

Abstract
The small GTPase RAP1 is critical for platelet activation and thrombus formation. RAP1 activity in platelets is controlled by the GEF CalDAG-GEFI and an unknown regulator that operates downstream of the adenosine diphosphate (ADP) receptor, P2Y12, a target of antithrombotic therapy. Here, we provide evidence that the GAP, RASA3, inhibits platelet activation and provides a link between P2Y12 and activation of the RAP1 signaling pathway. In mice, reduced expression of RASA3 led to premature platelet activation and markedly reduced the life span of circulating platelets. The increased platelet turnover and the resulting thrombocytopenia were reversed by concomitant deletion of the gene encoding CalDAG-GEFI. Rasa3 mutant platelets were hyperresponsive to agonist stimulation, both in vitro and in vivo. Moreover, activation of Rasa3 mutant platelets occurred independently of ADP feedback signaling and was insensitive to inhibitors of P2Y12 or PI3 kinase. Together, our results indicate that RASA3 ensures that circulating platelets remain quiescent by restraining CalDAG-GEFI/RAP1 signaling and suggest that P2Y12 signaling is required to inhibit RASA3 and enable sustained RAP1-dependent platelet activation and thrombus formation at sites of vascular injury. These findings provide insight into the antithrombotic effect of P2Y12 inhibitors and may lead to improved diagnosis and treatment of platelet-related disorders.
Introduction
Mammalian platelets are small anucleated blood cells specialized to continuously monitor and preserve the integrity of the cardiovascular system (hemostasis) (1–3). Once released from megakaryocytes, they circulate for 10 days in human blood and 5 days in mouse blood. If they are not consumed in the hemostatic process, senescent platelets are destroyed by the reticuloendothelial system in the spleen and the liver (4). Thrombus formation at sites of vascular injury depends on a high sensitivity of platelets toward agonists and the ability to shift from an antiadhesive to a proadhesive state. Aberrant platelet activation, however, can lead to premature platelet clearance or the formation of intravascular occlusive thrombi (thrombosis), as seen in myocardial infarction (heart attack) and ischemic stroke (1). Thus, platelet activation needs to be tightly regulated to facilitate vascular hemostasis and to prevent thrombocytopenia and thrombosis. Inhibitors of the purinergic receptor, P2Y12, are used widely to prevent thrombotic complications in patients with cardiovascular disease. Early studies demonstrated that P2Y12 mediates the amplifying effects of adenosine diphosphate (ADP) on platelet activation by various agonists (5, 6). Engagement of P2Y12 has been linked to several downstream signaling events, including inhibition of adenylate cyclase (7, 8) and activation of phosphoinositide 3-kinase (PI3K) (9), the serine/threonine PKB/AKT (10), and the small GTPase RAS-related protein 1 (RAP1) (11–13).
RAP proteins are small GTPases of the RAS family, which are expressed in various cell types, including endothelial cells, leukocytes, and platelets (14). The RAP family includes 5 members that are grouped into 2 subfamilies, RAP1 and RAP2. Small GTPases cycle between an inactive GDP-bound form and an active GTP-bound form. They are regulated tightly by GEFs, which stimulate GTP loading, and GAPs, which catalyze GTP hydrolysis. Our recent work and that of others demonstrated that RAP1 is a central signaling node, regulating platelet adhesion and thrombosis (15–17), and that CalDAG-GEFI (also known as RASGRP2) is a critical RAP-GEF expressed in platelets (18–21). Upon cellular stimulation, CalDAG-GEFI is important for the rapid, calcium-dependent (Ca2+-dependent) activation of RAP1 and integrin αIIbβ3 (22–26). RAP1 activation in the absence of Ca2+/CalDAG-GEFI is comparatively slow but sustained (17) and requires signaling via PKC (23, 27), P2Y12 (11, 13, 17), and PI3K (11, 28).
Based on these differences in the kinetics of RAP1 activation, we proposed that the P2Y12 signaling axis leads to sustained activation of RAP1 and αIIbβ3 integrin by negatively regulating a putative RAP-GAP. In previous work, Smolenski and colleagues suggested a role for RAP1GAP2 in platelet activation (29). However, RNA and protein expression profiling demonstrated that RAP1GAP2 is very weakly expressed in human platelets and virtually absent in mouse platelets (30–32). The same studies identified the dual specificity GAP, RASA3, as the most abundant RAP-GAP expressed in platelets, with protein expression levels comparable to that of CalDAG-GEFI. An important confirmation that RASA3 may be a critical regulator of platelet function came from our findings that a G125V mutation in Rasa3 (Rasa3scat) is associated with severe thrombocytopenia in mice (33) and from recent studies by Molina-Ortiz et al., who described increased RAP1 activation in megakaryocytes from mice expressing a catalytically inactive mutant of RASA3 (34).
Here, we present evidence strongly suggesting that the GAP RASA3 (35) is the missing link between P2Y12 and RAP1 in platelets and that inhibitors of P2Y12, used clinically, prevent thrombosis mainly via their effect on RASA3/RAP1 signaling. Our studies also demonstrate that mutations in RASA3 cause spontaneous platelet activation, CalDAG-GEFI/RAP1-dependent platelet clearance, and severe thrombocytopenia in mice, indicating that RASA3 is critical for maintaining circulating platelets in a quiescent state.
Results
Platelet preactivation and increased platelet clearance in Rasa3 mutant mice.
In our search for novel RAP regulators in platelets, we identified RASA3 as the most abundant RAP-GAP based on recent transcriptomic and proteomic studies (30–32). Studies on the role of RASA3 in the activation response of human platelets are not possible currently, as specific inhibitors do not exist and patients with mutations in the gene have not been identified. Recent studies in mice demonstrated that expression of RASA3 mutants leads to high embryonic lethality and severe thrombocytopenia (33, 34). To test whether the embryonic lethality of Rasa3 mutant mice is caused by defective platelet function, we deleted Rasa3 both systemically (Rasa3–/–) and specifically in the megakaryocyte lineage (Rasa3fl/flPF4-Cre+) (Supplemental Figure 1, A and B; supplemental material available online with this article; doi:10.1172/JCI77993DS1). Compared with their respective controls, both Rasa3–/– and Rasa3fl/flPF4-Cre+ mice exhibited high lethality at P21 (Figure 1A). Peripheral platelet counts in embryos (data not shown) and in the few surviving Rasa3–/– mice (Figure 1B) were markedly decreased when compared with those of controls. Blood-filled lymphatic vessels were observed in Rasa3–/– and Rasa3fl/flPF4-Cre+ embryos but not Rasa3+/+ and Rasa3+/+PF4-Cre+ embryos (Figure 1C). Immunohistochemistry studies confirmed the presence of rbc in lymphatic vessels of Rasa3–/– and Rasa3fl/flPF4-Cre+ embryos (Figure 1D), including cutaneous and jugular lymphatics and the thoracic duct (Supplemental Figure 2), in which platelets and the lymphovenous valve are required to prevent backflow of blood into the lymphatic vasculature (36). These findings are consistent with those of previous studies, demonstrating a critical role for platelets in lymphatic vascular development and survival of mice (37–39). However, we consistently observed a more severe hemorrhagic phenotype in Rasa3–/– embryos when compared with Rasa3fl/flPF4-Cre+ embryos, suggesting that RASA3-expressing cells other than platelets also contribute to vascular integrity in the developing mouse embryo. While characterizing these knockout mice, we also identified a recessive Rasa3 allele causing a missense mutation (H794L) in another thrombocytopenic mouse strain, hlb381 (Rasa3hlb), derived from a forward genetic screen in C57BL/6J mice (Supplemental Figure 1C). Rasa3hlb/hlb mice were born at expected Mendelian ratios (data not shown), and their peripheral platelet counts were significantly higher than those observed in Rasa3 knockout mice or mice with a different missense mutation in Rasa3 (i.e., Rasa3scat mice) (Figure 2A, Supplemental Figure 3, and ref. 33). Platelets in Rasa3hlb/hlb mice were larger in size (Supplemental Figure 4 and Table 1). wbc counts in Rasa3hlb/hlb mice were also significantly decreased, while their rbc counts and hematocrit were normal (Table 1). Platelet and leukocyte counts were normal in Rasa3+/hlb mice as well as Rasa3+/scat mice (ref. 33 and Table 1). Severe thrombocytopenia, however, was observed in compound heterozygous Rasa3hlb/scat mice (Supplemental Figure 3), confirming that the mutations in Rasa3 are causative for the low peripheral platelet counts observed in Rasa3scat and Rasa3hlb homozygotes. Interestingly, RASA3 expression was reduced strongly in lysates from Rasa3hlb/hlb platelets when compared with WT cells (Figure 2A). Furthermore, RASA3 expression in platelets from mice heterozygous for the H794L mutation (Rasa3+/hlb) was comparable to that in platelets from Rasa3+/– or Rasa3+/flPF4-Cre+ heterozygous mice (Supplemental Figure 5, A and B). Thus, our data strongly suggest that the H794L mutation markedly impairs expression or stability of RASA3 protein and that the very low level of expression explains the hypomorphic phenotype of Rasa3hlb/hlb mice. Consistently, bioinformatics analyses using the SIFT, Panther, and MutPred algorithms predict that the RASA3(H794L) mutation is not tolerated among various species (40, 41).
Figure 1. Severe thrombocytopenia and embryonic lethality in Rasa3 knockout mice.

(A) P21 survival of F1 generation offspring from crosses of Rasa3+/– or Rasa3+/flPF4-Cre+ mice. (B) Peripheral platelet count in Rasa3+/+ (n = 18), Rasa3+/– (n = 17), and Rasa3–/– (n = 2) mice. Due to the high mortality, statistical significance for the lower platelet counts observed in Rasa3–/– mice could not be established. (C and D) Severe thrombocytopenia in Rasa3 knockout animals results in a blood-lymphatic mixing phenotype during midgestation. (C) Representative images of Rasa3+/+ (E15.5), Rasa3–/– (E15.5), Rasa3+/+PF4-Cre+ (E17.5), and Rasa3fl/flPF4-Cre+ (E17.5) embryos showing hemorrhage (black arrows) and cutaneous blood-lymph mixing (blue arrowheads). The bottom row shows high-magnification images of the area around the eye. Scale bar: 1 mm (top row), 500 μm (bottom row). (D) Immunohistochemical analysis revealed the abnormal presence of rbc in the jugular lymph sac of Rasa3–/– and Rasa3fl/flPF4-Cre+ embryos. Sections were either stained with H&E (top rows) or with antibodies to LYVE-1 (green) and DAPI (blue) (bottom rows). Autofluorescent rbc (yellow) confirmed blood-filled lymphatic vessels. CA, carotid artery; JV, jugular vein; JLS, jugular lymph sac. Scale bar: 100 μm. Images in C and D are representative of 3 independent experiments.
