

Abstract
In cultured human osteosarcoma (OS) cells, we recently demonstrated that insulin-like growth factors (IGF-1)-induced MG-63 and 143B human OS cells proliferation were consistent with increasing ClC-3 expression, and ClC-3 was up-regulated in cells with high metastatic potency. Blockade of ClC-3 greatly suppressed the phosphorylation activation of Akt/GSK3β. We also found that blockade of ClC-3 effectively down-regulated the expression of cyclin D1 and cyclin E, and caused activation of p27KIP and p21CIP. The synthesized effects on these proteins which play a major role in cell cycle regulation bring about G0/G1 cell cycle arrest in MG-63 cells, and finally abrogate the cell proliferation. Besides, ClC-3 deletion attenuates OS cell migration via down-regulation the expression of MMP-2 and MMP-9. Such information suggests that ClC-3 might be a potential target for anti-OS.
Keywords: ClC-3 chloride channel, osteosarcoma, proliferation and migration, AKT/GSK3β signaling pathway
Introduction
Osteosarcoma (OS) is one of the world widespread malignant diseases, which constitutes a serious threat to human life and health. As there is no obvious symptom at the early stage, the metastasis is still the primary reason for death in patients with OS, Therefore, further study in tumorigenesis, metastasis and invasion molecular mechanism of OS and trying to identify novel therapeutic targets and elucidating mechanisms are important for treatment of OS patients. In recent years, a series of molecules have been identified to be involved in tumorigenesis and progression of OS, such as Stro-1, CXCR4, Smoothened and etc. [1-3].
As we know, excessive proliferation of cells is closely related to tumorigenesis. The vast majority of cell proliferation accompany with changes in cell volume and the activity of VRCC (volume regulated chloride channel, VRCC). Lots of studies in many kinds of cells support the idea that ClC-3 is the molecular basis of VRCC which is involved in the activation or regulation of Icl,vol. Besides, gene-targeting studies attest that ClC-3 play important roles of in cell proliferation, apoptosis, and migration [4]. Recent studies have shown that ClC-3 is highly correlated to many kinds of malignant tumor cells, including human glioma cells, human endometrial cancer cells and nasopharyngeal carcinoma cells [5-7]. And our previous study showed that IGF-1-induced MG-63 cells proliferation was consistent with increasing endogenous ClC-3 expression. We hypothesized that ClC-3 is also involved in OS, and it may provide novel insight into identifying therapeutic targets for OS.
Therefore, we investigated the function of ClC-3 chloride channel in the proliferation and migration of OS cells, and further clarify the signal pathway that maybe involved in OS.
Materials and methods
Reagents
Antibody targeting ClC-3 was purchased from Alamone. Antibody targeting Cyclin D1, Cyclin E, p27, P21 and GAPDH were purchased from Santa Cruz Biotech (Santa Cruz, CA). Antibodies targeting MMP-2 and MMP-9, Akt and phospho-Akt, GSK-3β and phospho-GSK-3β were purchased from Cell Signaling Technology (Beverly, MA). Insulin-like growth factor 1 (IGF-1) was purchased from Sigma (Sigma-Aldrich, USA). Cell Counting Assay Kit-8 (CCK-8) was purchased from Dojindo Molecular Technologies (Japan). Fetal bovine serum (FBS) and DMEM medium were purchased from Gibco (Gibco, USA).
Cell culture
Osteosarcoma cell line MG-63 and 143B were purchased from the American Type Culture Collection (ATCC, Rockville, MD). Cells were cultured in DMEM medium contenting 10% fetal bovine serum (FBS), 100 U/ml penicillin and streptomycin. Cells were subcultured in a humidified atmosphere containing 5% CO2 at 37°C.
Cell counting kit-8 (CCK-8) assay for cell viability
Cell viability was determined using cell counting assay kit-8 incorporation as the manufacturer’s protocol. 100 μL of MG-63 and 143B cells were plated into 96-well plates with a density of 1-1.5 × 104 cells/ml for 24 h followed with a synthronization before adding stimulates. Then each well was incubated with 10 μL CCK-8 solution for another 2 h, and then measured the absorbance at 450 nm using a microplate reader (Bio-Tek, Winooski, VT, USA).
