

Abstract
Influenza A virus poses a major threat to human health, causing outbreaks from time to time. Currently available vaccines employ inactivated viruses of different strains to provide protection against influenza virus infection. However, high mutation rates of influenza virus hemagglutinin (H) and neuraminidase (N) glycoproteins give rise to vaccine escape mutants. Thus, an effective vaccine providing protection against all strains of influenza virus would be a valuable asset. The ectodomain of matrix 2 protein (M2e) was found to be highly conserved despite mutations of the H and N glycoproteins. Hence, one to five copies of M2e were fused to the carboxyl-terminal end of the recombinant nodavirus capsid protein derived from Macrobrachium rosenbergii. The chimeric proteins harboring up to five copies of M2e formed nanosized virus-like particles approximately 30 nm in diameter, which could be purified easily by immobilized metal affinity chromatography. BALB/c mice immunized subcutaneously with these chimeric proteins developed antibodies specifically against M2e, and the titer was proportional to the copy numbers of M2e displayed on the nodavirus capsid nanoparticles. The fusion proteins also induced a type 1 T helper immune response. Collectively, M2e displayed on the nodavirus capsid nanoparticles could provide an alternative solution to a possible influenza pandemic in the future.
Keywords: matrix 2 ectodomain, nodavirus capsid, virus-like particle, fusion protein, subunit vaccine, immunogenicity
Introduction
Influenza A virus is responsible for the death of millions in human history. The first recorded influenza pandemic, commonly known as “Spanish flu”, claimed 50 million lives worldwide in 1918. The most recent pandemic, in 2009, also known as “swine flu”, was first detected in the USA. Nucleotide sequence analysis of the virus genome revealed that it contained genes from humans, birds, and pigs. A statistical study by Dawood et al1 estimated that there were 284,500 deaths during the swine flu pandemic. H1N1 was the culprit virus responsible for both these pandemic outbreaks. Unlike other strains of influenza virus, which preferentially infect the elderly, H1N1 also infects healthy young individuals, which makes it particularly dangerous. At present, there are still approximately 3–5 million people worldwide infected by influenza virus and develop severe illness annually, of which 250,000–500,000 die each year.2
Influenza A virus belongs to the Orthomyxoviridae family and has a genome comprising eight segmented, negative sense, single-stranded RNA molecules. In total, the genome encodes 14 proteins, of which three are of particular interest in vaccine development, ie, the hemagglutinin (H), neuraminidase (N), and matrix (M) proteins. Current licensed influenza vaccines are trivalent, and consist of inactivated influenza A virus H1N1, H3N2, and an influenza B virus. These seasonal vaccines are revised yearly, and their protective efficacy is about 60%–90%.3 A recently released quadrivalent influenza vaccine comes with an addition of influenza B subtype to the trivalent vaccine.4
These influenza vaccines provide protection in immunized individuals mainly through induction of specific antibodies against the H and N glycoproteins. The vaccines are highly effective, provided that the virus strains included in the vaccines are equivalent to the virus strains being protected against. However, due to the unusually high mutation rates of the H and N glycoproteins, a phenomenon known as “drifting”, these vaccines sometimes fail to provide protection against influenza virus infection. In addition, “shifting” may occur, whereby gene re-assortment takes place in a common host, between viruses which have infected two or more different species.5 With birds serving as an influenza gene reservoir, new strains of influenza virus can be introduced into the human gene pool at any time, which dramatically reduces the effectiveness of the current vaccines. In the worst scenario, a new pandemic outbreak can occur at any time, representing a “time bomb” for the human population.
Matrix 2 (M2) protein, originally discovered by Lamb et al6 is a type III transmembrane protein existing in a form of tetramer, which functions as a pH-regulated proton channel. M2 protein is crucial in the virus replication cycle, as it facilitates the acidic dissociation of RNA from matrix 1 protein during viral infection. It also stabilizes the budding site and membrane scission during release of virus progeny.7–9 Matrix 2 ectodomain (M2e), containing 23 amino acids, was found to be highly conserved in M2 protein, regardless of virus subtype.10 Thus, M2e is a potential candidate for development of a universal vaccine against the never-ending series of outbreaks of influenza virus.
