

Abstract
Ulcerative colitis (UC) is an inflammatory bowel disease, and its pathogenesis involves a variety of genetic, environmental, and immunological factors such as T helper cells and their secreted cytokines. B and T lymphocyte attenuator (BTLA) is an immunoregulatory receptor that has a strong suppressive effect on T-cell function. However the role of BTLA in UC remains poorly understood. Here we demonstrated that the frequency of BTLA-expressing CD3+ T cells, especially CD4+ T cells, increased in blood and mucosa in mice with DSS-induced colitis. The frequency of Foxp3-expressing cells in BTLA+ CD4+ T cell from lamina propria mononuclear cells (LPMCs) was much higher in DSS-treated mice than that in controls. Similarly, the proportion of IL-17+ cells in BTLA+ CD4+ T cells from LPMCs in DSS-treated mice is much higher than that in controls, while no perceptible difference for the proportion of IFN-γ+ cells in BTLA+ CD4+ T cells was noted between DSS-treated mice and controls. Treatment of mesalazine, an anti-ulcerative colitis drug, down-regulated Foxp3 and IL-17 expression in BTLA positive T cells along with attenuated severity for colitis. Our findings indicate that BTLA may be involved in the control of inflammatory responses through increasing Foxp3 expression, rather than attenuating IL-17 production, in DSS-induced colitis.
Keywords: Colitis, dextran sulfate sodium, B and T lymphocyte attenuator, Foxp3, IL-17, IFN-γ
Introduction
Inflammatory bowel diseases (IBD), including ulcerative colitis (UC) and Crohn’s disease (CD), are chronic gastrointestinal disorders characterized by chronic intestinal inflammation. Although it is widely accepted that genetic, environmental, and immunological factors are involved [1,2], the exact mechanisms underlying IBD pathoetiologies are yet to be addressed, T cells and their cytokines are the main effectors in the induction and perpetuation of intestinal inflammation [3]. It was not until a few years ago that Th1 and Th2 paradigm was used to differentiate the underlying immunological conditions of CD and UC [4,5]. More recently, a third subset of T helper cells, Th17 cells, has been described contributing to IBD pathogenesis [6,7]. Th17 cells produce IL-17, IL-22, and IL-23. IL-17 levels both in mucosa and serum were increased in active IBD patients [8,9]. Unlike effector T cells, Foxp3+ Treg cells (regulatory T cells) play a nonredundant role in immune homeostasis by preventing pathological inflammatory responses to environmental and self-antigens [10,11]. Loss-of-function mutations in the Foxp3 gene lead to impaired Treg function and the development of a severe multi-organ inflammatory disease [12]; illustrating the importance of Treg cells in the control of intestinal immune function. Mouse models of intestinal inflammation have also pinpointed a key role for Treg cells in intestinal homeostasis [13].
Several models of experimental colitis have been reported to demonstrate various pathophysiological aspects of human IBD [1]. Dextran sulfate sodium (DSS)-induced colitis is a well-established animal model of mucosal inflammation for the study of IBD pathogenesis [24,25]. DSS-induced colitis is known as a UC model, and many studies have described UC as a Th2 disease [26,27]. However, there were also several studies suggested that DSS colitis is dependent on Th1- or Th17-mediated inflammation [28-31]. Thus, the roles of T helper cells in DSS-induced colitis are unclear.
B and T lymphocyte attenuator (BTLA), a recently discovered inhibitory receptor belonging to the CD28 family, is expressed by most lymphocytes and shares structural and functional similarities with cytotoxic T-lymphocyte antigen-4 (CTLA-4) and programmed death 1 (PD1) [14]. BTLA interacts with the herpesvirus entry mediator (HVEM; TNFRSF14), a TNFR super family member found on T, B, natural killer (NK), dendritic (DC) and myeloid cells [15]. The function of BTLA was described in several models of inflammatory disorders and autoimmune diseases [16-18], suggesting that BTLA is crucial for dampening overreactive immune responses. BTLA-deficient mice exhibit enhanced specific antibody responses and sensitivity to EAE [14], rapid rejection of partially MHC-mismatched cardiac allograft [16], and acceleration of experimental colitis [17]. It has been shown that the deficiency of BTLA also disturbed self-tolerance, resulting in the development of an autoimmune hepatitis like disease and lymphocytic infiltration in multiple organs [18]. More recent studies further demonstrated emerging evidence suggesting that BTLA could also initiate pro-survival signals for activated or effector T cells relevant to the generation of immune memory other than its regulatory role in diverse immune responses [19,20-23]. Therefore, BTLA expression on T cells might correlate with T-cell dysfunction and disease pathogenic events. However, the impact of BTLA on the modulation of T-cell responses during the course of UC is yet to be fully addressed. We thus in the present study employed an animal model with DSS-induced colitis to dissect the role of BTLA in T cells and their implication in the pathogenesis of ulcerative colitis.
Materials and methods
Mice
C57BL/6 mice (6- to 8 wk-old) were purchased from the Experimental Animal Center of Guangdong Province (Guangzhou, China). The mice were housed in the Experimental Animal Center at Guangdong Medical College for at least 1 wk before inclusion in experiments. All of the studies were approved by Committees on Use and Care of Animals at the Guangdong Medical College.
Collection of human colon tissues
Human colon tissues were collected from patients with UC via endoscopic colon biopsy. All procedures were approved by the Human Institutional Review Board of the Guangdong Medical College. Informed consent was obtained in writing and a copy was inserted in the medical records of the patients.
Antibodies and reagents
Anti-mouse CD4-FITC (GK1.5), CD8a-PE (53-6.7), CD272 (BTLA)-APC (8F4), mouse/rat IL-17A-PE (eBio17B7), IFN-gamma- PerCP-Cy5.5 (XMG1.2), Foxp3 (clone FJK-16s), and PE-Cy5.5 Foxp3 (FJK-16s) were purchased from eBioscience Inc. (San Diego, CA). Anti-mouse CD3e-PE-Cy7 (17A2) was obtained from BioLegend (San Diego, CA). Dextran sulfate sodium (DSS) was derived from MP Biomedicals (Shanghai, China). Fecal occult blood kits were originated from Nanjing Jiancheng Bioengineering Institute (Nanjing, China).