Figure 2. Platelet preactivation and clearance in Rasa3hlb/hlb mice.

(A) RASA3 and RAP1 protein levels evaluated by immunoblotting in lysates of Rasa3+/+ and Rasa3hlb/hlb platelets (representative of 3 independent experiments). Peripheral platelet count in Rasa3+/+ (n = 23) and Rasa3hlb/hlb (n = 18) mice. (B) Rasa3hlb/hlb platelets circulate in a preactivated state. RAP1 (representative of 3 experiments) and integrin αIIbβ3 activation (n = 5) in unstimulated (none) or ADP-activated (10 μM) platelets isolated from Rasa3+/+ or Rasa3hlb/hlb mice. (C) Higher percentage of thiazole orange–positive reticulated platelets in Rasa3hlb/hlb blood when compared with Rasa3+/+ blood (n = 6). (D) Decreased life span of Rasa3hlb/hlb platelets. Whole blood samples were obtained from Rasa3+/+ and Rasa3hlb/hlb mice at the indicated time points after injection of Alexa Fluor 488–labeled antibodies to GPIX, and the fraction of labeled to unlabeled platelets was determined (n = 6). (E) Thrombocytopenia in Rasa3hlb/hlb mice is caused by a defect in the hematopoietic system. Peripheral platelet count in lethally irradiated WT mice reconstituted with bone marrow cells from Rasa3+/+ or Rasa3hlb/hlb mice (n = 6). (F and G) Significantly increased numbers of mature, acetylcholinesterase-positive megakaryocytes were observed in the (F) spleens and (G) bone marrow of Rasa3hlb/hlb mice. (H) Normal colony-forming ability (CFU) of megakaryocyte (Mk) and nonmegakaryocyte progenitors in bone marrow cells (BMC) isolated from Rasa3hlb/hlb mice (n = 6). (I) Normal proplatelet (PP) formation in Rasa3hlb/hlb megakaryocytes when compared with controls. Quantification of the percentage of megakaryocytes forming proplatelets in culture and representative images. Scale bar: 50 μm (n = 6). *P < 0.05, **P < 0.01, ***P < 0.0001, 2-tailed Student’s t test.
Table 1. Peripheral blood cell counts and spleen weights in Rasa3hlb mutant adult male mice carrying the H794L mutation in all cell types.
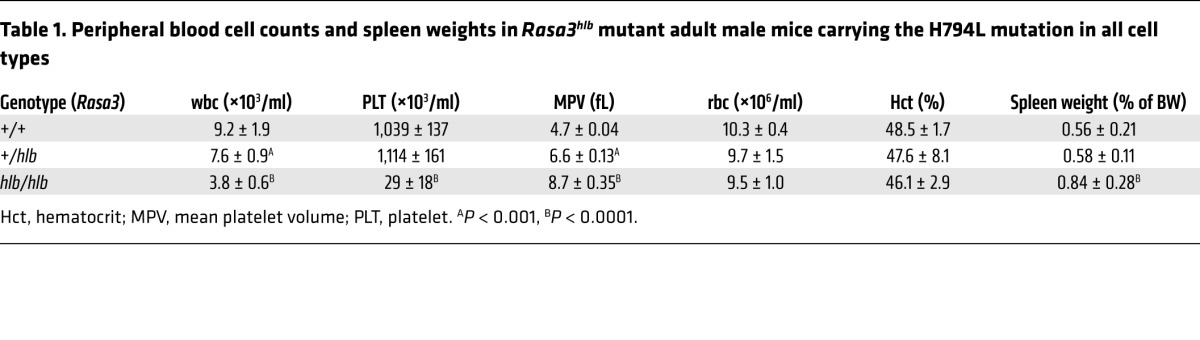
The incomplete thrombocytopenia and the Mendelian birth rates observed in Rasa3hlb/hlb mice provided us with a unique model system to isolate Rasa3 mutant platelets for functional studies. Rasa3hlb/hlb platelets exhibited increased levels of activated RAP1 (RAP1-GTP) and activated integrin αIIbβ3, both at baseline and in response to agonist stimulation (Figure 2B). Surface expression levels of αIIbβ3 were not significantly different between Rasa3hlb/hlb and control platelets (data not shown). Furthermore, the percentage of reticulated (young) platelets in the peripheral blood was markedly higher in Rasa3hlb/hlb mice (Figure 2C), suggesting that the thrombocytopenia in these mice may result from increased platelet clearance but not impaired platelet production. Consistent with this hypothesis, we observed a markedly reduced half-life for circulating Rasa3hlb/hlb platelets compared with that for Rasa3+/+ platelets (t1/2 ~14 hours and t1/2 ~55 hours, respectively) (Figure 2D). Severe thrombocytopenia (Figure 2E) and reduced platelet survival (data not shown) were also observed in lethally irradiated Rasa3+/+ mice reconstituted with bone marrow cells from Rasa3hlb/hlb mice, excluding the possibility that the increased clearance rate resulted from a hyperactive phagocytic system. Rasa3hlb/hlb mice displayed splenomegaly (Table 1), effacement of the splenic architecture (Supplemental Figure 6A), and a significant expansion of megakaryocytes in the red pulp (Figure 2F and Supplemental Figure 6B). Megakaryocyte numbers were also significantly higher in the bone marrow of Rasa3hlb/hlb mice (Figure 2G and Supplemental Figure 6B), but no other gross morphological changes were observed in bone sections when compared with those of controls (Supplemental Figure 6A). Rasa3hlb/hlb megakaryocytes stained positive for acetylcholinesterase (Supplemental Figure 6B), a marker for mature megakaryocytes (42), and exhibited an ultrastructure with a clear distinction between the perinuclear, intermediate, and ectoplasmic compartments (Supplemental Figure 6, C and D). No significant difference was observed in a CFU assay for megakaryocyte progenitors in bone marrow of Rasa3+/+ and Rasa3hlb/hlb mice (Figure 2H). Importantly, megakaryocytes isolated from Rasa3hlb/hlb mice were indistinguishable from Rasa3+/+ megakaryocytes in their ability to form proplatelets ex vivo (Figure 2I). Together, these data suggest increased platelet clearance as a major contributor to the thrombocytopenia observed in Rasa3hlb/hlb mice.
Loss of CalDAG-GEFI prolongs platelet life span and ameliorates thrombocytopenia in Rasa3 mutant mice.
To test the hypothesis that RASA3 restrains RAP1-dependent platelet activation and clearance, we crossed Rasa3hlb/hlb mice with CalDAG-GEFI knockout mice (resulting in Caldaggef1–/– Rasa3hlb/hlb mice). As shown in Figure 3, deletion of Caldaggef1 led to a dose-dependent increase in the peripheral platelet count of Rasa3hlb/hlb mice (Figure 3A). Importantly, loss of CalDAG-GEFI also normalized the life span of Rasa3hlb/hlb platelets in circulation (Figure 3B). A significant increase in platelet life span and peripheral platelet counts was also observed in Rasa3scat/scat mice lacking CalDAG-GEFI (Supplemental Figure 7A). Platelets isolated from Caldaggef1–/– Rasa3scat/scat or Caldaggef1–/– Rasa3hlb/hlb mice did not show signs of preactivation of αIIbβ3 integrin (Supplemental Figure 7, B and C). Importantly, platelets isolated from Caldaggef1+/– Rasa3hlb/hlb mice had a significantly shorter life span than control platelets when transfused into WT recipient mice (Figure 3C), confirming that the reduced platelet half-life observed in Rasa3 mutant mice is a platelet-intrinsic defect. Twenty-four hours after transfusion, increased numbers of Caldaggef1+/– Rasa3hlb/hlb platelets were detectable in the spleens and the livers of WT recipient mice (Supplemental Figure 8A). However, splenectomy did not lead to a recovery of the peripheral platelet count in Rasa3hlb/hlb mice (Supplemental Figure 8B).
Figure 3. Platelet number and life span in Rasa3hlb/hlb mice are normalized by reducing CalDAG-GEFI expression.

(A and B) Deletion of CalDAG-GEFI restores the peripheral platelet count and platelet life span in Rasa3hlb/hlb mice. (A) Whole blood samples were obtained from controls and Caldaggef1+/+ Rasa3hlb/hlb, Caldaggef1+/– Rasa3hlb/hlb, and Caldaggef1–/– Rasa3hlb/hlb mice, and platelet counts per ml blood were determined (n = 6). (B) Whole blood samples were obtained from WT, Caldaggef1+/+ Rasa3hlb/hlb, Caldaggef1+/– Rasa3hlb/hlb, and Caldaggef1–/– Rasa3hlb/hlb mice at the indicated time points after injection of anti-GPIX Alexa Fluor 488, and the fraction of labeled to unlabeled platelets was determined (n = 6). *P < 0.05, ***P < 0.0001, 2-way ANOVA with Bonferroni post-test. (C) Reduced platelet half-life in Rasa3hlb/hlb mice is caused by a defect that is intrinsic to platelets. The bar graph shows the survival of Caldaggef1+/– Rasa3+/+ or Caldaggef1+/– Rasa3hlb/hlb platelets after transfusion into WT recipient mice (n = 6). ***P < 0.0001, 2-tailed Student’s t test.
Rasa3 mutant platelets are insensitive to inhibitors of P2Y12 and PI3K.
Since RASA3 is critical to maintain circulating platelets in a resting state, we hypothesized that (a) RASA3 activity would be a limiting factor for platelet adhesion and hemostatic plug formation at sites of vascular injury and (b) RASA3 would be part of the CalDAG-GEFI–independent RAP1 activation pathway controlled by P2Y12 signaling. To test these hypotheses and to circumvent the severe thrombocytopenia observed in Rasa3 mutant mice, we primarily used Caldaggef1–/– Rasa3hlb/hlb platelets to evaluate how RASA3 affects agonist-induced platelet activation in vitro and in vivo. No significant difference in the expression levels of major adhesion receptors (Figure 4A) or RAP1 protein (Figure 4B) was observed among WT, Caldaggef1–/–, and Caldaggef1–/– Rasa3hlb/hlb platelets. RASA3 protein expression, however, was strongly reduced in lysates from Caldaggef1–/– Rasa3hlb/hlb platelets when compared with WT or Caldaggef1–/– cells (Figure 4B), confirming results obtained with Rasa3hlb/hlb platelets (Figure 2A). Consistent with the critical role of CalDAG-GEFI in platelet signaling (16–18, 24), both Caldaggef1–/– and Caldaggef1–/– Rasa3hlb/hlb platelets exhibited impaired αIIbβ3 activation in response to cellular stimulation (Figure 4C). Agonist-induced activation of αIIbβ3, however, was significantly increased in Caldaggef1–/– Rasa3hlb/hlb platelets when compared with that in Caldaggef1–/– controls. In addition, agonist stimulation of Caldaggef1–/– Rasa3hlb/hlb platelets led to a stronger aggregation response (Figure 4D, Figure 5A, and Supplemental Figure 9) and increased RAP1 activation (Figure 5B) when compared with Caldaggef1–/– controls.
