

Abstract
Macrocytic anemia with abnormal erythropoiesis is a common feature of megaloblastic anemias, congenital dyserythropoietic anemias, and myelodysplastic syndromes. Here, we characterized a family with multiple female individuals who have macrocytic anemia. The proband was noted to have dyserythropoiesis and iron overload. After an extensive diagnostic evaluation that did not provide insight into the cause of the disease, whole-exome sequencing of multiple family members revealed the presence of a mutation in the X chromosomal gene ALAS2, which encodes 5′-aminolevulinate synthase 2, in the affected females. We determined that this mutation (Y365C) impairs binding of the essential cofactor pyridoxal 5′-phosphate to ALAS2, resulting in destabilization of the enzyme and consequent loss of function. X inactivation was not highly skewed in wbc from the affected individuals. In contrast, and consistent with the severity of the ALAS2 mutation, there was a complete skewing toward expression of the WT allele in mRNA from reticulocytes that could be recapitulated in primary erythroid cultures. Together, the results of the X inactivation and mRNA studies illustrate how this X-linked dominant mutation in ALAS2 can perturb normal erythropoiesis through cell-nonautonomous effects. Moreover, our findings highlight the value of whole-exome sequencing in diagnostically challenging cases for the identification of disease etiology and extension of the known phenotypic spectrum of disease.
Introduction
Macrocytic anemia and abnormal dysplastic erythropoiesis (herein referred to as dyserythropoiesis) are features found in a number of congenital and acquired conditions, including congenital dyserythropoietic anemias (CDAs), specific mitochondrial disorders, myelodysplastic syndromes (MDS), and megaloblastic anemias (1, 2). While numerous etiologies are known for this class of disorders, some cases lack a clear explanation (3). An improved understanding of the etiology of these disorders will provide additional insight into the mechanisms of abnormal blood cell production in humans (4).
We hypothesized that certain cases of this type of anemia either represent new genetic conditions or, alternatively, are due to the phenotypic variation in genes previously associated with anemia. We identified a female proband with a macrocytic dyserythropoietic anemia since childhood and evidence of iron overload, who had undergone an extensive, but unrevealing, clinical evaluation. The proband’s sister and mother were also noted to have macrocytic anemia, supporting a potential genetic etiology. We therefore performed whole-exome sequencing and functional studies to identify and understand the basis of the anemia observed in this family.
Results and Discussion
The proband (family member II-2) (Figure 1E) was a 32-year-old woman with a history of atrial septal defect who had been noted to have an anemia in childhood of unclear etiology. She had come to clinical attention 7 years earlier, when a routine blood count during her first pregnancy revealed an anemia with a hematocrit of 24% to 26% and a mean corpuscular volume of 114 to 122 femtoliters (Supplemental Table 1; supplemental material available online with this article; doi:10.1172/JCI78619DS1) that was unresponsive to iron and vitamin B12 supplementation. Delivery occurred at full term and was complicated by placenta accreta with concomitant blood loss, and the proband required an rbc transfusion for the first time. Three years later, the proband delivered healthy twins and experienced a massive hemorrhage 2 weeks after the delivery, resulting in a drop of her hematocrit to 17% that required an additional transfusion.
Figure 1. Identification of an ALAS2 mutation in a family with macrocytic anemia and dyserythropoiesis.

(A) BM aspirate from the proband (II-2) with erythroid hyperplasia. (B) Prussian blue staining revealed rare erythroblasts with siderotic granules. (C) BM aspirate with dyserythropoiesis. (D) Core biopsy demonstrating erythroid hyperplasia and dyserythropoiesis. (E) Pedigree of the family, with affected individuals highlighted by half-filled circles. Arrows highlight those individuals who underwent whole-exome sequencing. (F) Sanger sequencing traces around X chromosome position 55042086 (hg19 coordinates) in 2 affected individuals. Original magnification, ×100 (A and B) and ×2000 (C and D).
At that time, it was noted that the proband had a macrocytic anemia and an elevated serum ferritin level (800 ng/ml), prompting referral to a hematologist. A BM biopsy revealed trilineage hematopoiesis with dyserythropoiesis (Figure 1, A–D). The proband had a normal BM karyotype and MDS/acute myeloid leukemia FISH panel (evaluating chromosomes 5, 7, 8, 13, 20, and X). According to the report, Prussian blue reaction on the initial BM aspirate showed no excess iron or sideroblasts. A liver biopsy revealed an iron index of 11 (normal, <1), and HFE genetic testing revealed no mutations. Therapeutic phlebotomy was attempted, but was limited by symptomatic anemia.