DSS-induced colitis and mesalazine treatment
Experimental colitis was induced by administration of DSS (36-50kDa) for 7 days. Briefly, mice in DSS-treated group were orally administered 5.0% DSS in drinking water, while mice provided with acidified water were served as controls. A portion of DSS-induced animals were orally treated with mesalazine (MSLZ, 0.0026/0.02 Kg/day) for 1 wk. The activity, coat color changes, stool, color and the presence of hemorrhage were monitored on daily basis.
Clinical evaluation of DSS-induced colitis
Body weight loss during the course for induction of colitis was normalized by the body weight on day 1. Stool consistency was checked for diarrhea and feces were screened for blood detection using an aforementioned fecal occult blood kit. The disease activity index (DAI) was as following [35]: (a) weight loss (0 point = none, 1 point = 1-5% weight loss, 2 points = 5-10% weight loss, 3 points = more than 10% weight loss), (b) stool consistency (0 point = normal, 1 point = soft stools, 2 points = very soft stools, 3 points = watery stools), and (c) the date of presence for blood in stool (0 point = no blood in stool, 1 point = day 7, 2 points = day 6 or day 5, 3 points = within day 4). The DAI was calculated as the sum of weight loss, stool consistency, and day of bleeding, with the total DAI score ranging from 0 (unaffected) to 9 (severe colitis).
Histopathological analysis and immunohistochemistry
Paraffin-embedded sections were stained with hematoxylin and eosin. The degree of colonic inflammation was analyzed according to the scoring system described previously [36]. For serial and double immunohistochemical staining, frozen sections were incubated with indicated primary antibodies overnight at room temperature, followed by 30 min of incubation with appropriate secondary antibodies. Slides were then counterstained with 4’,6’-diamidino-2-phenylindole (DAPI) and analyzed using a Zeiss confocal microscope (Carl Zeiss Microimaging, Oberkochen, Germany) as instructed.
Mononuclear cell isolation
Gut LPMCs were isolated as described below. Intestinal tissue was washed extensively with RPMI 1640 medium and all the patches were removed with a scissor, followed by a longitudinal incision, and then cut into 1 mm2 pieces. Processed tissue debris was incubated for 30 min at 37°C in calcium-and magnesium-free Hanks’s balanced salt solution containing 5% FCS and 1 mM EDTA. After washed, tissue debris were incubated for 1 h at 37°C in 30 ml complete RPMI-5 (RPMI1640 + 5% FCS) containing 1 mg/ml collagenase (GIBICO). A 20 cc syringe with a 16 g feeding needle was next employed to, aspirate tissue pieces in and out of syringe (moderately slowly) 10 times to disrupt the tissue and release remaining cells. The LPMCs preparations were washed through a nylon mesh screen into fresh 15 ml tubes. The cells were resuspended in RPMI-5 after extensive washed and then applied to Ficoll solution gently, followed by centrifugation at 3000 rpm for 20 min. LPMCs were collected from the white interface and subjected to washed with RPMI-5. The number and viability of isolated LPMCs were assessed by Trypan blue staining.
Flow cytometric analysis
The isolated cells were stimulated with PMA for 4 h and BFA was added into the culture after 2 h of stimulation. For cytofluorometric analysis, the cells were stained for 15 min at room temperature using the saturating concentrations of antibodies against CD3, CD4, CD8, and CD272 (BTLA), respectively. After washed, the cells were incubated for 15 min at room temperature in Fix/PERM A (MuLTISCIENCES Biotech Co. Ltd), followed by 20 min incubation in Fix/PERM B (MuLTISCIENCES Biotech Co. Ltd), and stained for 20 min with following antibodies: IL-17A mAb, IFN-γ mAb, and Foxp3 mAb. After washed, cells were analyzed on a FACS 440 flow cytometer (BD Biosciences, Mountain View, CA).
Statistical analysis
All data were analyzed using GraphPad Prism 5.0 software. Data were shown as mean ± SD and were analyzed by the Mann Whitney test or the Wilcoxon matched-pairs signed-rank test. An unpaired Student t test (two-tailed) was used to assess differences between two groups when the data met the assumptions of a t test, depending on the experiment. A P value < 0.05 was considered with statistical significance.
Results
Increased expression of BTLA in CD4+ T cells in DSS-induced colitis
To assess the role of BTLA in pathogenesis of ulcerative colitis, colon biopsies were obtained in patients with UC, and then subjected to immunohistochemical analysis of BTLA expression. Interestingly, high levels of BTLA were detected in colon mucosal area of all examined patients with active UC (Figure 1A, 1B). Con-immunostaining further revealed that BTLA was detected on both infiltrated CD4+ and CD4- T cells (Figure 1C, 1D).
Figure 1.