Figure 4. RASA3 deletion leads to increased αIIbβ3 activation in stimulated platelets.

(A) Normal membrane expression of glycoprotein receptors in WT, Caldaggef1–/– Rasa3+/+, or Caldaggef1–/– Rasa3hlb/hlb platelets. Diluted whole blood was stained for 10 minutes with fluorophore-labeled antibodies (2 μg/ml) to the indicated antigens and analyzed by flow cytometry (n = 6). (B) RASA3 and RAP1 protein level evaluated by immunoblotting in lysates of WT, Caldaggef1–/– Rasa3+/+, or Caldaggef1–/– Rasa3hlb/hlb platelets. Results are representative of 3 independent experiments. (C) Increased integrin αIIbβ3 activation (JON/A-PE binding) in activated Caldaggef1–/– Rasa3hlb/hlb platelets when compared with Caldaggef1–/– controls. Platelets were stimulated for 10 minutes with increasing concentrations of Par4-activating peptide (Par4p) or the GPVI-specific agonist convulxin (Cvx), stained with JON/A-PE, and immediately analyzed by flow cytometry. MFI, mean fluorescence intensity. *P < 0.05, ***P < 0.0001, 2-way ANOVA with Bonferroni post-test (n = 6, 3 independent experiments). (D) Aggregation response of washed WT, Caldaggef1–/– Rasa3+/+, or Caldaggef1–/– Rasa3hlb/hlb platelets stimulated with low (left) or high (right) doses of collagen, thrombin, or the thromboxane analog U46619. Results are representative of 3 independent experiments.
Figure 5. RAP1 and integrin activation in Rasa3 mutant platelets is insensitive to inhibitors of the P2Y12/PI3K signaling pathway.

(A) Aggregation of WT, Caldaggef1–/–, or Caldaggef1–/– Rasa3hlb/hlb platelets stimulated with 250 μM Par4-activating peptide in the presence or absence or 100 μM 2-MeSAMP or 200 nM wortmannin. Aggregation was monitored for 3 minutes. (B) Levels of activated RAP1 (RAP1-GTP) and AKT (phosphorylated AKT [AKT-P]) in WT, Caldaggef1–/–, or Caldaggef1–/– Rasa3hlb/hlb platelets stimulated for 3 minutes with Par4p in the presence of vehicle, 2-MeSAMP, or wortmannin. Total RAP1 and AKT are shown for loading controls. (C) Real-time flow cytometry analysis of αIIbβ3 activation in WT, Caldaggef1–/–, or Caldaggef1–/– Rasa3hlb/hlb platelets stimulated with Par4p. The blue box highlights delayed αIIbβ3 activation in Caldaggef1–/– and Caldaggef1–/– Rasa3hlb/hlb platelets. (D) Effect of ADP or 2-MeSAMP on Par4p-induced αIIbβ3 activation kinetics in Caldaggef1–/– Rasa3+/+ or Caldaggef1–/– Rasa3hlb/hlb platelets. (E) Effect of P2Y12 inhibition on αIIbβ3 activation in Rasa3hlb/hlb platelets stimulated for 10 minutes with the indicated concentrations of Par4p. Data represent MFI ± SD in activated blood samples after subtraction of MFI measured in nonactivated cells. ***P < 0.0001, 2-way ANOVA with Bonferroni post-test (n = 6). (F) Aggregation of Caldaggef1+/– Rasa3+/hlb or Caldaggef1+/– Rasa3hlb/hlb platelets (in PRP at 1.5 × 108 platelets/ml) stimulated with 3 μM ADP, 0.1 μM MRS-2365 (P2Y1 agonists), or MRS-2365 plus 1 μM epinephrine (α2A-R agonist). Results in A–D and F are representative of 4 independent experiments.
Notably, agonist-induced aggregation (Figure 5A) and RAP1 activation (Figure 5B) were sensitive to inhibition of P2Y12 in Caldaggef1–/– platelets but not in Caldaggef1–/– Rasa3hlb/hlb platelets, even at threshold concentrations of agonists (Supplemental Figure 9). Similarly P2Y12 inhibition had no significant effect on αIIbβ3 activation in stimulated Caldaggef1–/– Rasa3scat/scat (Supplemental Figure 7B) or Caldaggef1–/– Rasa3hlb/hlb platelets (Supplemental Figure 7C). Signaling via P2Y12 leads to the activation of PI3K, and both RAP1 and the serine/threonine protein kinase AKT are known to be downstream of the P2Y12/PI3K signaling axis (10, 13). We next evaluated whether PI3K signaling affects RAP1 activation in a RASA3-dependent manner. As expected, inhibition of PI3K by wortmannin inhibited RAP1 activation and αIIbβ3-mediated aggregation of WT or Caldaggef1–/– platelets (Figure 5, A and B). However, saturating concentrations of PI3K inhibitors had little effect on the activation of RAP1 or αIIbβ3 in Caldaggef1–/– Rasa3hlb/hlb platelets. Importantly, inhibition of P2Y12 or PI3K abolished AKT phosphorylation in Caldaggef1–/– Rasa3hlb/hlb and control platelets (Figure 5B). Thus, our studies suggest that RASA3 activity toward RAP1 is under the control of P2Y12 and PI3K, independently of AKT. Impaired function of RASA3 leads to a state of continuous P2Y12 signaling, which makes RAP1-dependent platelet activation independent of feedback activation by ADP. This latter conclusion was confirmed when we studied integrin activation kinetics in platelets using a flow cytometry–based assay. As shown in Figure 5C, agonist-induced αIIbβ3 activation in Caldaggef1–/– Rasa3hlb/hlb platelets was more robust than that in Caldaggef1–/– controls. Furthermore, αIIbβ3 activation in Caldaggef1–/– platelets was modulated by the presence of an exogenous P2Y12 agonist (ADP) or P2Y12 inhibitor (2-methylthio-AMP triethylammonium salt hydrate [2-MeSAMP]) (Figure 5D), while no such modulation was observed in Caldaggef1–/– Rasa3hlb/hlb cells (Figure 5D). Consistent with the documented role for Ca2+/CalDAG-GEFI in the rapid activation of RAP1 (17, 24), αIIbβ3 activation in Caldaggef1–/– Rasa3hlb/hlb platelets occurred with a delay when compared with that in WT controls (Figure 5C, blue box).
Due to their marked thrombocytopenia, we were unable to perform comprehensive aggregation or RAP1 activation studies with platelets from Rasa3hlb/hlb mice. However, our flow cytometry studies demonstrated that αIIbβ3 activation in Rasa3hlb/hlb platelets was not significantly reduced in the presence of an inhibitor of P2Y12 (Figure 5E), even though there was a trend to lower αIIbβ3 activation in these cells. We also studied the aggregation response of Caldaggef1+/– Rasa3hlb/hlb platelets and control platelets to ADP and to an agonist specific for the Gαq-coupled receptor for ADP, P2Y1 (MRS-2365) (43). As shown previously, ADP-induced platelet aggregation requires concomitant signaling by both the Gαq-coupled receptor P2Y1 and the Gαi-coupled receptor P2Y12 (44). Consistently, Caldaggef1+/– Rasa3+/hlb control platelets aggregated in response to stimulation with ADP but not the P2Y1-specific agonist MRS-2365 (Figure 5F). Aggregation was restored, however, when platelets were stimulated with MRS-2365 and epinephrine, a platelet agonist that stimulates a Gαi signaling response similar to that of P2Y12 (44). Importantly, aggregation of Caldaggef1+/– Rasa3hlb/hlb platelets required only MRS-2365, further demonstrating that impaired RASA3 function constitutes a state of continuous P2Y12 signaling.
Platelet accumulation and hemostatic plug formation at sites of vascular injury are insensitive to P2Y12 inhibitors in Caldaggef1–/– Rasa3hlb/hlb mice.
To validate our findings in vivo, we studied the hemostatic response in WT, Caldaggef1–/–, and Caldaggef1–/– Rasa3hlb/hlb mice, i.e., mice with similar peripheral platelet counts. Hemostatic plug formation was assessed in mice treated or not with the P2Y12 inhibitor clopidogrel bisulfate (Figure 6 and Supplemental Videos 1–6). Using a laser ablation system, an injury to the endothelium with a diameter of approximately 50 to 100 μm was generated in the saphenous vein, and platelet accumulation (Figure 6, A and B) and time to cessation of blood loss (Figure 6C) were quantified. In agreement with our in vitro data, we observed significantly increased platelet adhesion and a shortened bleeding time in Caldaggef1–/– Rasa3hlb/hlb mice when compared with those of Caldaggef1–/– controls. However, platelet accumulation and hemostatic plug formation were delayed in Caldaggef1–/– Rasa3hlb/hlb mice compared with those in WT controls, consistent with the documented role for CalDAG-GEFI in the rapid activation of platelet integrins (Figure 5C and ref. 17). In spite of their slow adhesion kinetics, more Caldaggef1–/– Rasa3hlb/hlb platelets than WT platelets accumulated over time at the site of injury, as platelet adhesion was more stable in Caldaggef1–/– Rasa3hlb/hlb mice compared with that observed in WT mice (45).
Figure 6. Hemostatic plug formation in Rasa3hlb/hlb mice is insensitive to P2Y12 inhibitors.