Transfection of MG-63 and 143B with siRNA
Oligonucleotide sequences of siRNA against human ClC-3 gene were synthesized by Invitrogen Corporation. And the ClC-3 siRNA corresponded to the following sequence: 5’-CTGCTTGACCTATGATTAA-3’. The ClC-3 siRNA was transfected to cells according to the manufacturer. A negative stealth siRNA sequence was used as the negative control. ClC-3 protein expression was detected by Western blot analysis.
Cell cycle analysis
Cell cycle status was assessed by flow cytometry. Briefly, cells were plated into 100 cm2 plate with 10% FBS for 24 hours followed by a synthronization. After transfected with siRNA targeting ClC-3 for 48 h, cells were collected and fixed with 75% ethanol for 24 h. Then the cells were stained with PI buffer (PBS containing 50 μg/mL of propidium iodide, 10 μg/mL RNase A, 0.1% sodium citrate and 0.1 TritonX-100). Cell cycle distribution was detected using flow cytometry system.
Transwell assay
We used transwell assay to assessing the invasive and migratory potential of cells. Briefly, cells cultured in DMEM medium with 0.1% BSA for 12 h were washed with PBS twice, and digested with 0.2% trypsin. Then resuspend the cells in DMEM medium containing 0.1% BSA. 1 × 105 cells and the same amount of control cells were added to the upper chamber; and 500 μl DMEM medium contained with 0.1% BSA was added to the lower chamber. Then the cells were allowed to migrate for 10 h in a 37°C cell incubator. After incubation, invaded cells in the lower chamber were fixed with 4% paraformaldehyde, stained with 0.5% crystal violet solution, and then photographed and counted under microscope.
Western blot analysis
After washed with ice-cold PBS for three times, cells were lysed in lysis buffer (50 mM Tris-HCl, 150 mM NaCl, 0.02% NaN3, 1% nonidet P-40, sodium deoxycholate, 0.1% sodium dodecyl sulfate, and 1% protease inhibitor cocktail) for approximately 30 minutes on ice. Then gently scraped the cells with a rubber policeman and centrifugated at 12000 × g for 12 min at 4°C. Protein concentration was measured by BCA method (Bio-Rad Laboratories, Hercules, CA, USA). The bovin serum albumin (BSA) was used as the standard.
60 μg protein for per sample was separated by 8%~12% SDS-PAGE and transferred onto PVDF membranes. Then membranes were blocked for 1h at room temperature in 5% non-fat dry milk (150 mM NaCl, 10 mM Tris pH 7.5, 0.1% Tween 20), incubated with primary antibodies initially overnight at 4°C and then with the appropriate secondary peroxidase-conjugated antibodies for another 1.5 h at room temperature. Blot were developed using a chemiluminescence substrates (Beyotime, Jiangsu, China). Finally, membranes were visualized by exposure to Kodak X-ray film. Scan the result and analyze using Image J software. Primary antibodies used include anti-ClC-3, anti-cyclin D1, anti-cyclin E, anti-P27, anti-P21, anti-MMP-2 and anti-MMP-9, anti-Akt and anti-phospho-Akt, anti-GSK-3β and anti-phospho-GSK-3β.
Statistical analysis
All of the data was represented as the mean ± SD. Statistical significance between 2 groups was assessed using Student’s t test and among 3 groups using ANOVA -test (SPSS 13.0). The P values less than 0.05 was considered statistically significant.
Results
ClC-3 expression parallels with rate of Osteosarcoma cells proliferation
We first examined whether ClC-3 expression correlates with the rate of cell proliferation. MG-63 and 143B cells were stimulated with various concentrations (30 ng/ml, 100 ng/ml, 300 ng/ml) of IGF-1 for 48 h, then cell viability was assessed by CCK-8 kit and the endogenous ClC-3 protein expression was examined by western blot (WB). As shown in Figure 1A-C, after treatment with IGF-1 for 48 h, the cell viability was significantly faster and parallel to an effective increase in the expression of endogenous ClC-3 protein in MG-63 cells. And in 143B cells, those results were reproducible, as shown in Figure 1D-F. And, it may demonstrate that ClC-3 was a necessity protein for Osteosarcoma cells proliferation.
Figure 1.