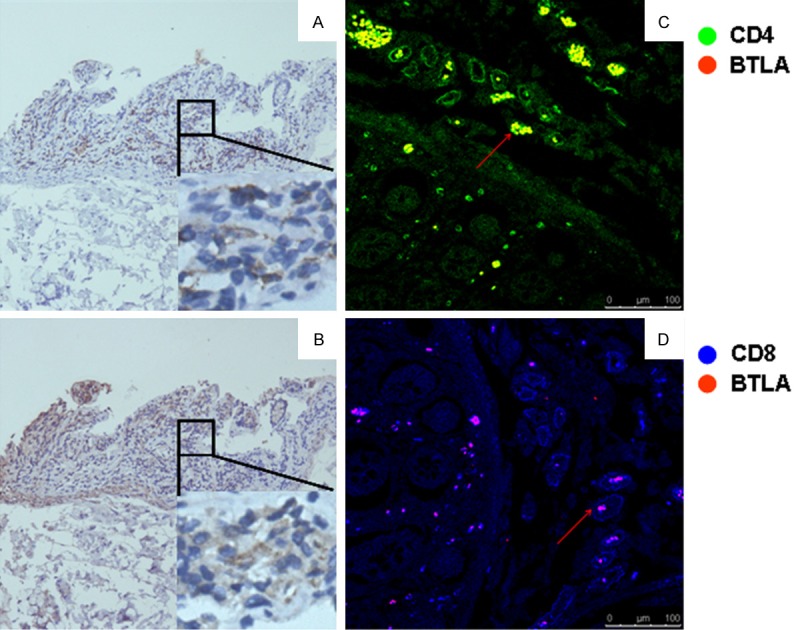
BTLA expression in colon tissue from the patient with UC. Colon tissues from UC patients were stained with enzyme- or fluorescence-labeled antibodies, and then observed under a light microscopy or Laser Scanning Confocal Microscopy. Same area in colon tissue were stained with anti-human CD3 (A) or anti-human BTLA (B) respectively, immunohistochemically. (C) and (D) show co-staining of BTLA (red) with CD4 (green) or CD8 (blue).
To confirm these results, we next induced colitis by feeding mice with DSS water. During the course of colitis development, body weight and stool occult blood (OB) were tested daily, colon tissues and peripheral blood were harvested at day 8 of induction. After 4 days of DSS treatment, mice displayed a progressive disease severity as manifested by the acceleration of weight loss, presence of diarrhea, reduction of mobility, stool OB, and piloerection of the fur. Notably, DSS-treated mice showed a reduction of ~15% of initial starting weight (Figure 2A) and increased DAI scores (Figure 2B), which were attenuated by MSLZ treatment. Consistently, DSS-treated mice showed severe intestinal inflammation as evidenced by the shorter length of colon (Figure 2C), H & E staining revealed prominent epithelial cell hyperplasia, absence of goblet cells, and a massive infiltration of mononuclear cells (Figure 2D). Histological analysis further demonstrated significant pathological scores in DSS-induced mice (Figure 2E).
Figure 2.
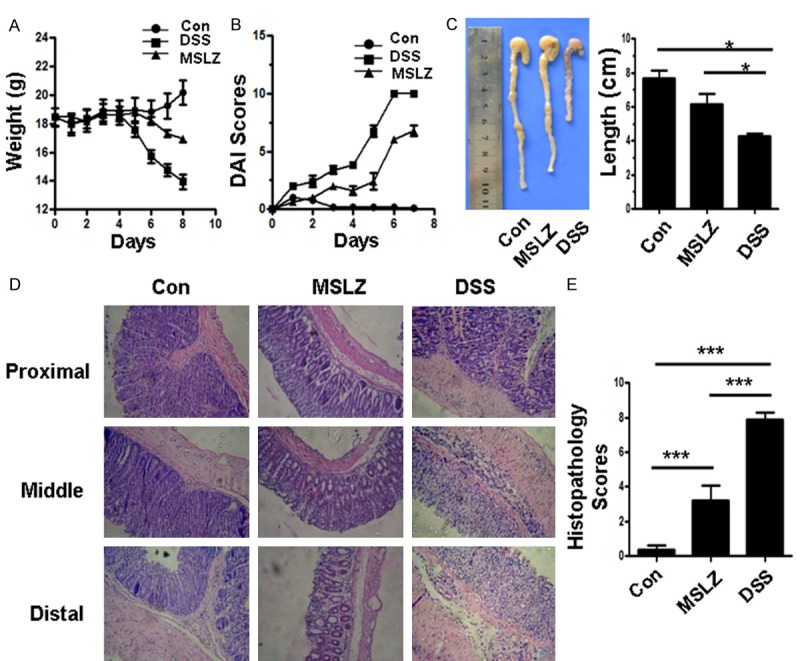
DAI scores and histopathology scores of mice. (A) and (B) show body weight changes and DAI scores, respectively. (C) shows the colon length of each group. (D) shows representative images for histopathological changes in each group, (E) is statistical analysis of histopathological changes. *P < 0.05; **P < 0.01; ***P < 0.001.
BTLA expression in peripheral blood T cells or in colon infiltrated T cells was then analyzed by flow cytometry. A 2-fold increase for BTLA expression was noted both in peripheral blood CD3+ T cells (Figure 3A) and colon infiltrated CD3+ T cells (Figure 3D). Furthermore, a significant increase for the number of BTLA+ CD4+ T cells was characterized both in PBMCs (Figure 3B) and LPMCs (Figure 3E), while perceptible difference for BTLA+ CD8+ T cells was detected between DSS-induced mice and controls (Figure 3C and 3F). Laser Scanning Confocal Microscopy also showed an abundant expression of BTLA in DSS-induced mice (Figure S1). Together these data demonstrate that during the course of colitis development, T cells, predominantly CD4+ T cells, express high levels of BTLA, which may contribute to UC progression.
Figure 3.