Laboratory testing of the proband, her sister (family member II-4), and mother (family member I-2) demonstrated rbc macrocytosis (Figure 1E and Supplemental Table 1). The brother and daughters of the proband had normal complete blood counts (Figure 1E). T2* MRI was performed on the proband and revealed an estimated liver iron concentration of greater than 350 μmol/g (normal, <35 μmol/g). In addition, the proband had a horseshoe kidney and a mildly enlarged spleen, measuring 14.8 cm. A trial with the iron chelator deferasirox was attempted but was not tolerated, and therefore another trial of phlebotomy with a target hemoglobin level of 10 g/dl was initiated. Since the clinical features of this patient suggested the possibility of CDA, targeted sequencing of CDAN1, SEC23B, and KLF1 was performed, but no mutations were identified (5). The proband and family members then consented to participate in a whole-exome sequencing study (6, 7).
We used an automated pipeline to identify novel coding and the loss-of-function (LOF) variants found in the 3 affected individuals from this family (I-1, II-2, and II-4), but not in the unaffected daughter (III-1) (6). Each individual had a total of approximately 200 novel coding and LOF variants (Supplemental Table 2). A single notable variant fit the model of complete penetrance and was found to be a coding mutation in the X chromosomal gene ALAS2. This A-to-G variant was found at position 55042086 on the X chromosome (hg19 coordinates), resulting in a coding change of Y365C in the ALAS2 protein (Figure 1F and Supplemental Figure 1).
The enzyme encoded by the ALAS2 gene plays a critical role in heme biosynthesis, and mutations in this gene are known to cause a microcytic sideroblastic anemia in male carriers and in females with skewed X inactivation (X-linked sideroblastic anemia [XLSA]; Online Mendelian Inheritance in Man [OMIM] entry no. 300571) (8–15). However, mutations in this gene have only rarely been associated with macrocytic anemia or dyserythropoiesis (14). We confirmed that this mutation was found in all affected individuals through Sanger sequencing (Figure 1F), while the unaffected individuals in this family lacked this mutation, further supporting its causality for the phenotype. Additionally, another BM aspirate from the proband revealed occasional cells with siderotic granules, while nearly all erythroid cells showed dyserythropoietic features, even in the absence of such granules (Figure 1, A–D).