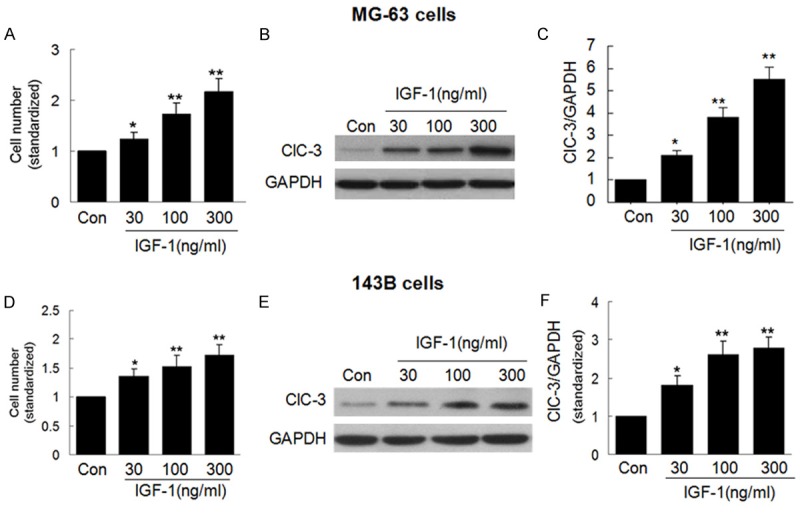
ClC-3 expression parallels with OS cells proliferation. A. Effect of 30-300 ng/ml IGF-1 on the cell viability in MG-63 cells. B and C. Effect of 30-300 ng/ml IGF-1 on the expression of ClC-3 in MG-63 cells. (Data represent mean ± SD (n = 4), Con: control, *P < 0.05 vs. con; **P < 0.01 vs. con. ANOVA test was used). D. Effect of 30-300 ng/ml IGF-1 on the cell viability in 143B cells. E and F. Effect of 30-300 ng/ml IGF-1 on the expression of ClC-3 in 143B cells. (Data represent mean ± SD (n = 4), Con: control, *P < 0.05 vs. con; **P < 0.01 vs. con. ANOVA test was used).
ClC-3 siRNA arrests cell cycle in G0/G1 phase
To further investigate the potential interaction between ClC-3 and proliferation, we determined that ClC-3 deletion on the effects of cells cycle in MG-63 cells. As shown in Figure 2A and 2B, after transfection with 20-80 nM ClC-3 siRNA for 48 h, ClC-3 protein expression was reduced in a concentration-dependent manner (**P < 0.01 vs. con. N = 4). Whereas ClC-3 expression stayed still after transfection with lipofectamine (Lipo) or negative siRNA control (NS) (n = 4).
Figure 2.
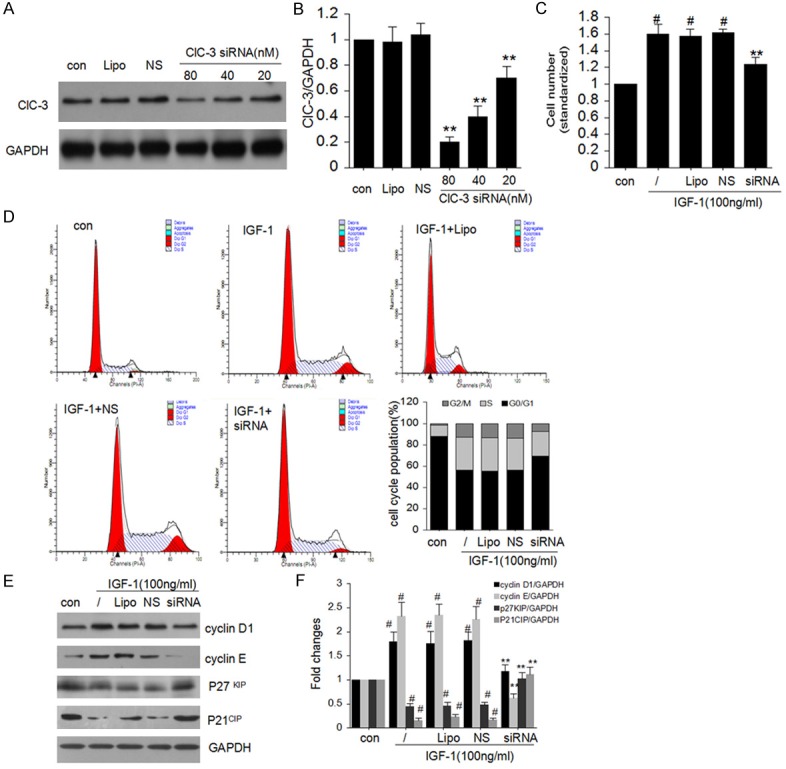
Effect of ClC-3 siRNA on cell cycle in MG-63 cells. A and B. Effect of 20-80 nM ClC-3 siRNA on the expression of endogenous ClC-3 protein in MG-63 cells detected by western blot analysis. (Data represent mean ± SD (n = 4), con: control, Lipo: Liposomal, NS: Negative siRNA, **P < 0.01 vs. con. ANOVA test was used). C. Knock-down of ClC-3 reduces the IGF-1-induced cell viability in MG-63 cells. D. Knock-down of ClC-3 arrests cell cycle in G0/G1 phase in MG-63 cells. E and F. ClC-3 siRNA restrained cyclin D1, cyclin E and enhanced p21, p27 protein expression induced by 100 ng/ml IGF-1 respectively in MG-63 cells. (Data represent mean ± SD (n = 4), con: control, Lipo: Liposomal, NS: Negative siRNA, #P < 0.05 vs. con; **P < 0.01 vs. IGF-1 only. ANOVA test was used).
The effect of ClC-3 deletion on cell proliferation was detected using CCK-8 assay (Figure 2C). Transfection of ClC-3 siRNA could inhibit IGF-1-induced cell growth (**P < 0.01 vs. IGF-1 only. N = 4). Whereas transfection of negative siRNA control had no distinguished effect on IGF-1-induced cell viability.
To figure out the regulating effect of ClC-3 on cancer cell proliferation, we investigated whether ClC-3 deletion affects the cell cycle distribution of the Osteosarcoma cell line MG-63. And found that knockdown of ClC-3 increase cell population in G0/G1 phase, in contrast to decrease cell population in S phase and G2/M phase (Figure 2D). And these data demonstrate that ClC-3 siRNA knockdown could cause MG-63 cell cycle G1/S arrest.