M2 protein is present in minimal amounts in the virion, thus the seasonal influenza vaccines often do not induce a sufficient amount of protective antibodies against this protein. Feng et al11 demonstrated that influenza A virus infection induced a poor M2e antibody response. Therefore, in the present study, M2e was displayed on the capsid protein of Macrobrachium rosenbergii nodavirus (MrNV), which has been previously shown by Goh et al to self-assemble into nanosized virus-like particles (VLPs).12 However, the potential of MrNV VLPs as a vaccine carrier for an influenza virus epitope has yet to be studied. Therefore, the purpose of this study was to display M2e on MrNV nanoparticles and study the immune response in BALB/c mice.
Materials and methods
Synthesis of five copies of M2e coding region
Five copies of M2e (M2ex5) coding sequence was synthesized by overlapping extension polymerase chain reaction (PCR) with DreamTaq DNA polymerase (0.6 U, Thermo Scientific, Waltham, MA, USA). Four primers (5 pmol each, fragments 1–4, Table 1) were mixed in 20 μL of PCR reaction (10× DreamTaq buffer, 1 mM dNTP mix). The reaction was heated at 95°C (10 minutes), followed by 25 cycles of 95°C (1 minute) and 72°C (1 minute 20 seconds). A final extension was performed at 72°C for 10 minutes. The PCR product was analyzed on agarose gel (1.5%, w/v) and the PCR product of 400 bp was excised from the gel and purified using a Qiaquick gel extraction kit (Qiagen, Hilden, Germany).
Table 1.
Primer sequences
| Primer | Nucleotide sequences (5′ to 3′) |
|---|---|
| Fragment 1 | GGAGGAGGAAGCCTTCTAACCGAGGTCGAAACGCCTATCAGAAACGAATGGGGGTGCAGATGCAACGATTCAAGTGACGGTGGTGGTTCACTATTAACCGAAGTAGAAACACCAATAAGGAATGAG-3′ |
| Fragment 2 | CATTACAACGACAACCCCATTCATTACGAATTGGAGTCTCGACTTCCGTCAATAAAGATCCACCGCCGTCGCTAGAGTCATTGCAACGACATCCCCACTCATTCCTTATTGGTGTTTCTACTTCGGT-3′ |
| Fragment 3 | TTCGTAATGAATGGGGTTGTCGTTGTAATGATAGTTCTGATGGAGGTGGCAGTCTACTTACGGAGGTGGAAACTCCAATACGCAACGAGTGGGGCTGTCGCTGCAATGATAGCTCAGACGG-3′ |
| Fragment 4 | GCCCTCCACCGTCTGAACTGTCGTTACAACGGCAGCCCCATTCATTCCGGATCGGGGTCTCTACCTCTGTGAGCAGCGAACCGCCCCCGTCTGAGCTATCATTGCAGCGACAGCC-3′ |
| M2ex5-forward | GGAGGAGGAAGCCTTCTAA-3′ |
| M2ex5-reverse | GCCCTCCACCGTCTGAAC-3′ |
| *M2e1c-forward | CAATGATAGCGAATTCGGGGGCGGTTCGCTGCTC-3′ |
| *M2e3c-forward | CAATGACTCTGAATTCGGCGGTGGATCTTTATTGAC-3′ |
| *M2e5c-forward | CCATGGCCGCGAATTCGGAGGAGGAAGCCTTCTAAC-3′ |
| **M2enc-reverse | GGCCGCACTAAAGCTTGCCCTCCACCGTCTGAAC-3′ |
Notes:
Underlined sequences indicate EcoRI recognition site.
Underlined sequence indicates HindIII recognition site.