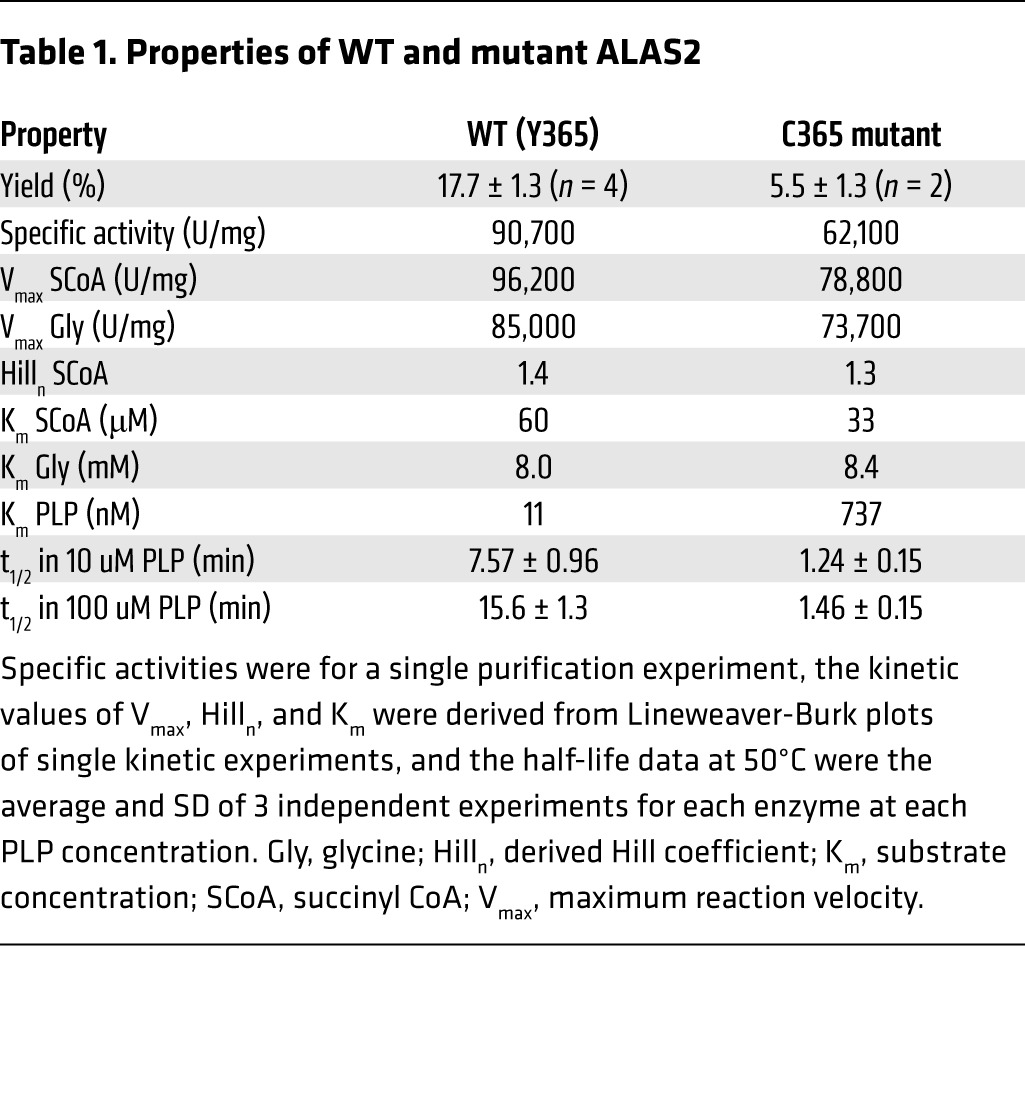
By modeling this novel ALAS2 Y365C mutation in the structure of the Rhodobacter capsulatus homolog, we noted that Y365 fits within a hydrophobic core critical for binding the essential cofactor pyridoxal 5′-phosphate (PLP) (Figure 2A and ref. 16). The C365 mutation would disrupt this hydrophobic core and would therefore be predicted to reduce the ability of ALAS2 to bind to PLP (Figure 2A). Consistent with this idea, we found that the ALAS2 C365 mutant purified to homogeneity had a 70-fold lower affinity for PLP than did the WT enzyme (Table 1). The ALAS2 C365 mutant had enzyme kinetics similar to that of the WT enzyme (Table 1). However, the mutant enzyme was unstable, consistent with the notion that PLP binding plays a key role in stabilizing ALAS2 (17, 18). When the enzymes were expressed as maltose-binding protein (MBP) fusions, the yield of the mutant enzyme in the initial crude extract was only 3.3% of the WT enzyme yield (Figure 2B). After removal of the MBP and purification by liquid chromatography (Figure 2C), only 5.5% of the mutant enzyme activity was recovered compared with 17.9% of the WT enzyme activity (Table 1). Overall, the yield relative to that of WT was only 0.2% — lower than the residual activities in classic cases of XLSA (15). Furthermore, the mutant enzyme was 6-fold less stable than the WT enzyme at 50°C (Table 1). Under conditions of increased PLP, the mutant was 11-fold less stable because the PLP stabilized the WT, but not the mutant, enzyme (Table 1). Together, these findings indicate that the Y365C mutation markedly impairs PLP binding, which may account for some or all of the substantially reduced stability of the ALAS2 enzyme. Severe LOF due to this mutation may explain why no male carriers were identified in the family (Figure 1E) and why the proband’s anemia did not improve with pyridoxine therapy.
Figure 2. Severe LOF with the ALAS2 Y365C mutation and lack of highly skewed X inactivation in female mutation carriers.