Intravital microscopy studies to monitor hemostatic plug formation after laser injury to the saphenous vein in WT (also see Supplemental Videos 1 and 4), Caldaggef1–/– (also see Supplemental Videos 2 and 5), and Caldaggef1–/– Rasa3hlb/hlb mice (also see Supplemental Videos 3 and 6). Where indicated, mice were treated with clopidogrel bisulfate 24 and 3 hours prior to the experiment at a dosage of 75 mg/kg bodyweight to inhibit signaling via the platelet P2Y12 receptor. Prior to laser injury, animals were injected with Alexa Fluor 488–labeled antibodies to GPIX, a receptor expressed exclusively on the platelet surface. (A) Representative images taken 30 seconds and 240 seconds after laser injury (except where indicated by an asterisk). Arrows highlight blood loss at the site of injury. Scale bar: 50 μm. (B) Sum fluorescence intensity ± SEM recorded at the site of injury over time in WT, Caldaggef1–/–, and Caldaggef1–/– Rasa3hlb/hlb mice (n = 8–19). (C) Time to stable occlusion (no leakage of blood for more than 60 seconds) of the vascular lesion in WT (circles), Caldaggef1–/– (triangles), and Caldaggef1–/– Rasa3hlb/hlb (squares) mice. White symbols indicate mice treated with clopidogrel bisulfate. Dotted line represents the time point when recordings were stopped. *P < 0.05, ***P < 0.0001, 2-way ANOVA with Bonferroni post-test.
Clopidogrel bisulfate (Plavix) is one of the most widely used antiplatelet drugs (46). We demonstrated recently that administration of clopidogrel bisulfate significantly reduces the stability of thrombi in injured arterioles and venules of WT mice, while it virtually abolishes thrombus formation in Caldaggef1–/– mice (17). Here, we confirmed these findings in the laser injury hemostasis model (Figure 6). Consistent with our in vitro data, however, administration of clopidogrel bisulfate did not significantly impair platelet accumulation (Figure 6, A and B) or hemostasis (Figure 6C) at sites of laser injury in Caldaggef1–/– Rasa3hlb/hlb mice. Together, our findings strongly suggest that P2Y12 signaling downregulates RASA3 activity during thrombus formation.
Discussion
Based on our results, we propose the following model for the regulation of RAP1 signaling (Figure 7). In circulating platelets, CalDAG-GEFI activity is low and RASA3 is active in the plasma membrane (PM) (47–49) to limit RAP1 activation. At sites of vascular injury, platelets are activated via engagement of ITAM receptors and/or GPCRs. Activation of PLC is a downstream signaling event common to most of these receptors. Activated PLC leads to the generation of two important second messengers: Ca2+ and diacylglycerol (DAG). Ca2+ activates CalDAG-GEFI, which mediates rapid nucleotide exchange on RAP1. Activated RAP1-GTP in turn promotes various platelet responses, including TALIN-dependent αIIbβ3 activation (50). CalDAG-GEFI signaling, however, is reversible and returns to baseline when cytosolic Ca2+ levels normalize. DAG is critical for the activation of PKC, which affects RAP1 signaling in two ways. First, PKC signaling can induce nucleotide exchange in RAP1 by a CalDAG-GEFI-independent mechanism. In addition, PKC signaling is critical for the release of ADP-containing storage granules from activated platelets. ADP, via binding to P2Y12, induces the activation of PI3K, which leads to decreased RASA3 function and prolonged RAP1 signaling. Inhibitors to P2Y12 prevent the inactivation of RASA3, prohibit prolonged RAP1 signaling, and destabilize the growing thrombus.
Figure 7. Schematic illustrating the regulation of RAP1 signaling in platelets.

RAP1 activation in platelets is tightly controlled by the antagonistic balance between the calcium-sensitive RAP-GEF, CalDAG-GEFI, and the RAP-GAP, RASA3. In quiescent platelets, RASA3 restrains unwanted CalDAG-GEFI/RAP1 signaling to ensure platelet homeostasis. At sites of vascular injury, platelet stimulation through ITAM-coupled (collagen receptor GPVI, podoplanin receptor CLEC-2, and IgG receptor FcγRIIA) and G protein–coupled (thrombin receptors PAR1/3/4, ADP receptor P2Y1, and TxA2 receptors) receptors leads to the activation of PLCγ2 and PLCβ2/3, respectively. PLCs convert PIP2 to DAG and inositol 1,4,5-trisphosphate (IP3). IP3 mediates Ca2+ store release and a rise in the cytosolic Ca2+ concentration, which triggers rapid CalDAG-GEFI-dependent RAP1 activation. DAG leads to PKC activation, which contributes to nucleotide exchange on RAP1 and the release of platelet granules. CalDAG-GEFI signaling eventually subsides and RASA3 must be inactivated in order to maintain RAP1 in an activated state. RASA3 inactivation depends on PI3 kinase signaling, which in platelets is under the control of Gαi-coupled receptors, such as P2Y12 (ADP) or α2A-R (epinephrine). Both rapid and sustained RAP1 signaling are critical for integrin inside-out activation and the formation of a hemostatic plug. P2Y12 inhibitors prevent the inactivation of RASA3 and thus destabilize the growing thrombus.
RASA3 is positioned perfectly to prevent unwanted RAP1-dependent platelet activation, as it is targeted to the PM of resting cells by the interaction of its unique PH/BTK domain with phosphatidylinositol 4,5-bisphosphate (PIP2) (47–49). At sites of vascular injury, however, RASA3 activity in platelets must be reduced to allow for the formation of a hemostatic plug. Our studies suggest that this process is mediated by PI3K, a family of enzymes capable of phosphorylating the 3′ position hydroxyl group of the inositol ring of phosphoinositol. Activated PI3K mediates the conversion of PIP2 to phosphatidylinositol 3,4,5-trisphosphate (PIP3) and/or phosphatidylinositol 3,4-biphosphate. Studies by Lova et al. demonstrated that stimulation of a Gαi-dependent signaling pathway leads to sustained RAP1 activation through the action of the PI3K product PIP3 but not phosphatidylinositol 3,4-biphosphate (28). The remaining question is whether PIP3, which also binds the PH/BTK domain of RASA3 with high affinity, affects RASA3 function by modulating its enzymatic activity and/or the subcellular localization of RASA3. Answering this question, however, has proven to be extremely difficult, even in heterologous cell systems (35, 48). Adding to the complexity, recent findings suggest that segregation of PIP2 and PIP3 into distinct nanoscale regions in the PM (51) could be another mechanism for regulating the function of RASA3 in resting and in activated platelets.
Our findings that Caldaggef1–/– Rasa3hlb/hlb platelets show no signs of RAP1 activation unless stimulated with agonist further demonstrate that RAP1 activation in the absence of CalDAG-GEFI requires RASA3 inactivation and a signaling event that leads to increased nucleotide exchange in the GTPase. Our work (18, 23) and that of others (21) suggest that this CalDAG-GEFI-independent mechanism is mediated by PKC, as aggregation in the absence of functional CalDAG-GEFI could be induced with an agonist for PKC. Downstream of PKC, the nucleotide exchange could be facilitated by a different RAP-GEF, such as CalDAG-GEFIII, PDZ-GEF1/2, or C3G, which are expressed at low levels in platelets (29, 31, 52). Alternatively, nucleotide exchange may be regulated by posttranslational modifications of RAP1, such as oxidation (53), ubiquitination (54), or phosphorylation (55).
Our studies also identified a significant expansion of megakaryocytes in the spleens and the bone marrow of Rasa3 mutant mice. Rasa3hlb/hlb megakaryocytes exhibited largely normal morphology and ultrastructure, and we did not observe a defect in their ability to proliferate in vitro. Similar findings were described recently for chimeric mice expressing a catalytically inactive mutant of RASA3 in blood cells only (34). However, Molina-Ortiz et al. also provided ex vivo evidence suggesting that impaired function of RASA3 leads to thrombocytopenia in mice due to impaired ability of megakaryocytes to form proplatelets (34). Using a similar ex vivo assay, we did not observe a defect in proplatelet formation for megakaryocytes isolated from Rasa3hlb/hlb mice. One potential explanation for these discrepant results derives from the fact that the RASA3 protein level is markedly reduced in Rasa3hlb/hlb mice, while the deletion of the catalytic domain results in the expression of a truncated RASA3 protein that may affect megakaryocyte function independent of its GAP activity. While we were unable to detect a defect in platelet production, we provided strong evidence for increased platelet turnover in Rasa3hlb/hlb mice. First, the life span of circulating platelets was dramatically shortened in Rasa3hlb/hlb mice. Our studies further demonstrated that the decreased life span is a defect that is intrinsic to the platelet, as we observed increased turnover for endogenous platelets in Rasa3hlb/hlb mice as well as for Rasa3hlb/hlb platelets transfused into WT recipient mice. Importantly, a reduction in the expression of the RAP-GEF, CalDAG-GEFI, significantly increased both the platelet life span and the peripheral platelet count in Rasa3 mutant mice. Together, these studies suggest that RASA3 is required to protect circulating platelets from CalDAG-GEFI/RAP1-mediated activation and clearance. Consistent with this, Grosse et al. recently described a very similar platelet clearance phenotype for mice expressing a gain-of-function mutant of stromal interaction molecule 1 (Stim1Sax) (56). STIM1 is an important regulator of store-operated calcium entry (SOCE), the predominant mechanism in the regulation of calcium influx into platelets (57). Platelets from Stim1+/Sax mice exhibit constitutive SOCE, preactivation, and markedly reduced life span in circulation. Thus, both elevation of cytosolic Ca2+, a critical regulatory step in the activation of CalDAG-GEFI (18, 24, 58), and inactivation of RASA3 lead to platelet activation and markedly increased platelet turnover.
As indicated by the significantly increased volume of circulating platelets in both Stim1+/Sax and Rasa3hlb/hlb mice, the rapid turnover of platelets likely affects the process of proplatelet formation from maturing megakaryocytes. This has previously been demonstrated for megakaryocytes in mice that were rendered thrombocytopenic by infusion of antiplatelet serum or neuraminidase (59, 60). In both cases, induction of severe thrombocytopenia was followed by an increase in sectional areas of platelets. Our observation that megakaryocytes from Rasa3hlb/hlb mice show a more intact ultrastructure than those from Rasa3scat/scat animals correlates well with the degree of thrombocytopenia observed in these mice. At this point, we have only limited information on how Rasa3 mutant platelets are cleared from circulation. Our studies suggest that clearance occurs mainly in the spleen and liver, consistent with the removal of platelets by phagocytic cells of the reticuloendothelial system (4). Interestingly, however, platelet counts did not recover in splenectomized Rasa3hlb/hlb mice, suggesting that impaired RASA3 function may cause platelet clearance via more than one mechanism, maybe involving the formation and clearance of microaggregates.