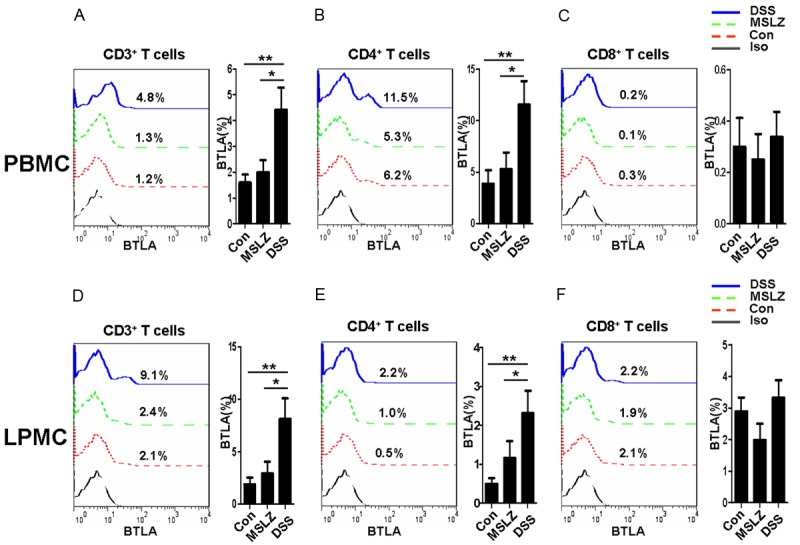
BTLA expression in T cell subsets. Mononuclear cells were isolated from peripheral blood and colon tissues. LPMCs and PBMC were then stained with anti-CD3, anti-CD4, anti-CD8, and anti-BTLA, and analyzed by flow cytometry. Left column, representative flow cytometric graph; Right column, statistical analysis. (A) and (D) show BTLA expressed in CD3+ T cell from PBMCs (A) or LPMCs (D); (B) and (E) show BTLA expressed in CD3+CD4+ T cell subset from PBMCs (B) or LPMCs (E); (C) and (F) show BTLA expressed in CD3+CD8+ T cell subset from PBMCs (B) or LPMCs (E). *P < 0.05; **P < 0.01; ***P < 0.001.
BTLA associates Foxp3 expression in colon tissue
As Tregs are crucial in maintaining intestinal mucosal homeostasis and take part in UC progression, we thus next sought to determine the impact of BTLA on Foxp3 expression. Interestingly, about 35% of CD4+ BTLA+ T cells from LPMCs expressed Foxp3 in DSS-treated mice, which were much higher than those in control mice. In addition, Foxp3+ cells in CD4+ BTLA+ LPMCs was higher than that in PBMCs (Figure 4A). Surprisingly, over 50% of CD8+ BTLA+ T cells in LPMCs from DSS-treated mice expressed Foxp3, which were two times higher than those in PBMCs. However, no significant difference in terms of the frequency for Foxp3+ CD8+ BTLA+ cells was observed between DSS-treated and control mice (Figure 3B). The expression of Foxp3 in BTLA+ and in BTLA- T cells was also analyzed. In LPMCs of DSS-treated mice, higher frequency of Foxp3+ cells was found in CD4+ BTLA+ T cells compared to CD4+ BTLA- counterparts (Figure 4C). In control mice, however, there was a lower frequency of Foxp3+ cells in CD4+ BTLA+ T cells compared to that in CD4+ BTLA- T cells (Figure 4C). In contrast, the number of Foxp3+ cells in CD8+ BTLA+ T cells was much higher than that in CD8+ BTLA- T cells in LPMCs from all groups of mice examined (Figure 4D). Similarly, much higher number of Foxp3+ cells was noted in both CD4+ (Figure 4E) and CD8+ (Figure 4F) in PBMCs of all group of mice examined. Collectively, these data suggest that BTLA expression is likely associated with induction of Foxp3 expression in the setting of DSS-induced colitis.
Figure 4.
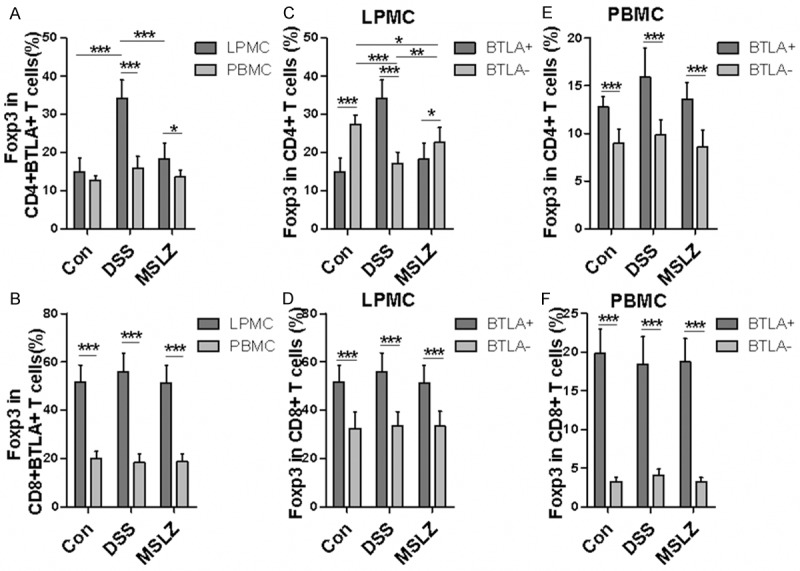
Foxp3 expression in BTLA-expressing T cells. LPMCs and PBMCs were stained with surface marker and intracellular Foxp3 as indicated, followed by flow cytometric analysis. (A and B) show Foxp3 expression in CD4+ BTLA+ and CD8+ BTLA+ T cells. (C and D) are comparative analyses of Foxp3 expression in BTLA+ and BTLA- T cells from LPMCs. (E and F) Foxp3 expression in BTLA+ and BTLA- T cells from PBMC. *P < 0.05; **P < 0.01; ***P < 0.001.
BTLA expression does not attenuate IL-17 production
Because IL-17 and IFN-γ are important inflammatory cytokines implicated in UC pathoetiology, we further explored the effect of BTLA on IL-17 and IFN-γ expression. LPMCs manifested higher frequencies for IL-17- and IFN-γ-producing cells both in CD4+ BTLA+ (Figure 5A) and CD4- BTLA+ T cells (Figure 5B) as compared with that of PBMCs. Importantly, DSS-treatment induced much more IL-17+ cells in CD4+ BTLA+ T cells from LPMCs as compared to controls (Figure 5A). Unexpectedly, DSS-treatment failed to show perceptible effect on IL-17 production in BTLA- T cells, and similarly, no discernable difference was noted in terms of IFN-γ+ cells between BTLA+ and BTLA- T cells (Figures 5C, 5E, 6A, 6C and 6E). In DSS-treated mice, higher proportion of IL-17+ cells was observed in BTLA+ T cells compared to BTLA- counterparts. In contrast, higher proportion of IFN-γ+ cells was found in BTLA- T cells than in BTLA+ counterpart in PBMCs (Figure 6E). Collectively, these results demonstrate that BTLA expression may not impact the production of IL-17+ cells in the setting of DSS-induced colitis.