The purified PCR product was used as a template for another PCR reaction (25 μL) containing DreamTaq DNA polymerase (0.7 U, Thermo Scientific). M2ex5 forward primer (50 pmol; Table 1) and M2ex5 reverse primer (50 pmol, Table 1) were added to the reaction mixture (1 μL template, 10× DreamTaq buffer, 1 mM dNTP mix). The initial denaturation step was carried out at 95°C (10 minutes), followed by 25 cycles of 95°C (1 minute), 48°C (45 seconds), and 72°C (1 minute). Final extension was done at 72°C for 10 minutes. The 400 bp PCR product was analyzed on agarose gel and purified as described above. The purified PCR product was ligated to pGEM-T vector (Promega, WI, USA) using T4 DNA ligase (Promega) at 4°C overnight. The recombinant plasmid, namely pGEM-T-M2ex5, harboring five copies of the M2e coding region, was amplified in Escherichia coli XL-1 Blue.
Construction of expression vectors carrying one, three, and five copies of M2e coding regions
pGEM-T-M2ex5 was used as a template for synthesis of one copy (M2ex1), three copies (M2ex3), and five copies (M2ex5) of M2e coding regions. These coding regions contained EcoRI and HindIII restriction sites at their 5′ and 3′ ends, respectively. All the coding regions shared a common sequence at their 3′ ends, so were synthesized using the same reverse primer, ie, M2enc-reverse (Table 1). M2ex1, M2ex3, and M2ex5 were synthesized separately using PCR with forward primers, ie, M2e1c-forward, M2e3c-forward, and M2e5c-forward, respectively (Table 1). The PCR reactions (25 μL: 0.5 μL pGEM-T-M2ex5, 50 pmol primers; 10× DreamTaq buffer, 1 mM dNTP mix) were mixed with DreamTaq DNA polymerase (0.7 U). Initial denaturation was done at 95°C (10 minutes), with 30 cycles of 95°C (1 minute), 55°C (45 seconds), and 72°C (1 minute). A final extension was performed at 72°C for 10 minutes.
The PCR products were then extracted with phenol: chloroform (1:1), followed by EcoRI and HindIII digestion. pTrcHis-TARNA2, which encodes the nodavirus capsid (NvC) protein,12 was also digested with the same restriction endonucleases. The digested PCR products (inserts) and pTrcHis-TARNA2 (vector) were electrophoresed on 1% (w/v) agarose gel, purified using the Qiaquick gel extraction kit (Qiagen), and ligated with T4 DNA ligase at 4°C overnight. The ligation mixtures were then introduced into E. coli TOP10-competent cells via heat shock. The nucleotide sequences of all the three recombinant plasmids, ie, pNvC-M2ex1 (carries M2ex1), pNvC-M2ex3 (carries M2ex3), and pNvC-M2ex5 (carries M2ex5), were verified by DNA sequencing.
Expression and purification of fusion proteins
Single bacterial colonies harboring pTrcHis-TARNA2, pNvC-M2ex1, pNvC-M2ex3, and pNvC-M2ex5 were inoculated individually into Luria-Bertani broth (10 mL) containing ampicillin (100 μg/mL) and incubated at 37°C and 220 rpm. After overnight incubation, the cultures were transferred individually into Luria-Bertani broth (500 mL) with continued shaking under the same conditions until A600 reached 0.6–0.8. Expression of the recombinant proteins, NvC, NvC-M2ex1, NvC-M2ex3, and NvC-M2ex5, was induced by adding isopropyl β-D-1-thiogalactopyranoside (IPTG, 1 mM) at room temperature for 5 hours and 220 rpm. The cells were then centrifuged at 8,000× g for 5 minutes, and the cell pellets were resuspended in lysis buffer (10 mL, 25 mM 2-(4-[2-hydroxyethyl] piperazine-1-yl) ethanesulfonic acid (HEPES), 500 mM NaCl; pH 7.4). MgCl2 (4 mM) and freshly prepared lysozyme (0.2 mg/mL) were added to the cell suspensions and incubated at room temperature for 1 hour. The cells were sonicated at 30 MHz (30 seconds for 15 cycles, with 30-second intervals for cooling).13 The cell lysates were centrifuged at 12,000× g for 10 minutes and the supernatants were then filtered through 0.45 μm syringe filters (Millipore, Billerica, MA, USA) prior to purification with immobilized metal affinity chromatography.