In addition to the severe thrombocytopenia, both Rasa3scat/scat and Rasa3hlb/hlb mice exhibited a marked reduction in the peripheral lymphocyte count. Preliminary results obtained with the few surviving Rasa3 knockout mice confirmed these observations (data not shown). Lymphocytes express significant amounts of RAS (61), and the duration and subcellular localization of RAS/ERK signaling was shown to dictate whether lymphocytes proliferate or differentiate (62). This is in contrast to platelets, in which physiological relevance for the low levels (63) of expressed RAS isoforms has not been documented yet (64). RAP1 is also expressed in lymphocytes and regulates the homing of these cells to lymph nodes (65). It will be interesting to see whether mutations in RASA3 affect peripheral lymphocyte counts by modulating the activity of RAS, RAP, or both. While Rasa3scat/scat mice also exhibit a profound anemia (33), the defect in rbc is very mild in Rasa3hlb/hlb mice (L.L. Peters, unpublished observations). Factors that may contribute to this phenotypic difference between the Rasa3scat and Rasa3hlb mutant models likely include the difference in the mutant allele and the genetic background of these mutant strains (BALB/cByJ-scat; C57BL/6J-hlb381). Furthermore, conditional deletion of Rasa3 will be required to identify whether any changes documented for lymphocytes or erythrocytes in Rasa3 mutant mice are explained solely by defects intrinsic to these cell types or whether some of the phenotype is secondary to the marked developmental defects caused by the severe thrombocytopenia.
To date, no patients with impaired RASA3 expression or function have been described, and one has to be careful when extrapolating findings in mice to humans. However, for the following reasons, we believe that a similar RASA3-dependent mechanism controls RAP1-dependent platelet activation in humans. First, transcriptome and proteome analysis of human platelets identified RASA3 as the predominant RAP-GAP in these cells (30–32). Second, we and others identified two independent yet synergistic pathways critical to RAP1 activation in mouse and human platelets (11, 21, 23, 27). Fast but reversible RAP1 activation is dependent on calcium and CalDAG-GEFI, while sustained RAP1 activation requires signaling by P2Y12 and PI3K (11, 13, 17, 24). Significantly, the importance of a CalDAG-GEFI-dependent and -independent pathway to RAP1 activation was confirmed recently in patients with impaired CalDAG-GEFI function (21). Last, a recent genome-wide association study identified a single nucleotide polymorphism that is in linkage with RASA3 and correlates with mean platelet volume variations in humans (66). Similar to these findings, we observed a significant increase in mean platelet volume in Rasa3hlb mice (Table 1).
The P2Y12 receptor is the target of clinically successful antiplatelet drugs, such as ticagrelor, clopidogrel bisulfate, or prasugrel (46). Based on our results, we propose that P2Y12 signaling is critical for the inactivation of RASA3 in activated platelets and that P2Y12 inhibitors protect from thrombosis primarily due to their effect on the RASA3/RAP1 signaling module. These findings may have important implications for the development of safer antiplatelet therapies. For example, an antidote to P2Y12 inhibitors would greatly improve the safety of such therapies, as it could be used to manage bleeding complications in patients taking these drugs. Our work suggests inhibition of RASA3 as one such approach to antagonize P2Y12 inhibitors, although such a therapy would likely need to be carefully monitored to avoid complications, such as thrombocytopenia or excessive platelet reactivity. Our findings may also explain some of the interindividual variability in ADP-induced integrin activation, observed both in the presence and absence of P2Y12 inhibitors (67). Based on our studies, we suggest that variations in the expression levels of CalDAG-GEFI and RASA3, i.e., the antagonistic balance of a critical GEF and GAP for RAP1, may contribute to this variability in platelet reactivity.
In summary, we have identified the RAP-GAP RASA3 as a critical negative regulator of platelet activation. RASA3 maintains circulating platelets in a quiescent state as it antagonizes low-level RAP1 activation by the RAP-GEF, CalDAG-GEFI. At sites of vascular injury, continuous signaling through P2Y12 and PI3K is necessary to ensure RASA3 inhibition, sustained RAP1/integrin activation, and the formation of a stable hemostatic plug. Thus, our studies suggest that the antagonistic balance between CalDAG-GEFI and RASA3 is critical for platelet homeostasis and vascular hemostasis. Our findings may lead to improved diagnosis and treatment of platelet-related disorders.
Methods
Mice
Rasa3scat/scat (Rasa3G125V/G125V) (33), Caldaggef1–/– (18), and Pf4-Cre+ (68) mice have been described previously.
Rasa3hlb/hlb (Rasa3H794L/H794L) mice were derived from a N-ethyl-N-nitrosourea mutagenesis screen performed at The Jackson laboratory, as described in detail previously (69). For the generation of Rasa3 knockout mice, a targeting vector was obtained from the KOMP-CSD (Knockout Mouse Project–CHORI/Sanger/University of California, Davis) consortium, and targeting of C57BL/6 ES cells was performed by the Animal Models Core at the University of North Carolina at Chapel Hill. Megakaryocyte-specific Rasa3 knockout mice were generated by crossing Rasa3fl/fl mice with Pf4-Cre+ mice (for details see Supplemental Figure 1). The following primers were used for the genotyping of conventional and conditional knockout mice: FlpA 5′-AAAACACACTGAGGTTCAGACACGCTCC-3′, FlpS 5′-GTCATCCATGGGTTTCCTAAGCACTTCC-3′, and Neo3-30 5′-CGCATCGCCTTCTATCGCCTTCTTGACGA-3′.
Where indicated, mice were treated with clopidogrel bisulfate (Plavix, Sanofi-Aventis) 24 and 3 hours prior to the experiment at a dosage of 75 mg/kg bodyweight. P2Y12 inhibition was confirmed by standard aggregometry (impaired response to ADP, data not shown). Treatment with clopidogrel bisulfate did not affect platelet counts or counts of other blood cells (data not shown).
Peripheral cell counts
Whole blood was collected from the retro-orbital plexus into EDTA-containing 1.5 ml microtubes. Complete blood counts were determined using an automated hematology analyzer (Advia 120 Multispecies Hematology Analyzer, Bayer Diagnostics).
Bone marrow cell transplantation
Bone marrow cells were harvested from the tibias and femurs of adult donor mice. A total of 2 × 106 cells were transplanted by retro-orbital injection (100 ml per mouse) into (8- to 10-week-old) recipient animals conditioned with a lethal dose of 2 × 600 cGy total body irradiation.
Platelet preparation
Blood was drawn with heparin-coated capillaries (VWR) from the retro-orbital plexus into tubes containing low-molecular-weight Lovenox (enoxaparin sodium, Sanofi-Aventis). Platelet-rich plasma (PRP) was obtained by centrifugation at 100 g for 5 minutes. PRP was centrifuged at 700 g for 5 minutes at room temperature in the presence of 5 μM prostacyclin (PGI2) from Sigma-Aldrich. After 2 washing steps, pelleted platelets were resuspended in modified Tyrode’s buffer (137 mM NaCl, 0.3 mM Na2HPO4, 2 mM KCl, 12 mM NaHCO3, 5 mM N-2-hydroxyethylpiperazine-N′-2-ethanesulfonic acid, 5 mM glucose, pH 7.3) containing 1 mM CaCl2. PRP from Rasa3hlb/hlb and Rasa3+/+ control mice was obtained by centrifugation of whole blood with 2 mM EDTA at 100 g. Platelets were washed with modified Tyrode’s buffer containing 2 mM EDTA and PGI2, centrifuged at 700 g using a swinging bucket rotor, and, ultimately, resuspended in modified Tyrode’s buffer containing 1 mM CaCl2.
Flow cytometry
Platelet count.
8- to 10-week-old mice were bled retro-orbitally (50 μl), and platelets were labeled for 10 minutes with 2 μg/ml Alexa Fluor 488–labeled antibody to GPIX (clone Xia.B4, Emfret Analytics). Samples were diluted with PBS, and the number of GPIX-positive events per volume was determined with a BD Accuri C6 Flow Cytometer.
Reticulated (young) platelet count.
Blood was drawn from the retro-orbital plexus (50 μl) and diluted in rbc lysis buffer (10 mM KHCO3, 150 mM NH4Cl, 0.1 mM EDTA pH 8.0). After rbc lysis, platelets were washed with PBS, resuspended in PBS with 2% fetal bovine serum, and labeled with an α-CD41 antibody (clone MWReg30, BD Bioscience) conjugated to phycoerythrin (PE). Cells were washed in PBS and resuspended in Retic Count solution (thiazole-orange dye) (Becton Dickinson) (70). Platelets were gated according to the PE staining and analyzed for thiazole-orange staining.
Platelet life span.
Platelets were labeled in vivo by a single intravenous injection of 5 μg Alexa Fluor 488–conjugated α-GPIX antibody (clone Xia.B4, Emfret Analytics) in 100 μl PBS at t = 0. Whole blood (50 μl) was drawn every 24 hours, diluted, and incubated with a PE-conjugated antibody to GPIbα (clone Xia.G5, Emfret Analytics) for 10 minutes at room temperature. The ratio of Alexa Fluor 488–positive platelets to PE-positive platelets was determined.
Surface receptors.
2 × 106 platelets in diluted whole blood (modified Tyrode’s buffer) were stained for 10 minutes with 2 μg/ml fluorophore-conjugated antibodies to the indicated platelet surface glycoproteins and immediately analyzed by flow cytometry. Antibodies for GPIX (clone Xia.B4), GPIbα (clone Xia.G5), GPVI (clone JAQ1), integrin α2 (clone Sam.G4), and IgG controls were purchased from Emfret Analytics. FITC-labeled CLEC-2 antibody was provided by Bernhard Nieswandt (University Clinic of Wuerzburg and Rudolf Virchow Center, Wuerzburg, Germany).
αIIbβ3 Activation and α-granule secretion.
2 × 106 platelets in diluted whole blood (modified Tyrode’s buffer containing 1 mM CaCl2) were activated with ADP (Sigma-Aldrich), convulxin (purchased from Kenneth Clemetson, Theodor Kocher Institute, University of Berne, Bern, Switzerland), or PAR4-activating peptide (Par4p; GL Biochem Inc.) in the presence of 2 μg/ml JON/A-PE, an antibody directed toward the activated form of murine αIIbβ3 (Emfret Analytics) (71), and 2 μg/ml α-P-selectin-FITC (clone RB40.34, BD Biosciences). Following 10 minutes of incubation, samples were diluted with PBS and analyzed immediately.
Real-time αIIbβ3 activation assay.
Washed platelets were diluted (1.25 × 107 platelets/ml) in Tyrode’s buffer containing 1 mM CaCl2. Where indicated, platelets were incubated for 10 minutes with 100 μM 2-MeSAMP (BioLog) to inhibit signaling by P2Y12. After establishing a baseline with unlabeled platelets, JON/A-PE (5 μg/ml) and Par4p (600 μM) were added simultaneously in an equal volume of modified Tyrode’s buffer to allow efficient mixing. JON/A-PE binding was recorded continuously for 10 minutes with a BD Accuri C6 Flow Cytometer (72).