(A) Model of ALAS2 shows PLP highlighted in blue and the Y or C amino acid at position 365 highlighted in red. (B) SDS-PAGE gel of WT and mutant ALAS2. Lanes 1 and 8 contain the protein standards, while lanes 2–4 and 5–7 contain WT and mutant ALAS2 protein samples, respectively. Lanes 2 and 5 show partially purified samples after amylose affinity chromatography, lanes 3 and 6 show results after factor Xa digestion, and lanes 4 and 7 show results after gel filtration chromatography. Thin vertical lines in this composite figure separate noncontiguous lanes in the 2 original gels. (C) Chromatographic profiles for purification of WT and mutant ALAS2 by size exclusion (absorbance at 280 nm is shown in milliabsorbance units [mAU]). (D) Quantification of HUMARA results in all affected individuals showing WT and mutant X chromosomes. (E) Sanger sequencing traces of genomic DNA (gDNA) and cDNA derived from reticulocyte mRNA from the proband (II-2) for ALAS2, with an arrow highlighting the mutation.
Table 1. Properties of WT and mutant ALAS2.
As only female carriers were found to be affected, we reasoned that there may have been highly skewed X inactivation present in the individuals in this family. We were surprised to find that 62%–82% of the active X chromosomes in the proband and other affected family members contained the ALAS2 mutant, as assessed by the human androgen receptor gene polymorphism assay (HUMARA) for X inactivation in whole-blood genomic DNA (Figure 2D). These values were all within 2 SD of the mean X inactivation ratios observed in healthy adult females (19). We obtained independent confirmation for a lack of highly skewed X inactivation by digesting genomic DNA from the affected family members with the methylation-sensitive restriction enzyme HpaII and noted no change in the intensity of the ALAS2 mutation by Sanger sequencing (Figure 1F and Supplemental Figure 2). The proband, who was the most severely affected, had the greatest extent of skewing toward the mutant allele, at 82% (Figure 2D). Collectively, these findings suggest that the presence of some erythroid cells expressing WT ALAS2 in the patients’ BM was insufficient to compensate for the presence of erythroid cells expressing the ALAS2 mutation.
While most ALAS2 mutations causing sideroblastic anemia are partial LOF alleles affecting heme biosynthesis (12, 13, 15), complete ALAS2 LOF in mouse models results in an early block of erythropoiesis at approximately the proerythroblast or basophilic erythroblast stages (20). Consistent with these results, when we sequenced peripheral blood reticulocyte mRNA samples, we only detected the presence of transcripts with the WT allele, while the mutant allele was readily detectable in genomic DNA (Figure 2E). To confirm these findings, peripheral blood mononuclear cells from the proband were cultured in medium that promotes erythroid differentiation. We found that as the cells transitioned from the early progenitor stages to terminally differentiated erythroblasts, there was selection against cells expressing the mutant ALAS2 mRNA, leaving only cells expressing mRNA from the WT allele (Figure 3A). Thus, the X inactivation and mRNA sequencing studies show that while there is initially a mixed population of erythroid progenitors and early precursors that express both ALAS2 alleles, as cells undergo erythroid differentiation, there is a loss of nonviable cells expressing the mutant allele, with a survival advantage for cells expressing the WT allele (Figure 3B and ref. 14). This pressure to produce a normal output of erythroid cells may affect differentiation and result in skipped cell divisions to ensure adequate erythroid output, which would explain the macrocytosis and dyserythropoiesis observed among those cells expressing WT ALAS2 alleles (ref. 21 and Figure 3B).
Figure 3. X-linked dominant LOF ALAS2 mutations can result in macrocytosis and dyserythropoiesis through cell-nonautonomous effects.

(A) Sequencing of cDNA derived from cultured erythroid cells from the proband showed skewing toward the WT ALAS2 allele at the late stages of erythroid differentiation. Cells began as progenitors (day 5), became intermediate erythroblasts (day 9), and transitioned to orthochromatic erythroblasts (day 14). By late erythropoiesis, only a trace amount of the mutant allele was detectable. (B) Model showing how developing erythroid progenitors and precursors compete for space within the BM. The active X chromosome expressed in each group of cells (brown and pink for the mutant and WT, respectively) is shown in blue, with some cells expressing the mutant (red) and others expressing the WT (green) allele.