As we know, cell cycle transitions are regulated by the balance between cyclins and CDKIs. To pinpoint the molecular mechanisms how ClC-3 deletion induced G1/S arrest, we analyzed cyclin D1, cyclin E, p27KIP and p21CIP expression levels in the model. As shown in (Figure 1E and 1F), ClC-3 siRNA knockdown down-regulated the expression of cyclin D1 and cyclin E, but p27KIP and p21CIP expression was elevated in MG-63 cell.
ClC-3 is indispensable for cell migration
Cancer cells migration is the key reason for tumor invasion and metastasis, thus we explored the role of ClC-3 in the Osteosarcoma cell migration. We first analyzed the endogenous ClC-3 protein expression in MG-63 and 143B cell lines based on there difference in migration capacity. Western blotting analyses showed a higher expression level in MG-63 cell lines (Figure 3A and 3B). After transfection of ClC-3 siRNA, the expression of ClC-3 was both decreased in these two cell lines. The effect of ClC-3 knockdown on migration of MG-63 and 143B cells was examined next with transwell assay. The difference in migration capacity of MG-63 and 143B cells was also observed (Figure 3C and 3D). The average percentage of migrating cells was different in the two cell lines; and ClC-3 knockdown reduced the percentage of migrating cells in both cell lines.
Figure 3.

ClC-3 knockdown attenuate cell migration in OS cells. A and B. The endogenous expression of ClC-3 in cells with different metastatic potency (MG-63 and 143B) and effect of ClC-3 siRNA on the expression of ClC-3 protein in both cells detected by western blot analysis. (Data represent mean ± SD (n = 4), con: control, NS: Negative siRNA, *P < 0.05 vs. MG-63 con; #P < 0.05 vs. MG-63 con; *#P < 0.05 vs. 143B con. ANOVA test was used). C and D. ClC-3 knockdown attenuated the migration ability of MG-63 and 143B cells detected by transwell assay. (Data represent mean ± SD (n = 4), con: control, NS: Negative siRNA, *P < 0.05 vs. MG-63 con; #P < 0.05 vs. MG-63 con; *#P < 0.05 vs. 143B con. ANOVA test was used). E and F. ClC-3 knockdown suppressed expression of MMP-2 and MMP-9 in MG-63 and 143B cells. (Data represent mean ± SD (n = 4), con: control, NS: Negative siRNA, *P < 0.05 vs. MG-63 con; #P < 0.05 vs. MG-63 con; *#P < 0.05 vs. 143B con. ANOVA test was used).