CFU assays for megakaryocyte progenitors
CFU-megakaryocytes were detected in bone marrow using a MegaCult-C Kit, a collagen-based media growth system (Stem Cell Technologies). The media was supplemented with 50 ng/ml rhTpo, 10 ng/ml rmIL-3, 20 ng/ml rhIL-6, and 50 ng/ml rmIL-11. Bone marrow aspirates from 3 Rasa3+/+ and 3 Rasa3hlb/hlb mice, each in 4 technical replicates per mouse, were plated in slide chambers. Slides were incubated at 37°C. After 7 days, the slides were dehydrated and fixed in acetone. Slides were stained with acetylthiocholine iodide and counterstained with Harris’ hematoxylin solution. The slides were then examined under an inverted microscope, and colonies containing at least 3 brown cells were scored.
Differentiation of mouse megakaryocytes
Proplatelet formation was studied in mouse bone marrow–derived megakaryocytes as described recently (73). Mice (8 to 10 weeks of age) were euthanized, and cells were obtained from the bone marrow of femurs and tibias. Cells were homogenized by pipetting, followed by passage through a 100-μm filter. The cell population was resuspended in 10% fetal bovine serum–supplemented DMEM with 2 mM l-glutamine, 50 U/ml penicillin/streptomycin, and fibroblast-conditioned media containing thrombopoietin. The cells were cultured for 4 days (37°C and 5% CO2), and mature megakaryocytes were layered over a BSA gradient. Megakaryocytes were resuspended in culture media and placed on immobilized fibrinogen, and megakaryocytes with proplatelets were counted. A minimum of 100 megakaryocytes was analyzed to determine the percentage of proplatelet formation.
Histology, immunohistochemistry, and electron microscopy
Megakaryocytes in spleens and bones of Rasa3+/+ and Rasa3hlb/hlb mice.
Euthanized mice were weighed, and tissues were dissected for weighing and/or analysis. Spleen and hind leg tissue from 3 control and mutant mice were fixed in Bouin’s solution (Sigma-Aldrich). Hind legs were left in Bouin’s solution for 3 days in order to decalcify bones. Tissues were paraffin-embedded, sectioned at 3 to 5 μm, and stained with H&E for pathological analysis using standard techniques. For staining of acetylcholinesterase activity, unfixed spleens and femurs were frozen in OCT embedding compound (Tissue-Tek), cryostat sectioned at 10 μm, and stained as previously described (74, 75). For transmission electron microscopy, bone marrow was flushed from both femurs using PBS containing fetal calf serum and EDTA. Packed bone marrow cells were washed with the same PBS solution and fixed overnight in 2% glutaraldehyde/2% paraformaldehyde in 0.1 M cacodylate buffer (pH 7.4). Bone marrow cell pellets were embedded in agar, sectioned, and postfixed in osmium using standard techniques. A JEOL 100CXII transmission electron microscope was used for examination and image capturing.
Rasa3 knockout embryos.
Pregnant animals were dissected at gestational day E15.5 (Rasa3+/– × Rasa3+/–) or E17.5 (Rasa3+/flPF4-Cre+ × Rasa3+/flPF4-Cre+), and embryos were imaged using a Leica MZ16FA dissecting stereoscope. For some embryos, blood was collected by placing the entire embryo in 100 μl PBS containing heparin, cutting the umbilical cord, and gently palpating the embryo’s abdomen. Embryos were paraffin embedded, and H&E-stained sections were imaged using an Olympus BX61 with a QImaging RETIGA 4000R camera with Volocity software (Improvision, PerkinElmer). For immunohistochemistry, embryo sections were deparaffinized, hydrated, permeabilized, and blocked with 5% normal donkey serum. Slides were then stained with primary antibody, polyclonal rabbit anti-mouse LYVE-1 (catalog no. 70R-LR003, Fitzgerald Industries International), overnight at room temperature. Sections were rinsed, blocked, and incubated with donkey anti-rabbit Alexa Fluor 488 (catalog no. 711-545-152, Jackson ImmunoResearch) and DAPI (Sigma-Aldrich) in the dark for 90 minutes at room temperature. Images were acquired on a Nikon E800 microscope with a Hammamatsu camera with Metamorph software (Molecular Devices Corp.).
Clearance of Rasa3 mutant platelets
Radiolabeling of antibody.
The anti-GPXI antibody (clone Xia.B4, Emfret Analytics) was washed twice with borate buffer (pH 9.0) containing 50 mM EDTA to remove trace metals. The antibody was washed an additional 5 times with borate buffer to remove excess EDTA. The final antibody was concentrated to 2 mg/ml (100 μl) and mixed with the bifunctional metal chelator p-SCN-Bn-NOTA (Macrocyclics) for 24 hours at 4°C. The reaction mixture was placed into a fresh Amicon filter (30,000 NMWL) and excess chelator was removed by 5 subsequent washes with 0.1 M ammonium acetate. 37 MBq (1 mCi) of copper-64 (copper-64 [t1/2 = 12.7 hour] was purchased from Mallinckrodt Institute of Radiology at Washington University School of Medicine as a copper chloride solution [in 0.1 M HCl] and was neutralized with 0.1 M ammonia acetate buffer upon arrival) was added to the antibody and allowed to chelate over 20 minutes at room temperature. This reaction gave a radiochemical yield of 95%. The reaction mixture was placed into a fresh Amicon filter (30,000 NMWL) and washed twice with PBS containing EDTA (50 mM) followed by 3 washes with PBS. Centrifugal filtration gave radiolabeled antibody with >99% radiochemical purity. Radiochemical purity was determined using size exclusion chromatography on an Agilent 1260 HPLC system equipped with a BIO-SEC-5 100Å column (data not shown).
Animal studies.
Approximately 3 MBq (~80 μCi) of 64Cu-NOTA mAb was incubated with 1.2 × 108 platelets (15 minutes, room temperature). Before injection into mice, platelets were washed and resuspended in modified Tyrodes buffer. 4 × 107 Caldaggef1+/– Rasa3hlb/hlb platelets or Caldaggef1+/– Rasa3+/+ control platelets were injected into C57BL/6 mice (n = 3 per group). Organs and blood were harvested 24 hours after platelet transfusion. Each tissue was counted for radioactivity using a Capintec CRC-55tW gamma-counter.
Aggregometry
Washed platelets were resuspended at a concentration of 2 × 108 platelets/ml in modified Tyrode’s Buffer containing 0.35% BSA (fraction V, Sigma-Aldrich) and 1 mM CaCl2. The experiment was performed at 37°C in the presence of 50 μg/ml human fibrinogen type I (Sigma-Aldrich) and under stirring conditions (1,200 rpm). Where indicated, platelets were incubated for 10 minutes with 100 μM 2-MeSAMP (P2Y12 inhibitor) or 200 nM wortmannin (PI3K inhibitor, Cayman Chemical). Platelets were stimulated with fibrillar collagen type I from Chrono-log, α-thrombin from Enzyme Research Laboratories, U46619 from Cayman Chemical, ADP and epinephrine from Sigma-Aldrich, Par4p from GL Biochem Inc., and MRS 2365 from Tocris Bioscience. Light transmission was recorded until it reached a plateau on a Chrono-log 4-channel optical aggregation system (Chrono-log).
RAP1 and AKT activation assay
Washed platelets (4 × 108 platelets per sample) were stimulated with 250 μM Par4p for 3 minutes at 37°C in a standard aggregometer. Reactions were stopped with ice-cold 2x lysis buffer (100 mM Tris/HCl, pH 7.4, 400 mM NaCl, 5 mM MgCl2, 2% Nonidet P-40, 20% glycerol, and protease inhibitor cocktail lacking ethylenediaminetetraacetic acid). Cell lysis was completed on ice for 15 minutes. 50 μl of sample was immediately solubilized in sample buffer (75 mM Tris/HCl, pH 6.8, 10% sodium dodecyl sulfate, 5% 2-Mercaptoethanol, 0.004% Bromophenol blue) for the detection of phosphorylated AKT, total AKT, and total RAP1 levels (see below). The cell lysates were incubated for 45 minutes with RalGDS-RBD beads (Millipore) to pull-down RAP1-GTP (24). After 3 washing steps the pellets were solubilized in sample buffer for the detection of active RAP1 by immunoblot. Whole cell lysates for the detection of RASA3 were obtained by lysing washed platelets in ice-cold 2x lysis buffer.
Western blotting
Proteins were separated by SDS–polyacrylamide gel electrophoresis on 4%–20% gradient gels and transferred to polyvinylidene fluoride membranes (Millipore). Standard Western blotting procedures were used. RAP1 (α-RAP1 clone 121, Santa Cruz Biotechnology) and RASA3 (rabbit polyclonal α-mouse RASA3 was generated as described recently, ref. 33) proteins were detected using HRP-conjugated secondary antibodies and visualized with Enhanced Chemiluminescense (Pierce). AKT (α-AKT, clone 40D4, Cell Signaling Technology) and phospho-AKT (α-phospho-AKT Ser473, catalog no. 9271, Cell Signaling Technology) were detected using IRDye 680–conjugated goat anti-mouse and IRDye 800–conjugated goat anti-rabbit secondary antibodies (Li-Cor Biosystems), respectively, and visualized with the Odyssey Infrared Imaging System (Li-Cor Biosystems).
Laser-induced injury of the saphenous vein
Mice (8–12 weeks of age) were anesthetized by intraperitoneal injection of ketamine (100 mg/kg) and xylazine (10 mg/kg) (Med-Vet International). Alexa Fluor 488–labeled antibodies against GPIX were administered through retro-orbital injection. The saphenous vein was located, and baseline fluorescence was recorded for 30 seconds using a Zeiss Axio Examiner Z1 microscope (Intelligent Imaging Innovations) equipped with a 20x/1 numerical aperture water immersion objective lens. Injury to the endothelium was initiated using an Ablate! photoablation system equipped with an attenuable 532-nm pulse laser (Intelligent Imaging Innovations), and fluorescence intensity was recorded for 270 seconds. Throughout the experiment, constant perfusion drip was maintained on the exposed saphenous vein with a physiologic salt solution containing 132 mmol/l NaCl, 4.7 mmol/l KCl, 1.2 mmol/l MgSO4, 2 mmol/l CaCl2, and 18 mmol/l NaHCO3 (pH 7.4), which was bubbled with 5% CO2/95% N2 15 minutes prior to the start of the experiment. The physiological salt solution was maintained at 37°C via a Sloflo In-line solution heater (SF-28) and single channel heater controller (model TC-324B, Warner Instruments). All data were recorded and analyzed using Slidebook 5.0 software (Intelligent Imaging Innovations).