The clarified lysates were loaded onto 1 mL His-Trap HP columns (GE Healthcare, Little Chalfont, UK). For optimal protein purification, the bound proteins were eluted with lysis buffer containing 50, 100, 200, and 500 mM imidazole. The eluted proteins were then analyzed by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and Western blotting. For optimum purity, 10 column volumes of washing buffer A (25 mM HEPES, 500 mM NaCl, 50 mM imidazole, pH 7.4) and washing buffer B (25 mM HEPES, 500 mM NaCl, 200 mM imidazole, pH 7.4) were flowed through the column in succession. Finally, bound proteins were eluted with 3 column volumes of elution buffer (25 mM HEPES, 500 mM NaCl, 500 mM imidazole, pH 7.4).
SDS-PAGE and Western blotting
Samples were mixed with loading buffer (100 mM Tris-HCl; pH 6.8, 20% [v/v] glycerol, 4% SDS [w/v], 0.2% [w/v] bromophenol blue, 200 mM mercaptoethanol), boiled, and electrophoresed on 12% sodium dodecyl sulfate-polyacrylamide gels. The purities of these proteins were determined based on the method described by Yap et al.14 Quantity One quantitation software provided in an imaging system (Bio-Rad, Gel Doc XR+, Hercules, CA, USA) was used to quantify the amount of fusion proteins. After SDS-PAGE, the proteins were stained with Coomassie Brilliant Blue, the area of interest on the gel was scanned, and the purity of the purified fusion proteins was determined by calculating the relative quantity of the fusion protein bands on the gel against the total amount of protein determined by the Bradford assay.15
For Western blotting, proteins on polyacrylamide gels were electrotransferred onto a nitrocellulose membrane and blocked with skimmed milk (10%; Anlene®, Fonterra, Auckland, New Zealand) for an hour. The nitrocellulose membrane was then incubated in TBS (50 mM Tris-HCl, 150 mM NaCl; pH 7.4) buffer containing anti-His monoclonal antibody (1:5,000 dilution; Invitrogen, San Diego, CA, USA) for 1 hour. The membrane was rinsed three times with TBS buffer containing 0.01% (v/v) Tween-20 and incubated with anti-mouse antibody (1:5,000 dilution in TBS; KPL, Gaithersburg, MA, USA) conjugated to alkaline phosphatase for 1 hour. After washing three times with TBS buffer containing 0.01% (v/v) Tween-20, 5-bromo-4-chloro-3′-indolylphosphate p-toluidine salt/nitro-blue tetrazolium chloride was added to the membrane as a substrate for color development.
Enzyme-linked immunosorbent assay
Purified NvC, NvC-M2ex1, NvC-M2ex3, NvC-M2ex5, and a synthetic M2e peptide (KSLLTEVETPIRNEWGCRCNDSSD; Genemed Synthesis Inc, San Antonio, TX, USA) were diluted to 0.2, 0.5, 1, 2, 10, 20, 40, and 100 μg/mL with HEPES buffer and coated (100 μL) on a microtiter plate at 4°C overnight. The wells were washed with TBST buffer (50 mM Tris-HCl, 150 mM NaCl, 0.05% [v/v] Tween-20; pH 7.4) and blocked with milk diluents (200 μL; KPL) at room temperature for 2 hours. The wells were then rinsed once with TBST buffer. Anti-influenza A M2 antibody (100 μL; 1:1,000 dilution in TBS buffer; Santa Cruz Biotechnology, Dallas, TX, USA) was added and incubated at room temperature for 1 hour. The wells were then washed three times with TBST. Anti-mouse antibody conjugated to alkaline phosphatase (100 μL; 1:5,000 dilution in TBS buffer) was added, followed by incubation for 1 hour. The wells were then washed three times. Next, 100 μL of p-nitrophenyl phosphate was added, followed by incubation for 20 minutes. A405 was measured using an ELx800 microtiter plate reader (Bio-Tek Instruments, Winooski, VT, USA).