To further explore the suppression mechanism of ClC-3 knockdown on cell migration, we evaluated the expression of matrix metalloproteinase-2 (MMP-2) and matrix metalloproteinase-9 (MMP-9) in MG-63 and 143B cells. And our data showed that ClC-3 knockdown restrained cell migration with suppression of MMP-2 and MMP-9 protein expression in MG-63 and 143B cells (Figure 3E and 3F).
ClC-3 siRNA attenuates Akt/GSK-3β signaling pathway
The phosphorylation of Akt/GSK-3β is well-known to regulate the evolution of cell cycle G1/S transition in many types cells including Osteosarcoma cells [23-25]. Therefore, we investigated the phosphorylation status of Akt after silencing ClC-3 in MG-63 cells. As shown in Figure 4A and 4B, ClC-3 deletion effectively attenuated the phosphorylations of GSK-3β and Akt. Yet, there is no distinguished change on the expression of Akt and GSK-3β protein after ClC-3 knockdown. And the special inhibitor of Akt LY294002 could diminish the phosphorylations of GSK-3β and the expression of ClC-3 (Figure 4C and 4D).
Figure 4.
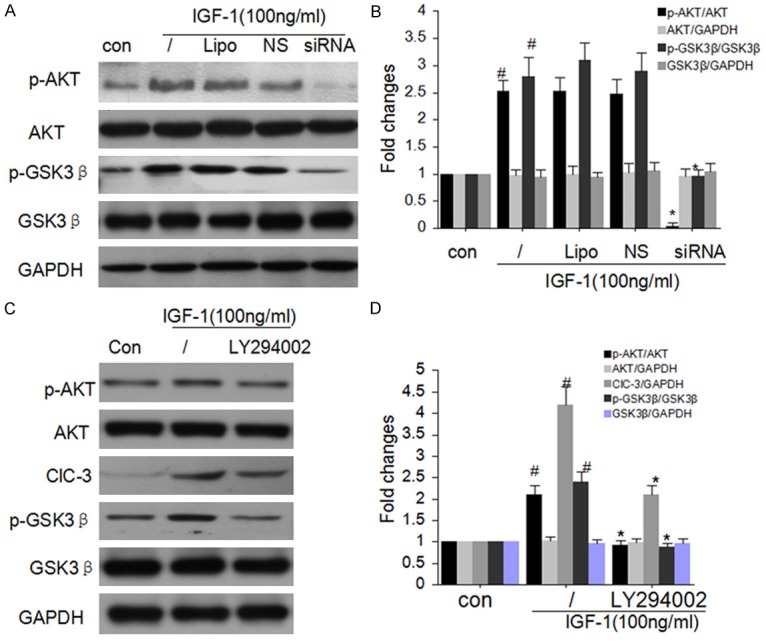
ClC-3 knockdown attenuates Akt signaling. A and B. ClC-3 knockdown significantly attenuated the phosphorylations of GSK-3β and Akt detected by western blot analysis. (Data represent mean ± SD (n = 4), con: control, Lipo: Liposomal, NS: Negative siRNA, #P < 0.05 vs. con; *P < 0.05 vs. IGF-1 only. ANOVA test was used). C and D. Akt inhibitor LY294002 diminished the expression of ClC-3 and the phosphorylations of GSK-3β. (Data represent mean ± SD (n = 4), con: control, Lipo: Liposomal, NS: Negative siRNA, #P < 0.05 vs. con; *P < 0.05 vs. IGF-1 only. ANOVA test was used).
Discussion
In our current study, we investigated the effects of ClC-3 chloride channel on regulation of osteosarcoma cell viability, migration, and invasion capacity, and the suppression mechanism. Our data showed that there is endogenous ClC-3 expression in human osteosarcoma cell lines MG-63 and 143B. IGF-1-induced cell proliferation parallels with significant increase in the expression of endogenous ClC-3 and ClC-3 was up-regulated in the cells with high metastatic potency. While knockdown of ClC-3 with transfection of ClC-3 siRNA inhibit MG-63 cell viability and the migration of OS cell line was also restrained. All these results demonstrated that ClC-3 is involved in the proliferation and migration of osteosarcoma cell.