It is unclear whether there is a connection between the ALAS2 mutation and the proband’s congenital defects in other tissues (i.e., atrial septal defect, horseshoe kidney) and her abnormal placentation, which were not present in other family members. We examined known genetic causes of iron overload in the proband, including mutations in the ALAS2, HAMP, HFE, HFE2, SLC40A1, and TFR2 genes, and only the mutation in ALAS2 was identified. We also observed low levels of hepcidin in the blood of the proband (Supplemental Table 1), consistent with the expected reduction of hepcidin that occurs in the context of ineffective erythropoiesis (22).
Our findings illustrate how cell-nonautonomous defects could arise as a result of a mutant ALAS2 allele on one X chromosome, even in the absence of significantly skewed X inactivation. Few examples exist of such mutations in human disease (23). We have only identified a single family with this phenotype, but as more cases of macrocytic dyserythropoietic anemia are studied, it is likely that other similar mutations in ALAS2 will be identified. While XLSA is typically a dimorphic, microcytic anemia in females, sufficiently deleterious ALAS2 mutations can result in macrocytic anemia without a dimorphic population in such females. We illustrate how defining such mutations using modern genomic sequencing can help lead to further insight into human disease and hematopoiesis (7).
Methods
Further information can be found in the Supplemental Methods.
The whole-exome sequencing data on the family members are available in the dbGaP database (http://www.ncbi.nlm.nih.gov/gap) under the accession number phs000474.v2.p1.
Statistics.
All pairwise comparisons were performed using the 2-tailed Student’s t test, unless otherwise indicated. Differences were considered significant if the P value was less than 0.05.
Study approval.
All family members provided written informed consent to participate in this study. The IRBs of the Dana-Farber Cancer Institute, Boston Children’s Hospital, and Massachusetts Institute of Technology approved the study protocols.
Supplementary Material
Acknowledgments
We are grateful to the family for their interest in this study. We thank D. Nathan, S. Lux, C. Brugnara, F. Briccetti, M. Westerman, J. Hirschhorn, G. Evrony, R. Do, D. Bennett, and M. Burns for their valuable advice and support. This work was supported by NIH grants R21 HL120791 and R01 DK103794 (to V.G. Sankaran) and by a grant from the Edward P. Evans Foundation (to D.P. Steensma).
Footnotes
Conflict of interest: The authors have delcared that no conflict of interest exists.
Reference information:J Clin Invest. 2015;125(4):1665–1669. doi:10.1172/JCI78619.
Rafael Bejar’s present address is: University of California, San Diego, La Jolla, California 92093, USA.
References
- 1.Hoffbrand V, Provan D. ABC of clinical haematology. Macrocytic anaemias. BMJ. 1997;314(7078):430–433. doi: 10.1136/bmj.314.7078.430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Davenport J. Macrocytic anemia. Am Fam Physician. 1996;53(1):155–162. [PubMed] [Google Scholar]
- 3.Younes M, Dagher GA, Dulanto JV, Njeim M, Kuriakose P. Unexplained macrocytosis. South Med J. 2013;106(2):121–125. doi: 10.1097/SMJ.0b013e3182824cdf. [DOI] [PubMed] [Google Scholar]
- 4.Sankaran VG, Orkin SH. Genome-wide association studies of hematologic phenotypes: a window into human hematopoiesis. Curr Opin Genet Dev. 2013;23(3):339–344. doi: 10.1016/j.gde.2013.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Iolascon A, Heimpel H, Wahlin A, Tamary H. Congenital dyserythropoietic anemias: molecular insights and diagnostic approach. Blood. 2013;122(13):2162–2166. doi: 10.1182/blood-2013-05-468223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sankaran VG, et al. Exome sequencing identifies GATA1 mutations resulting in Diamond-Blackfan anemia. J Clin Invest. 2012;122(7):2439–2443. doi: 10.1172/JCI63597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sankaran VG, Gallagher PG. Applications of high-throughput DNA sequencing to benign hematology. Blood. 2013;122(22):3575–3582. doi: 10.1182/blood-2013-07-460337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cotter PD, Baumann M, Bishop DF. Enzymatic defect in “X-linked” sideroblastic anemia: molecular evidence for erythroid delta-aminolevulinate synthase deficiency. Proc Natl Acad Sci U S A. 1992;89(9):4028–4032. doi: 10.1073/pnas.89.9.4028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bottomley SS, May BK, Cox TC, Cotter PD, Bishop DF. Molecular defects of erythroid 5-aminolevulinate synthase in X-linked sideroblastic anemia. J Bioenerg Biomembr. 1995;27(2):161–168. doi: 10.1007/BF02110031. [DOI] [PubMed] [Google Scholar]