Transmission electron microscopy
Purified protein samples (20 μg/mL; 15 μL) were absorbed onto carbon-coated grids for about 5 minutes. The grids were then negatively stained with freshly prepared uranyl acetate solution (1% [w/v]) for 5 minutes, dried, and viewed under an H7700 transmission electron microscope (Hitachi, Tokyo, Japan).
Immunization of BALB/c mice
Animal studies were approved by the Institutional Animal Care and Use Committee, Universiti Putra Malaysia (R058/2013). Female BALB/c mice aged about 4–5 weeks (mice in each group; n=7) were acclimatized for 2 weeks. The mice were then injected subcutaneously with purified protein samples (0.34 mg/mL; 100 μL) emulsified in Freund’s adjuvant. Purified NvC (0.34 mg/mL; 100 μL) with Freund’s adjuvant and HEPES buffer were also injected subcutaneously into the mice as controls. Two boosters were given to the mice at 3-week intervals. Serum samples were collected by submandibular bleeding before every injection and 1 week after the second booster.
Immunogenicity of fusion proteins
Synthetic M2e peptide (2 μg/mL; 100 μL) was coated onto microtiter plates and blocked with milk diluents (200 μL) as described above. Serum samples (100 μL; 1:2,000 dilution in TBS) collected during week 2, week 5, week 8, and week 9 of immunization were added to the wells and incubated for 1 hour at room temperature. After washing with TBST, anti-mouse antibody conjugated to alkaline phosphatase (100 μL; 1:5,000 dilution in TBS) was added and incubated for 1 hour. Color development and measurement of absorbance are as described above.
Immunophenotyping
At week 9, two mice from each group were euthanized. Their spleens were harvested and meshed through cell strainers (70 μm) in the presence of phosphate-buffered saline (137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, 1.5 mM KH2PO4; pH 7.4). The cell suspension was centrifuged at 300× g for 10 minutes and the pellet was resuspended in erythrocyte lysis buffer (155 mM NH4Cl, 10 mM KHCO3, 0.1 mM ethylenediaminetetraacetic acid; pH 7.4) followed by incubation at 4°C for 10 minutes. The cells were then centrifuged at 300× g for 10 minutes. The pellet was washed once and resuspended in phosphate-buffered saline (2 million cells/mL). Two sets of antibodies were added to the splenocytes: anti-CD3-FITC, CD4-PE, and CD8-APC; anti-F4/80-FITC (Abcam, Cambridge, UK). The mixtures were incubated for 2 hours on ice in the dark. The cells were then fixed with 1% (w/v) paraformaldehyde in phosphate-buffered saline for 24 hours at 4°C. The samples were then analyzed using a flow cytometer (FACSCalibur™; BD Biosciences, San Jose, CA, USA).
Sandwich ELISA for quantification of cytokines in peripheral blood
The concentration of interferon gamma (IFN-γ) and interleukin (IL)-10 in mouse peripheral blood was determined using an ELISA Max™ (BioLegend, San Diego, CA, USA) according to the manufacturer’s protocol. A microtiter plate was coated with cytokine capture antibodies (100 μL) at 4°C for overnight. After washing four times with PBST (137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, 1.5 mM KH2PO4, 0.05% [v/v] Tween-20; pH 7.4), the wells were blocked with phosphate-buffered saline-bovine serum albumin (137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, 1.5 mM KH2PO4, 1% [w/v] bovine serum albumin; pH 7.4; 200 μL) at room temperature for 2 hours. After washing with PBST, standards (IFN-γ and IL-10) and serum samples (100 μL) were added to the wells and incubated at room temperature for 2 hours. The wells were rinsed with PBST. Cytokine detection antibodies (100 μL) were then added and incubated at room temperature for 2 hours. After washing with PBST, avidin conjugated with horseradish peroxidase (100 μL) was added to the wells and incubated at room temperature for 30 minutes. The wells were then washed five times with PBST. Next, 100 μL of 3,3′,5,5′-tetramethylbenzidine was added and incubated for 5 minutes. Sulfuric acid (1 M; 100 μL) was then added to stop the reaction. A450 was measured with a microtiter plate reader.