To further explore how ClC-3 participates in the osteosarcoma cell proliferation and migration, we observed the effect of ClC-3 knockout on the cell cycle of MG-63 and 143B cells. We found that knockdown of ClC-3 arrested cell cycle in G0/G1 phase both in MG-63 and 143B cells. Although the study have reported that cell proliferation (especially in the G1/S restriction point) have significantly increased cell capacity, accompanying with VRCC activation caused RVD process, the molecular mechanisms is still not well understand [8,9]. The convert of the cell cycle is always regulated by the cell cycle regulatory proteins [10,11]. In our study, we analyzed the expression of these regulatory proteins, and our results showed that knockdown of ClC-3 decreased expression of cyclin D1 and cyclin E, while increased the expression of P27KIP and P21CIP, the two cyclin-dependent kinase inhibitors which arrested cell cycle in G0/G1 phase by reversibly inhibiting several cyclin-CDK complexes [11-13]. All these results demonstrated that ClC-3 knockdown could inhibit cell proliferation in osteosarcoma cell, and it is resulted from the block of G1/S transition in cell cycle and the restriction of G1/S transition is correlated to the downregulation of cyclins and the upregulation of CDKIs.
For patients with solid malignant tumors, the biggest threat to the therapy is tumor cell metastasis [13]. However, the specific molecular changes that promote the migration of cancer cells from the original site to the other part of our body are still unclear. Our results showed that the expression of endogenous ClC-3 protein was different in cells with different metastatic potency, and ClC-3 knockdown attenuated the migration ability of MG-63 and 143B cells. It hints that there is a potential link between ClC-3 protein and tumor metastasis in osteosarcoma. Increasing evidence suggests that matrix metalloproteinases (MMPs) promote tumor metastasis in many kinds of malignant tumors, such as human lung cancer [14], human osteosarcoma [15] and prostate cancer [16]. In our study, we also analyzed the relationship between ClC-3 chloride channel and MMPs. Our results demonstrated that ClC-3 knockdown down-regulated the expression of MMP-2 and MMP-9 in MG-63 and 143B cells. Collectively, our data indicated that ClC-3 knockdown attenuated osteosarcoma cell migration via down-regulation the expression of MMP-2 and MMP-9.
It is well known that deregulation of the PI3K/Akt/GSK3β pathway plays a key role in the development of human cancers [17-19]. And there is evidence showed that phosphorylation of Akt/GSK3β may increase the expression of cyclin and promote cell cycle G1/S transition in vascular smooth muscle cells [11]. Mutation of Akt at Ser473 and Thr308 could abrogate the stimulatory effect of serum on DNA synthesis and cell cycle G1/S transition [20,21]. And this effect has been documented to be interrelated to the cell cycle regulators such as cyclin and CDKIs in many cell types [22-24]. Recent works in breast cancer cells also suggested that attenuated the activation of Akt could mediated cell cycle procession and cellular apoptosis [25]. In this study, we found that the phosphorylations of Akt and GSK3β were attenuated by ClC-3 siRNA treatment in MG-63 cells. Interestingly, the PI3K/Akt inhibitor LY294002 could inhibit the activity of ClC-3 chloride channel and the phosphorylations of GSK3β. And these results demonstrated that the PI3K/Akt/GSK3β pathway is related to ClC-3 mediated proliferation and migration in osteosarcoma cell.
In summary, ClC-3 knockdown effectively suppressed the phosphorylation of Akt/GSK3β pathway, this inactivation lead to downregulation of the expression of cyclin D1 and cyclin E, caused activation of p27KIP and p21CIP. The combined effects of change on these cell cycle regulatory proteins lead to cell cycle arrested in G0/G1 phase, and finally abrogate the cell proliferation in MG-63 cells. Besides, ClC-3 knockdown attenuates osteosarcoma cell migration via down-regulation the expression of MMP-2 and MMP-9. Such information suggests that ClC-3 might be a potential target for anti-OS.
Disclosure of conflict of interest
None.