- 10.Cotter PD, et al. Four new mutations in the erythroid-specific 5-aminolevulinate synthase (ALAS2) gene causing X-linked sideroblastic anemia: increased pyridoxine responsiveness after removal of iron overload by phlebotomy and coinheritance of hereditary hemochromatosis. Blood. 1999;93(5):1757–1769. [PubMed] [Google Scholar]
- 11.Cazzola M, May A, Bergamaschi G, Cerani P, Rosti V, Bishop DF. Familial-skewed X-chromosome inactivation as a predisposing factor for late-onset X-linked sideroblastic anemia in carrier females. Blood. 2000;96(13):4363–4365. [PubMed] [Google Scholar]
- 12.Bergmann AK, et al. Systematic molecular genetic analysis of congenital sideroblastic anemia: evidence for genetic heterogeneity and identification of novel mutations. Pediatr Blood Cancer. 2010;54(2):273–278. doi: 10.1002/pbc.22244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fleming MD. Congenital sideroblastic anemias: iron and heme lost in mitochondrial translation. Hematology Am Soc Hematol Educ Program. 2011;2011:525–531. doi: 10.1182/asheducation-2011.1.525. [DOI] [PubMed] [Google Scholar]
- 14.Aivado M, et al. X-linked sideroblastic anemia associated with a novel ALAS2 mutation and unfortunate skewed X-chromosome inactivation patterns. Blood Cells Mol Dis. 2006;37(1):40–45. doi: 10.1016/j.bcmd.2006.04.003. [DOI] [PubMed] [Google Scholar]
- 15.Ducamp S, et al. Sideroblastic anemia: molecular analysis of the ALAS2 gene in a series of 29 probands and functional studies of 10 missense mutations. Hum Mutat. 2011;32(6):590–597. doi: 10.1002/humu.21455. [DOI] [PubMed] [Google Scholar]
- 16.Astner I, Schulze JO, van den Heuvel J, Jahn D, Schubert WD, Heinz DW. Crystal structure of 5-aminolevulinate synthase, the first enzyme of heme biosynthesis, and its link to XLSA in humans. EMBO J. 2005;24(18):3166–3177. doi: 10.1038/sj.emboj.7600792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bishop DF, Tchaikovskii V, Hoffbrand AV, Fraser ME, Margolis S. X-linked sideroblastic anemia due to carboxyl-terminal ALAS2 mutations that cause loss of binding to the β-subunit of succinyl-CoA synthetase (SUCLA2). J Biol Chem. 2012;287(34):28943–28955. doi: 10.1074/jbc.M111.306423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cotter PD, Rucknagel DL, Bishop DF. X-linked sideroblastic anemia: identification of the mutation in the erythroid-specific Δ-aminolevulinate synthase gene (ALAS2) in the original family described by Cooley. Blood. 1994;84(11):3915–3924. [PubMed] [Google Scholar]
- 19.Amos-Landgraf JM, et al. X chromosome-inactivation patterns of 1,005 phenotypically unaffected females. Am J Hum Genet. 2006;79(3):493–499. doi: 10.1086/507565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Harigae H, et al. Aberrant iron accumulation and oxidized status of erythroid-specific Δ-aminolevulinate synthase (ALAS2)-deficient definitive erythroblasts. Blood. 2003;101(3):1188–1193. doi: 10.1182/blood-2002-01-0309. [DOI] [PubMed] [Google Scholar]
- 21.Sankaran VG, et al. Cyclin D3 coordinates the cell cycle during differentiation to regulate erythrocyte size and number. Genes Dev. 2012;26(18):2075–2087. doi: 10.1101/gad.197020.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kautz L, Jung G, Valore EV, Rivella S, Nemeth E, Ganz T. Identification of erythroferrone as an erythroid regulator of iron metabolism. Nat Genet. 2014;46(7):678–684. doi: 10.1038/ng.2996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Migeon BR. The role of X inactivation and cellular mosaicism in women’s health and sex-specific diseases. JAMA. 2006;295(12):1428–1433. doi: 10.1001/jama.295.12.1428. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.