Figure 5.
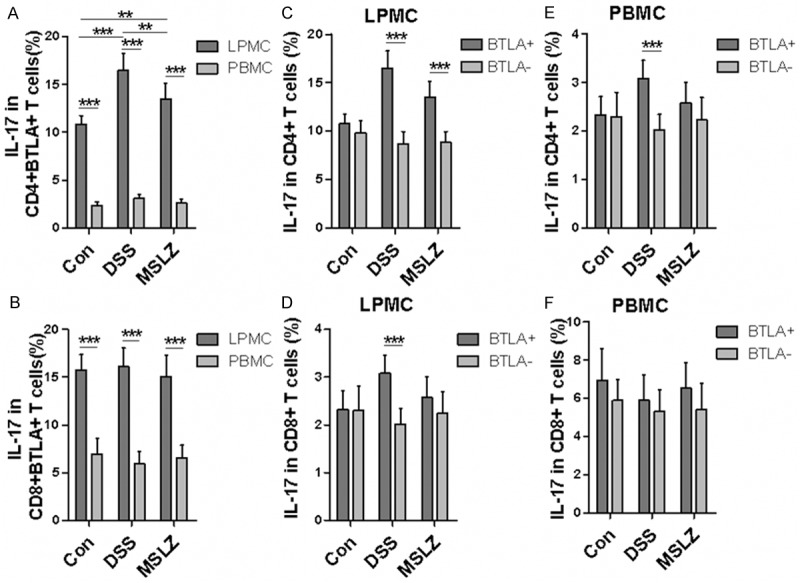
Intracellular cytokine staining of IL-17 in BTLA+ T cells. LPMCs and PBMCs were stained with surface marker and intracellular IL-17. A and B. Show IL-17 production in CD4+ BTLA+ and CD8+ BTLA+ T cells. C and D. IL-17 production in BTLA+ and BTLA- T cells from LPMCs. E and F. IL-17 production in BTLA+ and BTLA- T cells from PBMC. *P < 0.05; **P < 0.01; ***P < 0.001.
Figure 6.
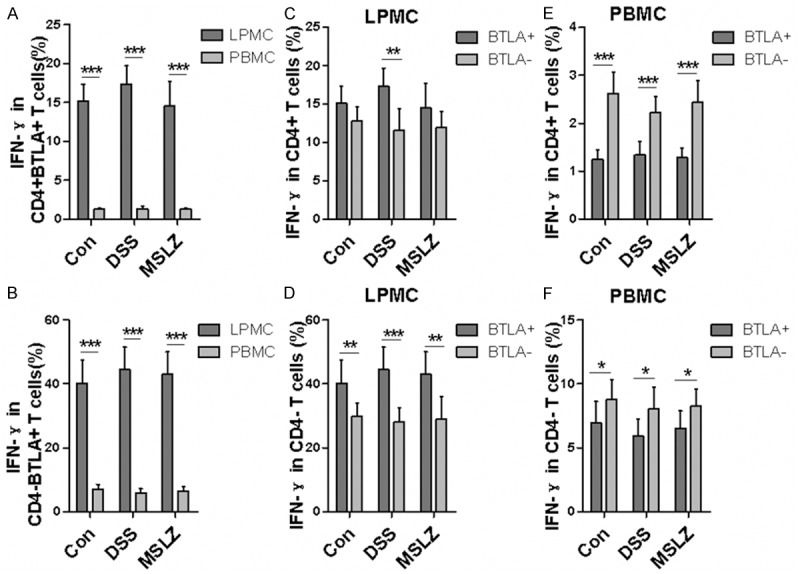
Intracellular cytokine stain of IFN-γ in BTLA+ T cells. LPMCs and PBMCs were stained with surface marker and intracellular IFN-γ. A and B. Show IFN-γ production in CD4+ BTLA+ and CD8+ BTLA+ T cells. C and D. IFN-γ production in BTLA+ and BTLA- T cells from LPMCs. E and F. IFN-γ production in BTLA+ and BTLA- T cells from PBMC. *P < 0.05; **P < 0.01; ***P < 0.001.
Mesalazine treatment inhibits BTLA expression along with attenuated production of Foxp3+ and IL-17+ T cells
Mesalazine (MSLZ) is an anti-inflammatory drug commonly used in the clinical setting for treatment of inflammatory bowel disease, especially UC. It is a bowel-specific aminosalicylate drug that acts locally in the gut, there by having few systemic side effects. We finally sought to investigate whether MSLZ attenuation of colitis symptom involves regulation of BTLA expression connected to the production of Foxp3+ or IL-17+ T cells. For this purpose, we conducted flow cytometry analysis of BTLA, Foxp3, and IL-17 expression in mice with MSLZ treatment. Indeed, MSLZ treatment significantly down-regulated BTLA expression in CD4+ T cells both in LPMCs and PBMCs (Figure 3A, 3B, 3D, 3E). In consistent with this observation, MSLZ treatment significantly decreased the proportion of Foxp3+ and IL-17+ cells in CD4+ BTLA+ T cells, but without detectable effect on the production of IFN-γ+ cells (Figure 5A). Collectively, those results suggest that MSLZ could modulate suppressive immune response and inflammatory response simultaneously.