References
- 1.Adhikari AS, Agarwal N, Wood BM, Porretta C, Ruiz B, Pochampally RR, Iwakuma T. CD117 and Stro-1 identify osteosarcoma tumor-initiating cells associated with metastasis and drug resistance. Cancer Res. 2010;70:4602–4612. doi: 10.1158/0008-5472.CAN-09-3463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Brennecke P, Arlt MJ, Campanile C, Husmann K, Gvozdenovic A, Apuzzo T, Thelen M, Born W, Fuchs B. CXCR4 antibody treatment suppresses metastatic spread to the lung of intratibial human osteosarcoma xenografts in mice. Clin Exp Metastasis. 2014;31:339–349. doi: 10.1007/s10585-013-9632-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hirotsu M, Setoguchi T, Sasaki H, Matsunoshita Y, Gao H, Nagao H, Kunigou O, Komiya S. Smoothened as a new therapeutic target for human osteosarcoma. Mol Cancer. 2010;9:5. doi: 10.1186/1476-4598-9-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Guan YY, Wang GL, Zhou JG. The ClC-3 Cl-channel in cell volume regulation, proliferation and apoptosis in vascular smooth muscle cells. Trends Pharmacol Sci. 2006;27:290–296. doi: 10.1016/j.tips.2006.04.008. [DOI] [PubMed] [Google Scholar]
- 5.Li M, Wu DB, Wang J. Effects of volume-activated chloride channels on the invasion and migration of human endometrial cancer cells. Eur J Gynaecol Oncol. 2013;34:60–4. [PubMed] [Google Scholar]
- 6.Su J, Xu Y, Zhou L, Yu HM, Kang JS, Liu N, Quan CS, Sun LK. Suppression of chloride channel 3 expression facilitates sensitivity of human glioma U251 cells to cisplatin through concomitant inhibition of Akt and autophagy. Anat Rec (Hoboken) 2013;296:595–603. doi: 10.1002/ar.22665. [DOI] [PubMed] [Google Scholar]
- 7.Wang L, Ma W, Zhu L, Ye D, Li Y, Liu S, Li H, Zuo W, Li B, Ye W, Chen L. ClC-3 is a candidate of the channel proteins mediating acid-activated chloride currents in nasopharyngeal carcinoma cells. Am J Physiol Cell Physiol. 2012;303:C14–23. doi: 10.1152/ajpcell.00145.2011. [DOI] [PubMed] [Google Scholar]
- 8.Shin HS, Lee HJ, Nishida M, Lee MS, Tamura R, Yamashita S, Matsuzawa Y, Lee IK, Koh GY. Betacellulin and amphiregulin induce upregulation of cyclin D1 and DNA synthesis activity through differential signaling pathways in vascular smooth muscle cells. Circ Res. 2003;93:302–310. doi: 10.1161/01.RES.0000086803.64109.9E. [DOI] [PubMed] [Google Scholar]
- 9.Hassan GS, Williams TM, Frank PG, Lisanti MP. Caveolin-1-deficient aortic smooth muscle cells show cell autonomous abnormalities in proliferation, migration, and endothelin-based signal transduction. Am J Physiol Heart Circ Physiol. 2006;290:H2393–2401. doi: 10.1152/ajpheart.01161.2005. [DOI] [PubMed] [Google Scholar]
- 10.Renaudo A, L’Hoste S, Guizouarn H, Borgese F, Soriani O. Cancer cell cycle modulated by a functional coupling between sigma-1 receptors and Cl-channels. J Biol Chem. 2007;282:2259–2267. doi: 10.1074/jbc.M607915200. [DOI] [PubMed] [Google Scholar]
- 11.Tang YB, Liu YJ, Zhou JG, Wang GL, Qiu QY, Guan YY. Silence of ClC-3 chloride channel inhibits cell proliferation and the cell cycle via G/S phase arrest in rat basilar arterial smooth muscle cells. Cell Prolif. 2008;41:775–785. doi: 10.1111/j.1365-2184.2008.00551.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jain AK, Raina K, Agarwal R. Deletion of p21/Cdkn1a confers protective effect against prostate tumorigenesis in transgenic adenocarcinoma of the mouse prostate model. Cell Cycle. 2013;12:1598–604. doi: 10.4161/cc.24741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Han J, Rong LF, Shi CB, Dong XG, Wang J, Wang BL, Wen H, He ZY. Screening of lymph nodes metastasis associated lncRNAs in colorectal cancer patients. World J Gastroenterol. 2014;20:8139–50. doi: 10.3748/wjg.v20.i25.8139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chen L, Zhou Q, Xu B, Liu J, Shi L, Zhu D, Wu C, Jiang J. MT2-MMP expression associates with tumor progression and angiogenesis in human lung cancer. Int J Clin Exp Pathol. 2014;7:3469–77. [PMC free article] [PubMed] [Google Scholar]