Statistical analysis
One-way analysis of variance using Duncan’s multiple range tests was performed to analyze the variations in production of antibodies, immunophenotyping of splenocytes, and cytokine concentration in peripheral blood, between different immunization groups. A P-value <0.05 was considered to be statistically significant, <0.001 as very significant, and <0.0001 as extremely significant. All data were analyzed using the Statistical Package for the Social Sciences software version 19 for Windows (IBM Corporation, Armonk, NY, USA).
Results
Synthesis of five copies of M2e coding region with PCR
The coding region for five copies of M2e (M2ex5) was synthesized by combining four primers in a single tube with overlapping PCR (Figure 1A). The 3′ regions of fragments 1 and 2 are reverse complementary to each other; the 5′ regions of fragments 2 and 3 are reverse complementary to each other; and the 3′ regions of fragments 3 and 4 are reverse complementary to each other. As fragments 1 and 2 anneal, the single-stranded regions are filled up by DNA polymerase. A portion of this PCR product is then denatured and annealed with fragment 3, or the PCR product yielded from fragments 3 and 4, and eventually this process gives rise to a full-length M2ex5 coding region. Figure 1B shows the sequence of the M2ex5 coding region, where three glycine codons were added in between each M2e sequence and also at both the terminal ends of the M2ex5 coding sequence as linkers. The nucleotide sequence was modified to have different codons encoding for the same amino acids, so that it could be used as a template for the synthesis of different M2e coding regions, from one copy up to five copies, in a simple PCR reaction. This M2ex5 coding sequence was then cloned into a pGEM-T vector for stable storage and amplification in bacteria.
Figure 1.

Synthesis of five copies of M2e coding region with PCR.
Notes: (A) Primer-overlapping PCR for generating the coding sequence of five copies of M2e. The overlapping of fragments 1, 2, 3, and 4 eventually gives rise to a full-length M2ex5 through multiple cycles of PCR reaction. (B) Nucleotide and amino acid sequences of the M2ex5 coding region and polypeptide, respectively. Three glycine residues (boxed) were added in between each M2e gene, and also at both terminal ends of the M2ex5 gene as linkers. Underlined nucleotide sequences indicate overlapping region of primers used for gene synthesis. The nucleotide sequence was modified to have different sequences encoding for the same amino acids.
Abbreviations: PCR, polymerase chain reaction; M2ex5, nucleotide sequence encoding five copies of matrix 2 ectodomain of influenza A virus; M2e, influenza A matrix 2 ectodomain.
Construction of plasmids carrying one, three, and five copies of M2e coding regions
M2ex1, M2ex3, and M2ex5 coding sequences were synthesized by PCR using pGEM-T-M2ex5 as the template. The PCR products contained EcoRI and HindIII restriction sites at their 5′ and 3′ ends, respectively. These PCR products were digested with EcoRI and HindIII endonucleases and inserted into plasmid pTrcHis-TARNA2 immediately after the MrNV capsid gene. Figure 2 depicts the primary structures of all the three recombinant proteins, NvC-M2ex1, NvC-M2ex3, and NvC-M2ex5, in comparison with NvC protein. Expression of these recombinant proteins is under the control of the IPTG-inducible trc promoter. The myc epitope and His-tag at the C-terminal end of these proteins are mainly for ease of detection and purification purposes.
Figure 2.

Primary structures and amino acid sequences of NvC, NvC-M2ex1, NvC-M2ex3, and NvC-M2ex5.