Discussion
In the present study, we demonstrate an increase of BTLA expression on CD3+ T cells (especially CD4+ subpopulation) from both LPMCs and PBMCs in DSS-induced colitis. Importantly, increased BTLA in CD4+ T cell is associated with the expression of Foxp3, a master regulator for Tregs, suggesting that BTLA might be important for suppressive function of Tregs. Particularly, we found lower IFN-γ production in BTLA+ T cells suggesting BTLA expression may suppress the production of IFN-γ. However, IL-17 production in CD4+ BTLA+ T cells was higher in LPMCs from DSS-treated mice than that in controls, indicating IL-17 production induced by DSS may not be attenuated by BTLA. These findings uncovered a previously unknown mechanism by which BTLA regulates T-cell immune responses in ulcerative colitis.
Foxp3 is an immune regulator found in Tregs and is important for the development and suppressive function of Tregs [33]. Loss-of-function mutations in the Foxp3 gene lead to severe multigrain inflammatory diseases [12]. Studies have shown that Foxp3+ Treg increased in inflamed mucosa of Crohn’s disease (CD) and ulcerative colitis (UC), despite the fact that they were decreased in PBMCs [34]. It has been shown that Foxp3+ Tregs express BTLA and BTLA signal displayed suppressive function on effector cells [35]. However, the relationship between BTLA and Foxp3 remains to be fully elucidated. Our study suggested that DSS treatment induces an increase of Foxp3+ Tregs in BTLA+ CD4+ T cells from LPMCs, while no difference was observed in BTLA+ CD4+ T cells from PBMCs. These results indicate that BTLA associates with Foxp3 expression in CD4+ T cells and it is likely that BTLA signal may enhance the suppressive function of Treg in DSS-induced colitis. Indeed, lower number of IFN-γ+ effector T cells was observed in BTLA4+ T cells compared with that of BTLA-counterparts.
Extensive studies have suggested a critical role for IL-17 in chronic inflammatory disease. It induces neutrophil migration and expansion in inflammatory sites. It also promotes dendritic cell maturation and secretion of inflammatory mediators such as TNF-α, IL-1β, IL-6, IL-8, GM-CSF, iNOS, PGE2, and metalloproteases [36,37]. BTLA has been reported to be able to negatively regulate IL-17 and TNF-α production in CD27(-) γδ T cells [38]. Interestingly, our current data showed that BTLA-expressing CD3+ CD4+ T cells and CD3+ CD8+ T cells produce much more IL-17 than that in BTLA negative CD3+ CD4+ T cells and CD3+ CD8+ T cells after DSS-treatment. Furthermore, BTLA-expressing CD4+ T cells produced much more IL-17 in LPMCs from DSS-treated mice than that from control mice, indicating that DSS-induced BTLA expression failed to attenuate IL-17 production in DSS-induced colitis.
Mesalazine, also known as 5-aminosalicylic acid (5-ASA), is an anti-inflammatory drug commonly used to treat inflammatory bowel disease, especially to UC by acting locally in the gut. As a derivative of salicylic acid, mesalazine is also thought to be an antioxidant that traps free radicals. Previous study has shown that TNBS induced high-level expression of Foxp3 in mucosa in a rat ileitis model, while 5-ASA administration could down-regulate IL-17 expression without affecting Foxp3 expression [39]. Interestingly, our study demonstrated that Mesalazine treatment down-regulates not only IL-17 but also Foxp3 in BTLA-expressing CD4+ T cells. This discrepancy is probably because we detected different cell population. We detected the expression of Foxp3 in BTLA+ T cells, while Requena et al detected Foxp3 in whole ileum samples in their reports [39]. Further studies are required to investigate the effect of mesalazine on Foxp3 expression and Treg suppressive function.
In summary, we demonstrated a previously unappreciated role for BTLA in T cells in UC. BTLA associates with increased Foxp3 expression and might be important for the suppressive function of Tregs in DSS-induced colitis. However, BTLA is not sufficient to attenuate IL-17 production in DSS-induced colitis. Mesalazine treatment decrease Foxp3 and IL-17 expression simultaneously. Our study indicates that BTLA may be involved in the control of inflammatory response in UC.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (81273237, 30972779), the Department of Science and Technology of Guangdong Province (2011B061300098), the Key Project of Science and Technology Innovation of Education Department of Guangdong Province (2012KJCX0059), the Science and Technology Project of Dongguan (2012105102016, 20131051010006), the Science and Technology Project of Zhanjiang (2013C03012) and the Science and Technology Innovation Fund of Guangdong Medical College (STIF201110, B2012078).
Disclosure of conflict of interest
None.