Statistics
Results are reported as mean ± SEM, and statistical significance was assessed by 2-tailed Student’s t test, unless indicated otherwise. A P value of less than 0.05 was considered significant.
Study approval
Mice were housed in the University of North Carolina at Chapel Hill, The Jackson Laboratory, and Case Western Reserve University animal facilities. Experimental procedures were approved by the University of North Carolina at Chapel Hill, The Jackson Laboratory, and Case Western Reserve University Institutional Animal Care and Use Committees.
Supplementary Material
Acknowledgments
We thank Dale Cowley and the Animal Models Core for the generation of Rasa3 knockout mice, Agnieszka Cholka for providing excellent animal husbandry services, Heather Lavender and Charlene Santos for skillful assistance with animal studies, and Pieter Haviernik for assistance with bone marrow transplants. This work was supported by the European Hematology Association and the International Society of Thrombosis and Hemostasis (joint fellowship to L. Stefanini), the American Heart Association (12POST12040088 to Y. Boulaftali and 14EIA18910004 to W. Bergmeier), and NIH grants F31 CA174194 (to D.O. Kechele), 1KL2TR001109 (to M.C. Parrott), R01 HL094594, HL106009, HL121650 (to W. Bergmeier), HL66611 (to L.L. Peters), DK099156 (to K.M. Caron), and HL112311 (to A.S. Weyrich).
Footnotes
Note regarding evaluation of this manuscript: Manuscripts authored by scientists associated with Duke University, The University of North Carolina at Chapel Hill, Duke-NUS, and the Sanford-Burnham Medical Research Institute are handled not by members of the editorial board but rather by the science editors, who consult with selected external editors and reviewers.
Conflict of interest: The authors have declared that no conflict of interest exists.
Reference information:J Clin Invest. 2015;125(4):1419–1432. doi:10.1172/JCI77993.
References
- 1.Jackson SP. Arterial thrombosis — insidious, unpredictable and deadly. Nat Med. 2011;17(11):1423–1436. doi: 10.1038/nm.2515. [DOI] [PubMed] [Google Scholar]
- 2.Ware J, Corken A, Khetpal R. Platelet function beyond hemostasis and thrombosis. Curr Opin Hematol. 2013;20(5):451–456. doi: 10.1097/MOH.0b013e32836344d3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jenne CN, Urrutia R, Kubes P. Platelets: bridging hemostasis, inflammation, and immunity. Int J Lab Hematol. 2013;35(3):254–261. doi: 10.1111/ijlh.12084. [DOI] [PubMed] [Google Scholar]
- 4.Grozovsky R, Hoffmeister KM, Falet H. Novel clearance mechanisms of platelets. Curr Opin Hematol. 2010;17(6):585–589. doi: 10.1097/MOH.0b013e32833e7561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Féliste R, et al. Broad spectrum anti-platelet activity of ticlopidine and PCR 4099 involves the suppression of the effects of released ADP. Thrombosis Research. 1987;48(4):403–415. doi: 10.1016/0049-3848(87)90398-7. [DOI] [PubMed] [Google Scholar]
- 6.Cattaneo M, et al. Ticlopidine selectively inhibits human platelet responses to adenosine diphosphate. Thromb Haemost. 1991;66(6):694–699. [PubMed] [Google Scholar]
- 7.Gachet C, et al. The thienopyridine ticlopidine selectively prevents the inhibitory effects of ADP but not of adrenaline on cAMP levels raised by stimulation of the adenylate cyclase of human platelets by PGE1. Biochemical Pharmacology. 1990;40(12):2683–2687. doi: 10.1016/0006-2952(90)90587-B. [DOI] [PubMed] [Google Scholar]
- 8.Mills DC, et al. Clopidogrel inhibits the binding of ADP analogues to the receptor mediating inhibition of platelet adenylate cyclase. Arterioscler Thromb. 1992;12(4):430–436. doi: 10.1161/01.ATV.12.4.430. [DOI] [PubMed] [Google Scholar]
- 9.Trumel C, et al. A key role of adenosine diphosphate in the irreversible platelet aggregation induced by the PAR1-activating peptide through the late activation of phosphoinositide 3-kinase. Blood. 1999;94(12):4156–4165. [PubMed] [Google Scholar]
- 10.Kim S, Jin J, Kunapuli SP. Akt activation in platelets depends on Gi signaling pathways. J Biol Chem. 2004;279(6):4186–4195. doi: 10.1074/jbc.M306162200. [DOI] [PubMed] [Google Scholar]
- 11.Woulfe D, Jiang H, Mortensen R, Yang J, Brass LF. Activation of Rap1B by G(i) family members in platelets. J Biol Chem. 2002;277(26):23382–23390. doi: 10.1074/jbc.M202212200. [DOI] [PubMed] [Google Scholar]
- 12.Larson MK, et al. Identification of P2Y12-dependent and -independent mechanisms of glycoprotein VI-mediated Rap1 activation in platelets. Blood. 2003;101(4):1409–1415. doi: 10.1182/blood-2002-05-1533. [DOI] [PubMed] [Google Scholar]
- 13.Lova P, Paganini S, Sinigaglia F, Balduini C, Torti M. A Gi-dependent pathway is required for activation of the small GTPase Rap1B in human platelets. J Biol Chem. 2002;277(14):12009–12015. doi: 10.1074/jbc.M111803200. [DOI] [PubMed] [Google Scholar]
- 14.Frische EW, Zwartkruis FJT. Rap1, a mercenary among the Ras-like GTPases. Dev Biol. 2010;340(1):1–9. doi: 10.1016/j.ydbio.2009.12.043. [DOI] [PubMed] [Google Scholar]
- 15.Chrzanowska-Wodnicka M, Smyth SS, Schoenwaelder SM, Fischer TH, White GC. Rap1b is required for normal platelet function and hemostasis in mice. J Clin Invest. 2005;115(3):680–687. doi: 10.1172/JCI22973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Stefanini L, et al. Rap1-Rac1 circuits potentiate platelet activation. Arterioscler Thromb Vasc Biol. 2012;32(2):434–441. doi: 10.1161/ATVBAHA.111.239194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Stolla M, et al. The kinetics of αIIbβ3 activation determines the size and stability of thrombi in mice: implications for antiplatelet therapy. Blood. 2011;117(3):1005–1013. doi: 10.1182/blood-2010-07-297713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Crittenden JR, et al. CalDAG-GEFI integrates signaling for platelet aggregation and thrombus formation. Nat Med. 2004;10(9):982–986. doi: 10.1038/nm1098. [DOI] [PubMed] [Google Scholar]
- 19.Boudreaux MK, Schmutz SM, French PS. Calcium diacylglycerol guanine nucleotide exchange factor I (CalDAG-GEFI) gene mutations in a thrombopathic Simmental calf. Vet Pathol. 2007;44(6):932–935. doi: 10.1354/vp.44-6-932. [DOI] [PubMed] [Google Scholar]
- 20.Boudreaux MK, Catalfamo JL, Klok M. Calcium-diacylglycerol guanine nucleotide exchange factor I gene mutations associated with loss of function in canine platelets. Transl Res. 2007;150(2):81–92. doi: 10.1016/j.trsl.2007.03.006. [DOI] [PubMed] [Google Scholar]
- 21.Canault M, et al. Human CalDAG-GEFI gene (RASGRP2) mutation affects platelet function and causes severe bleeding. J Exp Med. 2014;211(7):1349–1362. doi: 10.1084/jem.20130477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Franke B, Akkerman JW, Bos JL. Rapid Ca2+-mediated activation of Rap1 in human platelets. EMBO J. 1997;16(2):252–259. doi: 10.1093/emboj/16.2.252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cifuni SM, Wagner DD, Bergmeier W. CalDAG-GEFI and protein kinase C represent alternative pathways leading to activation of integrin αIIbβ3 in platelets. Blood. 2008;112(5):1696–1703. doi: 10.1182/blood-2008-02-139733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Stefanini L, Roden RC, Bergmeier W. CalDAG-GEFI is at the nexus of calcium-dependent platelet activation. Blood. 2009;114(12):2506–2514. doi: 10.1182/blood-2009-04-218768. [DOI] [PubMed] [Google Scholar]
- 25.Bergmeier W, Stefanini L. Novel molecules in calcium signaling in platelets. J Thromb Haemost. 2009;7(suppl 1):187–190. doi: 10.1111/j.1538-7836.2009.03379.x. [DOI] [PubMed] [Google Scholar]
- 26.Stefanini L, Bergmeier W. CalDAG-GEFI and platelet activation. Platelets. 2010;21(4):239–243. doi: 10.3109/09537101003639931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Franke B, et al. Sequential regulation of the small GTPase Rap1 in human platelets. Mol Cell Biol. 2000;20(3):779–785. doi: 10.1128/MCB.20.3.779-785.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lova P, et al. A selective role for phosphatidylinositol 3,4,5-trisphosphate in the Gi-dependent activation of platelet Rap1B. J Biol Chem. 2003;278(1):131–138. doi: 10.1074/jbc.M204821200. [DOI] [PubMed] [Google Scholar]
- 29.Schultess J, Danielewski O, Smolenski AP. Rap1GAP2 is a new GTPase-activating protein of Rap1 expressed in human platelets. Blood. 2005;105(8):3185–3192. doi: 10.1182/blood-2004-09-3605. [DOI] [PubMed] [Google Scholar]
- 30.Rowley JW, et al. Genome-wide RNA-seq analysis of human and mouse platelet transcriptomes. Blood. 2011;118(14):e101–e111. doi: 10.1182/blood-2011-03-339705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Burkhart JM, et al. The first comprehensive and quantitative analysis of human platelet protein composition allows the comparative analysis of structural and functional pathways. Blood. 2012;120(15):e73–e82. doi: 10.1182/blood-2012-04-416594. [DOI] [PubMed] [Google Scholar]
- 32.Simon LM, et al. Human platelet microRNA-mRNA networks associated with age and gender revealed by integrated plateletomics. Blood. 2014;123(16):e37–e45. doi: 10.1182/blood-2013-12-544692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Blanc L, et al. Critical function for the Ras-GTPase activating protein RASA3 in vertebrate erythropoiesis and megakaryopoiesis. Proc Natl Acad Sci U S A. 2012;109(30):12099–12104. doi: 10.1073/pnas.1204948109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Molina-Ortiz P, et al. Rasa3 controls megakaryocyte Rap1 activation, integrin signaling and differentiation into proplatelet. PLoS Genet. 2014;10(6):e1004420. doi: 10.1371/journal.pgen.1004420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cullen PJ, et al. Identification of a specific Ins(1,3,4,5)P4-binding protein as a member of the GAP1 family. Nature. 1995;376(6540):527–530. doi: 10.1038/376527a0. [DOI] [PubMed] [Google Scholar]
