- 18.Molnar A, Schwach F, Studholme DJ, Thuenemann EC, Baulcombe DC. miRNAs control gene expression in the single-cell alga Chlamydomonas reinhardtii. Nature. 2007;447:1126–9. doi: 10.1038/nature05903. [DOI] [PubMed] [Google Scholar]
- 19.Lewis BP, Burge CB, Bartel DP. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell. 2005;120:15–20. doi: 10.1016/j.cell.2004.12.035. [DOI] [PubMed] [Google Scholar]
- 20.Friedman RC, Farh KK, Burge CB, Bartel DP. Most mammalian mRNAs are conserved targets of microRNAs. Genome Res. 2009;19:92–105. doi: 10.1101/gr.082701.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bentwich I, Avniel A, Karov Y, Aharonov R, Gilad S, Barad O, Barzilai A, Einat P, Einav U, Meiri E Sharon E, Spector Y, Bentwich Z. Identification of hundreds of conserved and nonconserved human microRNAs. Nat Genet. 2005;37:766–70. doi: 10.1038/ng1590. [DOI] [PubMed] [Google Scholar]
- 22.Fineberg SK, Kosik KS, Davidson BL. MicroRNAs potentiate neural development. Neuron. 2009;64:303–9. doi: 10.1016/j.neuron.2009.10.020. [DOI] [PubMed] [Google Scholar]
- 23.Harraz MM, Dawson TM, Dawson VL. MicroRNAs in Parkinson’s disease. J Chem Neuroanat. 2011;42:127–30. doi: 10.1016/j.jchemneu.2011.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kocerha J, Kauppinen S, Wahlestedt C. microRNAs in CNS disorders. Neuromolecular Med. 2009;11:162–72. doi: 10.1007/s12017-009-8066-1. [DOI] [PubMed] [Google Scholar]
- 25.Maes OC, Chertkow HM, Wang E, Schipper HM. MicroRNA: Implications for Alzheimer Disease and other Human CNS Disorders. Curr Genomics. 2009;10:154–68. doi: 10.2174/138920209788185252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nelson PT, Wang WX, Rajeev BW. MicroRNAs (miRNAs) in neurodegenerative diseases. Brain Pathol. 2008;18:130–8. doi: 10.1111/j.1750-3639.2007.00120.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lukiw WJ. Micro-RNA speciation in fetal, adult and Alzheimer’s disease hippocampus. Neuroreport. 2007;18:297–300. doi: 10.1097/WNR.0b013e3280148e8b. [DOI] [PubMed] [Google Scholar]
- 28.Lukiw WJ, Zhao Y, Cui JG. An NF-kappaB-sensitive micro RNA-146a-mediated inflammatory circuit in Alzheimer disease and in stressed human brain cells. J Biol Chem. 2008;283:31315–22. doi: 10.1074/jbc.M805371200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang WX, Rajeev BW, Stromberg AJ, Ren N, Tang G, Huang Q, Rigoutsos I, Nelson PT. The expression of microRNA miR-107 decreases early in Alzheimer’s disease and may accelerate disease progression through regulation of beta-site amyloid precursor protein-cleaving enzyme 1. J Neurosci. 2008;28:1213–23. doi: 10.1523/JNEUROSCI.5065-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yao J, Hennessey T, Flynt A, Lai E, Beal MF, Lin MT. MicroRNA-related cofilin abnormality in Alzheimer’s disease. PLoS One. 2010;5:e15546. doi: 10.1371/journal.pone.0015546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shioya M, Obayashi S, Tabunoki H, Arima K, Saito Y, Ishida T, Satoh J. Aberrant microRNA expression in the brains of neurodegenerative diseases: miR-29a decreased in Alzheimer disease brains targets neurone navigator 3. Neuropathol Appl Neurobiol. 2010;36:320–30. doi: 10.1111/j.1365-2990.2010.01076.x. [DOI] [PubMed] [Google Scholar]
- 32.Gong J, Li J, Wang Y, Liu C, Jia H, Jiang C, Wang Y, Luo M, Zhao H, Dong L Song W, Wang F, Wang W, Zhang J, Yu J. Characterization of microRNA-29 family expression and investigation of their mechanistic roles in gastric cancer. Carcinogenesis. 2014;35:497–506. doi: 10.1093/carcin/bgt337. [DOI] [PubMed] [Google Scholar]
- 33.Hebert SS, Horre K, Nicolai L, Papadopoulou AS, Mandemakers W, Silahtaroglu AN, Kauppinen S, Delacourte A, De Strooper B. Loss of microRNA cluster miR-29a/b-1 in sporadic Alzheimer’s disease correlates with increased BACE1/beta-secretase expression. Proc Natl Acad Sci U S A. 2008;105:6415–20. doi: 10.1073/pnas.0710263105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang H, Liu J, Zong Y, Xu Y, Deng W, Zhu H, Liu Y, Ma C, Huang L, Zhang L Qin C. miR-106b aberrantly expressed in a double transgenic mouse model for Alzheimer’s disease targets TGF-beta type II receptor. Brain Res. 2010;1357:166–74. doi: 10.1016/j.brainres.2010.08.023. [DOI] [PubMed] [Google Scholar]