Supporting Information
References
- 1.Kawada M, Arihiro A, Mizoguchi E. Insights from advances in research of chemically induced experimental models of human inflammatory bowel disease. World J Gastroenterol. 2007;13:5581–5593. doi: 10.3748/wjg.v13.i42.5581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Vatn MH. Natural history and complications of IBD. Curr Gastroenterol Rep. 2009;11:481–487. doi: 10.1007/s11894-009-0073-8. [DOI] [PubMed] [Google Scholar]
- 3.Hundorfean G, Neurath MF, Mudter J. Functional relevance of T helper 17 (Th17) cells and the IL-17 cytokine family in inflammatory bowel disease. Inflamm Bowel Dis. 2012;18:180–186. doi: 10.1002/ibd.21677. [DOI] [PubMed] [Google Scholar]
- 4.Neurath MF, Finotto S, Glimcher LH. The role of Th1/Th2 polarization in mucosal immunity. Nat Med. 2002;8:567–573. doi: 10.1038/nm0602-567. [DOI] [PubMed] [Google Scholar]
- 5.Podolsky DK. Inflammatory bowel disease. N Engl J Med. 2002;347:417–429. doi: 10.1056/NEJMra020831. [DOI] [PubMed] [Google Scholar]
- 6.Park H, Li Z, Yang XO, Chang SH, Nurieva R, Wang YH, Wang Y, Hood L, Zhu Z, Tian Q, Dong C. A distinct lineage of CD4 T cells regulates tissue inflammation by producing interleukin 17. Nat Immunol. 2005;6:1133–1141. doi: 10.1038/ni1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Harrington LE, Hatton RD, Mangan PR, Turner H, Murphy TL, Murphy KM, Weaver CT. Interleukin 17-producing CD4+ effector T cells develop via a lineage distinct from the T helper type 1 and 2 lineages. Nat Immunol. 2005;6:1123–1132. doi: 10.1038/ni1254. [DOI] [PubMed] [Google Scholar]
- 8.Sanada Y, Mizushima T, Kai Y, Nishimura J, Hagiya H, Kurata H, Mizuno H, Uejima E, Ito T. Therapeutic effects of novel sphingosine-1-phosphate receptor agonist W-061 in murine DSS colitis. PLoS One. 2011;6:e23933. doi: 10.1371/journal.pone.0023933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fujino S, Andoh A, Bamba S, Ogawa A, Hata K, Araki Y, Bamba T, Fujiyama Y. Increased expression of interleukin 17 in inflammatory bowel disease. Gut. 2003;52:65–70. doi: 10.1136/gut.52.1.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sakaguchi S, Yamaguchi T, Nomura T, Ono M. Regulatory T cells and immune tolerance. Cell. 2008;133:775–77. doi: 10.1016/j.cell.2008.05.009. [DOI] [PubMed] [Google Scholar]
- 11.Shevach EM. Mechanisms of foxp3+ T regulatory cell-mediated suppression. Immunity. 2009;30:636–65. doi: 10.1016/j.immuni.2009.04.010. [DOI] [PubMed] [Google Scholar]
- 12.Bennett CL, Christie J, Ramsdell F, Brunkow ME, Ferguson PJ, Whitesell L, Kelly TE, Saulsbury FT, Chance PF, Ochs HD. The immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome (IPEX) is caused by mutations of FOXP3. Nat Genet. 2001;27:20–21. doi: 10.1038/83713. [DOI] [PubMed] [Google Scholar]
- 13.Izcue A, Hue S, Buonocore S, Arancibia-Cárcamo CV, Ahern PP, Iwakura Y, Maloy KJ, Powrie F. Interleukin-23 restrains regulatory T cell activity to drive T cell-dependent colitis. Immunity. 2008;28:559–570. doi: 10.1016/j.immuni.2008.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Watanabe N, Gavrieli M, Sedy JR, Yang J, Fallarino F, Loftin SK, Hurchla MA, Zimmerman N, Sim J, Zang X, Murphy TL, Russell JH, Allison JP, Murphy KM. BTLA is a lymphocyte inhibitory receptor with similarities to CTLA-4 and PD-1. Nat Immunol. 2003;4:670–679. doi: 10.1038/ni944. [DOI] [PubMed] [Google Scholar]
- 15.Sedy JR, Gavrieli M, Potter KG, Hurchla MA, Lindsley RC, Hildner K, Scheu S, Pfeffer K, Ware CF, Murphy TL, Murphy KM. B and T lymphocyte attenuator regulates T cell activation through interaction with herpesvirus entry mediator. Nat Immunol. 2005;6:90–98. doi: 10.1038/ni1144. [DOI] [PubMed] [Google Scholar]
- 16.Tao R, Wang L, Han R, Wang T, Ye Q, Honjo T, Murphy TL, Murphy KM, Hancock WW. Differential effects of B and T lymphocyte attenuator and programmed death-1 on acceptance of partially versus fully MHC-mismatched cardiac allografts. J Immunol. 2005;175:5774–582. doi: 10.4049/jimmunol.175.9.5774. [DOI] [PubMed] [Google Scholar]
- 17.Steinberg MW, Turovskaya O, Shaikh RB, Kim G, McCole DF, Pfeffer K, Murphy KM, Ware CF, Kronenberg M. A crucial role for HVEM and BTLA in preventing intestinal inflammation. J Exp Med. 2008;205:1463–1476. doi: 10.1084/jem.20071160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Oya Y, Watanabe N, Owada T, Oki M, Hirose K, Suto A, Kagami S, Nakajima H, Kishimoto T, Iwamoto I, Murphy TL, Murphy KM, Saito Y. Development of autoimmune hepatitis-like disease and production of autoantibodies to nuclear antigens in mice lacking B and T lymphocyte attenuator. Arthritis Rheum. 2008;58:2498–2510. doi: 10.1002/art.23674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Flynn R, Hutchinson T, Murphy KM, Ware CF, Croft M, Salek-Ardakani S. CD8 T cell memory to a viral pathogen requires trans cosignaling between HVEM and BTLA. PLoS One. 2013;8:e77991. doi: 10.1371/journal.pone.0077991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Steinberg MW, Huang Y, Wang-Zhu Y, Ware CF, Cheroutre H, Kronenberg M. BTLA interaction with HVEM expressed on CD8(+) T cells promotes survival and memory generation in response to a bacterial infection. PLoS One. 2013;8:e77992. doi: 10.1371/journal.pone.0077992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sakoda Y, Park JJ, Zhao Y, Kuramasu A, Geng D, Liu Y, Davila E, Tamada K. Dichotomous regulation of GVHD through bidirectional functions of the BTLA-HVEM pathway. Blood. 2011;117:2506–2514. doi: 10.1182/blood-2010-08-301325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gertner-Dardenne J, Fauriat C, Orlanducci F, Thibult ML, Pastor S, Fitzgibbon J, Bouabdallah R, Xerri L, Olive D. The co-receptor BTLA negatively regulates human Vγ9Vδ2 T-cell proliferation: a potential way of immune escape for lymphoma cells. Blood. 2013;122:922–931. doi: 10.1182/blood-2012-11-464685. [DOI] [PubMed] [Google Scholar]