- 15.Tsai HC, Su HL, Huang CY, Fong YC, Hsu CJ, Tang CH. CTGF increases matrix metallo proteinases expression and subsequently promotes tumor metastasis in human osteosarcoma through down-regulating miR-519d. Oncotarget. 2014;5:3800–12. doi: 10.18632/oncotarget.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gong Y, Chippada-Venkata UD, Oh WK. Roles of matrix metalloproteinases and their natural inhibitors in prostate cancer progression. Cancers (Basel) 2014;6:1298–327. doi: 10.3390/cancers6031298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sangawa A, Shintani M, Yamao N, Kamoshida S. Phosphorylation status of Akt and caspase-9 in gastric and colorectal carcinomas. Int J Clin Exp Pathol. 2014;7:3312–7. [PMC free article] [PubMed] [Google Scholar]
- 18.Vignarajan S, Xie C, Yao M, Sun Y, Simanainen U, Sved P, Liu T, Dong Q. Loss of PTEN stabilizes the lipid modifying enzyme cytosolic phospholipase A2α via AKT in prostate cancer cells. Oncotarget. 2014;5:6289–99. doi: 10.18632/oncotarget.2198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wen X, Zhu J, Dong L, Chen Y. The role of c2orf68 and PI3K/Akt/mTOR pathway in human colorectal cancer. Med Oncol. 2014;31:92. doi: 10.1007/s12032-014-0092-7. [DOI] [PubMed] [Google Scholar]
- 20.Gonzalez ME, DuPrie ML, Krueger H, Merajver SD, Ventura AC, Toy KA, Kleer CG. Histone methyltransferase EZH2 induces Akt-dependent genomic instability and BRCA1 inhibition in breast cancer. Cancer Res. 2011;71:2360–70. doi: 10.1158/0008-5472.CAN-10-1933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Héron-Milhavet L, Franckhauser C, Rana V, Berthenet C, Fisher D, Hemmings BA, Fernandez A, Lamb NJ. Only Akt1 Is Required for Proliferation, while Akt2 Promotes Cell Cycle Exit through p21 Binding. Mol Cell Biol. 2006;26:8267–8280. doi: 10.1128/MCB.00201-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Héron-Milhavet L, Franckhauser C, Rana V, Berthenet C, Fisher D, Hemmings BA, Fernandez A, Lamb NJ. Activation of phosphatidylinositol-3’-kinase/AKT signaling is essential in hepatoblastoma survival. Clin Cancer Res. 2009;15:4538–45. doi: 10.1158/1078-0432.CCR-08-2878. [DOI] [PubMed] [Google Scholar]
- 23.Palanivel K, Kanimozhi V amd Kadalmani B. Verrucarin A alters cell-cycle regulatory proteins and induces apoptosis through reactive oxygen species-dependent p38MAPK activation in the human breast cancer cell line MCF-7. Tumour Biol. 2014;35:10159–67. doi: 10.1007/s13277-014-2286-1. [DOI] [PubMed] [Google Scholar]
- 24.Loberg RD, Northcott CA, Watts SW, Brosius FC. PI3-kinase-induced hyperreactivity in DOCA-salt hypertension is independent of GSK-3 activity. Hypertension. 2003;41:898–902. doi: 10.1161/01.HYP.0000061762.58873.2F. [DOI] [PubMed] [Google Scholar]
- 25.Huang EW, Xue SJ, Zhang Z, Zhou JG, Guan YY, Tang YB. Vinpocetine inhibits breast cancer cells growth in vitro and in vivo. Apoptosis. 2012;17:1120–30. doi: 10.1007/s10495-012-0743-0. [DOI] [PubMed] [Google Scholar]