- 35.Zong Y, Wang H, Dong W, Quan X, Zhu H, Xu Y, Huang L, Ma C, Qin C. miR-29c regulates BACE1 protein expression. Brain Res. 2011;1395:108–15. doi: 10.1016/j.brainres.2011.04.035. [DOI] [PubMed] [Google Scholar]
- 36.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25:402–8. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 37.Han TS, Hur K, Xu G, Choi B, Okugawa Y, Toiyama Y, Oshima H, Oshima M, Lee HJ, Kim VN Chang AN, Goel A, Yang HK. MicroRNA-29c mediates initiation of gastric carcinogenesis by directly targeting ITGB1. Gut. 2014 doi: 10.1136/gutjnl-2013-306640. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ding DP, Chen ZL, Zhao XH, Wang JW, Sun J, Wang Z, Tan FW, Tan XG, Li BZ, Zhou F Shao K, Li N, Qiu B, He J. miR-29c induces cell cycle arrest in esophageal squamous cell carcinoma by modulating cyclin E expression. Carcinogenesis. 2011;32:1025–32. doi: 10.1093/carcin/bgr078. [DOI] [PubMed] [Google Scholar]
- 39.Xu F, Zhang Q, Cheng W, Zhang Z, Wang J, Ge J. Effect of miR-29b-1* and miR-29c knockdown on cell growth of the bladder cancer cell line T24. J Int Med Res. 2013;41:1803–10. doi: 10.1177/0300060513505266. [DOI] [PubMed] [Google Scholar]
- 40.Fang Y, Yu X, Liu Y, Kriegel AJ, Heng Y, Xu X, Liang M, Ding X. miR-29c is downregulated in renal interstitial fibrosis in humans and rats and restored by HIF-alpha activation. Am J Physiol Renal Physiol. 2013;304:F1274–82. doi: 10.1152/ajprenal.00287.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lv LL, Cao YH, Ni HF, Xu M, Liu D, Liu H, Chen PS, Liu BC. MicroRNA-29c in urinary exosome/microvesicle as a biomarker of renal fibrosis. Am J Physiol Renal Physiol. 2013;305:F1220–7. doi: 10.1152/ajprenal.00148.2013. [DOI] [PubMed] [Google Scholar]
- 42.Zhiyou C, Yong Y, Shanquan S, Jun Z, Liangguo H, Ling Y, Jieying L. Upregulation of BACE1 and beta-amyloid protein mediated by chronic cerebral hypoperfusion contributes to cognitive impairment and pathogenesis of Alzheimer’s disease. Neurochem Res. 2009;34:1226–35. doi: 10.1007/s11064-008-9899-y. [DOI] [PubMed] [Google Scholar]
- 43.Hong L, Koelsch G, Lin X, Wu S, Terzyan S, Ghosh AK, Zhang XC, Tang J. Structure of the protease domain of memapsin 2 (beta-secretase) complexed with inhibitor. Science. 2000;290:150–3. doi: 10.1126/science.290.5489.150. [DOI] [PubMed] [Google Scholar]
- 44.Vassar R, Bennett BD, Babu-Khan S, Kahn S, Mendiaz EA, Denis P, Teplow DB, Ross S, Amarante P, Loeloff R Luo Y, Fisher S, Fuller J, Edenson S, Lile J, Jarosinski MA, Biere AL, Curran E, Burgess T, Louis JC, Collins F, Treanor J, Rogers G, Citron M. Beta-secretase cleavage of Alzheimer’s amyloid precursor protein by the transmembrane aspartic protease BACE. Science. 1999;286:735–41. doi: 10.1126/science.286.5440.735. [DOI] [PubMed] [Google Scholar]
- 45.Willem M, Garratt AN, Novak B, Citron M, Kaufmann S, Rittger A, DeStrooper B, Saftig P, Birchmeier C, Haass C. Control of peripheral nerve myelination by the beta-secretase BACE1. Science. 2006;314:664–6. doi: 10.1126/science.1132341. [DOI] [PubMed] [Google Scholar]
- 46.Li Q, Chen M, Liu H, Yang L, Yang G. Expression of APP, BACE1, AChE and ChAT in an AD model in rats and the effect of donepezil hydrochloride treatment. Mol Med Rep. 2012;6:1450–4. doi: 10.3892/mmr.2012.1102. [DOI] [PubMed] [Google Scholar]
- 47.Yu F, Zhang Y, Chuang DM. Lithium reduces BACE1 overexpression, beta amyloid accumulation, and spatial learning deficits in mice with traumatic brain injury. J Neurotrauma. 2012;29:2342–51. doi: 10.1089/neu.2012.2449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Probst G, Xu YZ. Small-molecule BACE1 inhibitors: a patent literature review (2006-2011) Expert Opin Ther Pat. 2012;22:511–40. doi: 10.1517/13543776.2012.681302. [DOI] [PubMed] [Google Scholar]