- 23.Hurchla MA, Sedy JR, Murphy KM. Unexpected role of B and T lymphocyte attenuator in sustaining cell survival during chronic allostimulation. J Immunol. 2007;178:6073–6082. doi: 10.4049/jimmunol.178.10.6073. [DOI] [PubMed] [Google Scholar]
- 24.Okayasu I, Hatakeyama S, Yamada M, Ohkusa T, Inagaki Y, Nakaya R. A novel method in the induction of reliable experimental acute and chronic ulcerative colitis in mice. Gastroenterology. 1990;98:694–702. doi: 10.1016/0016-5085(90)90290-h. [DOI] [PubMed] [Google Scholar]
- 25.Perše M, Cerar A. Dextran sodium sulphate colitis mouse model: traps and tricks. J Biomed Biotechnol. 2012;2012:718617. doi: 10.1155/2012/718617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fuss IJ, Heller F, Boirivant M, Leon F, Yoshida M, Fichtner-Feigl S, Yang Z, Exley M, Kitani A, Blumberg RS, Mannon P, Strober W. Nonclassical CD1d-restricted NK T cells that produce IL-13 characterize an atypical Th2 response in ulcerative colitis. J Clin Invest. 2004;113:1490–1497. doi: 10.1172/JCI19836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Heller F, Florian P, Bojarski C, Richter J, Christ M, Hillenbrand B, Mankertz J, Gitter AH, Bürgel N, Fromm M, Zeitz M, Fuss I, Strober W, Schulzke JD. Interleukin-13 is the key effector Th2 cytokine in ulcerative colitis that affects epithelial tight junctions, apoptosis, and cell restitution. Gastroenterology. 2005;129:550–564. doi: 10.1016/j.gastro.2005.05.002. [DOI] [PubMed] [Google Scholar]
- 28.Alex P, Zachos NC, Nguyen T, Gonzales L, Chen TE, Conklin LS, Centola M, Li X. Distinct cytokine patterns identified from multiplex profiles of murine DSS and TNBS-induced colitis. Inflamm Bowel Dis. 2009;15:341–352. doi: 10.1002/ibd.20753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Brown JB, Cheresh P, Zhang Z, Ryu H, Managlia E, Barrett TA. P-selectin glycoprotein ligand-1 is needed for sequential recruitment of T-helper 1 (Th1) and local generation of Th17 T cells in dextran sodium sulfate (DSS) colitis. Inflamm Bowel Dis. 2012;18:323–32. doi: 10.1002/ibd.21779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Egger B, Bajaj-Elliott M, MacDonald TT, Inglin R, Eysselein VE, Büchler MW. Characterisation of acute murine dextran sodium sulphate colitis: cytokine profile and dose dependency. Digestion. 2000;62:240–24. doi: 10.1159/000007822. [DOI] [PubMed] [Google Scholar]
- 31.Ito R, Kita M, Shin-Ya M, Kishida T, Urano A, Takada R, Sakagami J, Imanishi J, Iwakura Y, Okanoue T, Yoshikawa T, Kataoka K, Mazda O. Involvement of IL-17A in the pathogenesis of DSS-induced colitis in mice. Biochem Biophys Res Commun. 2008;377:12–16. doi: 10.1016/j.bbrc.2008.09.019. [DOI] [PubMed] [Google Scholar]
- 32.Requena P, Daddaoua A, Martínez-Plata E, González M, Zarzuelo A, Suárez MD, Sánchez de Medina F, Martínez-Augustin O. Bovine glycomacropeptide ameliorates experimental rat ileitis by mechanisms involving downregulation of interleukin 17. Br J Pharmacol. 2008;154:825–832. doi: 10.1038/bjp.2008.138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhang L, Zhao Y. The regulation of Foxp3 expression in regulatory CD4(+)CD25(+)T cells: multiple pathways on the road. J Cell Physiol. 2007;211:590–597. doi: 10.1002/jcp.21001. [DOI] [PubMed] [Google Scholar]
- 34.Hanai H, Iida T, Ikeya K, Abe J, Maruyama Y, Shimura T, Sugimoto K, Watanabe F. A new paradigm in ulcerative colitis: regulatory T cells are key factor which induces/exacerbates UC through an immune imbalance. Mol Immunol. 2013;54:173–180. doi: 10.1016/j.molimm.2012.11.015. [DOI] [PubMed] [Google Scholar]
- 35.Xu H, Cao D, Guo G, Ruan Z, Wu Y, Chen Y. The intrahepatic expression and distribution of BTLA and its ligand HVEM in patients with HBV-related acute-on-chronic liver failure. Diagn Pathol. 2012;7:142. doi: 10.1186/1746-1596-7-142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ye P, Rodriguez FH, Kanaly S, Stocking KL, Schurr J, Schwarzenberger P, Oliver P, Huang W, Zhang P, Zhang J, Shellito JE, Bagby GJ, Nelson S, Charrier K, Peschon JJ, Kolls JK. Requirement of interleukin 17 receptor signaling for lung CXC chemokine and granulocyte colony-stimulating factor expression, neutrophil recruitment, and host defense. J Exp Med. 2001;194:519–527. doi: 10.1084/jem.194.4.519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yen D, Cheung J, Scheerens H, Poulet F, McClanahan T, McKenzie B, Kleinschek MA, Owyang A, Mattson J, Blumenschein W, Murphy E, Sathe M, Cua DJ, Kastelein RA, Rennick D. IL-23 is essential for T cell-mediated colitis and promotes inflammation via IL-17 and IL-6. J Clin Invest. 2006;116:1310–1316. doi: 10.1172/JCI21404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bekiaris V, Sedý JR, Macauley MG, Rhode-Kurnow A, Ware CF. The inhibitory receptor BTLA controls γδ T cell homeostasis and inflammatory responses. Immunity. 2013;39:1082–1094. doi: 10.1016/j.immuni.2013.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Requena P, Daddaoua A, Martínez-Plata E, González M, Zarzuelo A, Suárez MD, Sánchez de Medina F, Martínez-Augustin O. Bovine glycomacropeptide ameliorates experimental rat ileitis by mechanisms involving downregulation of interleukin 17. Br J Pharmacol. 2008;154:825–832. doi: 10.1038/bjp.2008.138. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.