- 36.Hess PR, et al. Platelets mediate lymphovenous hemostasis to maintain blood-lymphatic separation throughout life. J Clin Invest. 2014;124(1):273–284. doi: 10.1172/JCI70422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bertozzi CC, et al. Platelets regulate lymphatic vascular development through CLEC-2-SLP-76 signaling. Blood. 2010;116(4):661–670. doi: 10.1182/blood-2010-02-270876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Carramolino L, et al. Platelets play an essential role in separating the blood and lymphatic vasculatures during embryonic angiogenesis. Circ Res. 2010;106(7):1197–1201. doi: 10.1161/CIRCRESAHA.110.218073. [DOI] [PubMed] [Google Scholar]
- 39.Uhrin P, et al. Novel function for blood platelets and podoplanin in developmental separation of blood and lymphatic circulation. Blood. 2010;115(19):3997–4005. doi: 10.1182/blood-2009-04-216069. [DOI] [PubMed] [Google Scholar]
- 40.Kumar P, Henikoff S, Ng PC. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc. 2009;4(7):1073–1081. doi: 10.1038/nprot.2009.86. [DOI] [PubMed] [Google Scholar]
- 41.Li B, et al. Automated inference of molecular mechanisms of disease from amino acid substitutions. Bioinformatics. 2009;25(21):2744–2750. doi: 10.1093/bioinformatics/btp528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Shivdasani RA, Schulze H. Culture, expansion, and differentiation of murine megakaryocytes. Curr Protoc Immunol. 2005;Chapter 22:Unit 22F.6. doi: 10.1002/0471142735.im22f06s67. [DOI] [PubMed] [Google Scholar]
- 43.Chhatriwala M, et al. Induction of novel agonist selectivity for the ADP-activated P2Y1 receptor versus the ADP-activated P2Y12 and P2Y13 receptors by conformational constraint of an ADP analog. J Pharmacol Exp Ther. 2004;311(3):1038–1043. doi: 10.1124/jpet.104.068650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jin J, Kunapuli SP. Coactivation of two different G protein-coupled receptors is essential for ADP-induced platelet aggregation. Proc Natl Acad Sci U S A. 1998;95(14):8070–8074. doi: 10.1073/pnas.95.14.8070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Stalker TJ, et al. Hierarchical organization in the hemostatic response and its relationship to the platelet-signaling network. Blood. 2013;121(10):1875–1885. doi: 10.1182/blood-2012-09-457739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Michelson AD. P2Y12 antagonism: promises and challenges. Arterioscler Thromb Vasc Biol. 2008;28(3):s33–s38. doi: 10.1161/ATVBAHA.107.160689. [DOI] [PubMed] [Google Scholar]
- 47.Lockyer PJ, et al. Distinct subcellular localisations of the putative inositol 1,3,4,5-tetrakisphosphate receptors GAP1IP4BP and GAP1m result from the GAP1IP4BP PH domain directing plasma membrane targeting. Curr Biol. 1997;7(12):1007–1010. doi: 10.1016/S0960-9822(06)00423-4. [DOI] [PubMed] [Google Scholar]
- 48.Cozier GE, et al. GAP1IP4BP contains a novel group I pleckstrin homology domain that directs constitutive plasma membrane association. J Biol Chem. 2000;275(36):28261–28268. doi: 10.1074/jbc.M000469200. [DOI] [PubMed] [Google Scholar]
- 49.El-Daher SS, et al. Distinct localization and function of (1,4,5)IP(3) receptor subtypes and the (1,3,4,5)IP(4) receptor GAP1(IP4BP) in highly purified human platelet membranes. Blood. 2000;95(11):3412–3422. [PubMed] [Google Scholar]
- 50.Han J, et al. Reconstructing and deconstructing agonist-induced activation of integrin alphaIIbbeta3. Curr Biol. 2006;16(18):1796–1806. doi: 10.1016/j.cub.2006.08.035. [DOI] [PubMed] [Google Scholar]
- 51.Wang J, Richards DA. Segregation of PIP2 and PIP3 into distinct nanoscale regions within the plasma membrane. Biol Open. 2012;1(9):857–862. doi: 10.1242/bio.20122071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gutiérrez-Herrero S, et al. C3G transgenic mouse models with specific expression in platelets reveal a new role for C3G in platelet clotting through its GEF activity. Biochim Biophys Acta. 2012;1823(8):1366–1377. doi: 10.1016/j.bbamcr.2012.05.021. [DOI] [PubMed] [Google Scholar]
- 53.Mitchell L, Hobbs GA, Aghajanian A, Campbell SL. Redox regulation of Ras and Rho GTPases: mechanism and function. Antioxid Redox Signal. 2013;18(3):250–258. doi: 10.1089/ars.2012.4687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Baker R, et al. Site-specific monoubiquitination activates Ras by impeding GTPase-activating protein function. Nat Struct Mol Biol. 2013;20(1):46–52. doi: 10.1038/nsmb.2430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Edreira MM, et al. Phosphorylation-induced conformational changes in Rap1b: allosteric effects on switch domains and effector loop. J Biol Chem. 2009;284(40):27480–27486. doi: 10.1074/jbc.M109.011312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Grosse J, et al. An EF hand mutation in Stim1 causes premature platelet activation and bleeding in mice. J Clin Invest. 2007;117(11):3540–3550. doi: 10.1172/JCI32312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Varga-Szabo D, Braun A, Nieswandt B. STIM and Orai in platelet function. Cell Calcium. 2011;50(3):270–278. doi: 10.1016/j.ceca.2011.04.002. [DOI] [PubMed] [Google Scholar]
- 58.Iwig JS, et al. Structural analysis of autoinhibition in the Ras-specific exchange factor RasGRP1. eLife. 2013;2:e00813. doi: 10.7554/eLife.00813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Stenberg PE, Levin J. Ultrastructural analysis of acute immune thrombocytopenia in mice: dissociation between alterations in megakaryocytes and platelets. J Cell Physiol. 1989;141(1):160–169. doi: 10.1002/jcp.1041410124. [DOI] [PubMed] [Google Scholar]
- 60.Stenberg PE, Levin J, Baker G, Mok Y, Corash L. Neuraminidase-induced thrombocytopenia in mice: effects on thrombopoiesis. J Cell Physiol. 1991;147(1):7–16. doi: 10.1002/jcp.1041470103. [DOI] [PubMed] [Google Scholar]
- 61.Kortum RL, Rouquette-Jazdanian AK, Samelson LE. Ras and extracellular signal-regulated kinase signaling in thymocytes and T cells. Trends Immunol. 2013;34(6):259–268. doi: 10.1016/j.it.2013.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Yasuda T, Kurosaki T. Regulation of lymphocyte fate by Ras/ERK signals. Cell Cycle. 2008;7(23):3634–3640. doi: 10.4161/cc.7.23.7103. [DOI] [PubMed] [Google Scholar]
- 63.Shock DD, He K, Wencel-Drake JD, Parise LV. Ras activation in platelets after stimulation of the thrombin receptor, thromboxane A2 receptor or protein kinase C. Biochem J. 1997;321(pt 2):525–530. doi: 10.1042/bj3210525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Tulasne D, Bori T, Watson SP. Regulation of RAS in human platelets. Evidence that activation of RAS is not sufficient to lead to ERK1-2 phosphorylation. Eur J Biochem. 2002;269(5):1511–1517. doi: 10.1046/j.1432-1033.2002.02798.x. [DOI] [PubMed] [Google Scholar]
- 65.Chen Y, et al. A critical role of Rap1b in B-cell trafficking and marginal zone B-cell development. Blood. 2008;111(9):4627–4636. doi: 10.1182/blood-2007-12-128140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Gieger C, et al. New gene functions in megakaryopoiesis and platelet formation. Nature. 2011;480(7376):201–208. doi: 10.1038/nature10659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Michelson AD, et al. Evidence that pre-existent variability in platelet response to ADP accounts for ‘clopidogrel resistance’. J Thromb Haemost. 2007;5(1):75–81. doi: 10.1111/j.1538-7836.2006.02234.x. [DOI] [PubMed] [Google Scholar]
- 68.Tiedt R, Schomber T, Hao-Shen H, Skoda RC. Pf4-Cre transgenic mice allow the generation of lineage-restricted gene knockouts for studying megakaryocyte and platelet function in vivo. Blood. 2007;109(4):1503–1506. doi: 10.1182/blood-2006-04-020362. [DOI] [PubMed] [Google Scholar]
- 69.Svenson KL, Bogue MA, Peters LL. Invited review: Identifying new mouse models of cardiovascular disease: a review of high-throughput screens of mutagenized and inbred strains. J Appl Physiol. 2003;94(4):1650–1659. doi: 10.1152/japplphysiol.01029.2003. [DOI] [PubMed] [Google Scholar]
- 70.Kienast J, Schmitz G. Flow cytometric analysis of thiazole orange uptake by platelets: a diagnostic aid in the evaluation of thrombocytopenic disorders. Blood. 1990;75(1):116–121. [PubMed] [Google Scholar]
- 71.Bergmeier W, et al. Flow cytometric detection of activated mouse integrin αIIbβ3 with a novel monoclonal antibody. Cytometry. 2002;48(2):80–86. doi: 10.1002/cyto.10114. [DOI] [PubMed] [Google Scholar]
- 72.Stefanini L, et al. A talin mutant that impairs talin-integrin binding in platelets decelerates αIIbβ3 activation without pathological bleeding. Blood. 2014;123(17):2722–2731. doi: 10.1182/blood-2013-12-543363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Shi DS, et al. Proteasome function is required for platelet production. J Clin Invest. 2014;124(9):3757–3766. doi: 10.1172/JCI75247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Lojda Z, Gossrau R, Schiebler TH. Enzyme Histochemistry. New York, New York, USA: Springer-Verlag; 1979. [Google Scholar]
- 75. Sheehan DC, Hrapchak BB. Theory And Practice Of Histotechnology. Columbus, Ohio, USA: Battelle Press; 1987. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.