Notes: One, three, and five copies of M2e were fused to the C-terminal end of the MrNV capsid protein. Horizontal lines indicate the absence of amino acid sequence. The amino acid sequences of MrNV capsid protein, linker, M2e, myc epitope, and His-tag are shown.
Abbreviations: MrNV, Macrobrachium rosenbergii nodavirus; NvC, Macrobrachium rosenbergii nodavirus capsid protein; NvC-M2ex1, Macrobrachium rosenbergii nodavirus capsid protein displaying one copy of influenza A matrix 2 ectodomain; NvC-M2ex3, Macrobrachium rosenbergii nodavirus capsid protein displaying three copies of influenza A matrix 2 ectodomain; NvC-M2ex5, Macrobrachium rosenbergii nodavirus capsid protein displaying five copies of influenza A matrix 2 ectodomain; M2e, influenza A matrix 2 ectodomain.
Expression and purification of fusion proteins
For expression of NvC-M2ex1, NvC-M2ex3, and NvC-M2ex5, their respective plasmids were introduced into E. coli TOP 10 cells. Total cell proteins before and after induction with IPTG were analyzed. For cells harboring plasmid pNvC-M2ex1, a distinct band of approximately 50 kDa was observed on the polyacrylamide gel upon IPTG induction (Figure 3A). This corresponded well to the calculated molecular mass of 49 kDa for NvC-M2ex1. E. coli cells harboring pNvC-M2ex3 and pNvC-M2ex5 plasmids showed extra protein bands of approximately 56 kDa and 63 kDa, respectively, which are in good agreement with the calculated molecular mass of 54 kDa and 60 kDa for NvC-M2ex3 and NvC-M2ex5, respectively (Figure 3A). These extra bands were detected by anti-His antibody in Western blotting (Figure 3B), indicating that NvC-M2ex1, NvC-M2ex3, and NvC-M2ex5 were successfully expressed in E. coli. These fusion proteins were then purified and analyzed by SDS-PAGE (Figure 3C) and Western blotting (Figure 3D). Their purity was above 90% as quantified by gel imaging software. Apart from the expected proteins, smaller protein bands of approximately 48 kDa, 54 kDa, and 61 kDa, respectively, were observed in purified NvC-M2ex1, NvC-M2ex3, and NvC-M2ex5 on the polyacrylamide gel (Figure 3C). These smaller protein bands were detected by anti-His antibody in Western blot analysis (Figure 3D), suggesting that they were N-terminal degraded products of NvC-M2ex1, NvC-M2ex3, and NvC-M2ex5. The N-terminal degraded product was reported earlier by Goh et al12 for purified NvC, which assembled into nanoparticles.12 Our results showed that the N-terminal degraded products, along with the full-length NvC-M2ex1, NvC-M2ex3, and NvC-M2ex5, self-assembled into spherical nanoparticles approximately 30 nm in diameter as analyzed by transmission electron microscopy (Figure 4), closely resembling the VLPs formed by NvC.12
Figure 3.

Expression and purification of the recombinant proteins.
Notes: Total cell lysate proteins were analyzed on (A) SDS-PAGE, and (B) Western blotting. “−” refers to non-induced cell lysate samples, whereas “+” refers to IPTG-induced cell lysate samples from Escherichia coli expressing the NvC-M2ex1, NvC-M2ex3, and NvC-M2ex5. On both SDS-PAGE and Western blot, protein bands of approximately 50 kDa, 56 kDa, and 63 kDa can be observed upon induction (indicated by arrow heads). Purified NvC-M2ex1, NvC-M2ex3, and NvC-M2ex5 proteins were analyzed with (C) SDS-PAGE and (D) Western blotting.
Abbreviations: SDS-PAGE, sodium dodecyl sulfate polyacrylamide gel electrophoresis; IPTG, isopropyl β-D-1-thiogalactopyranoside; NvC-M2ex1, Macrobrachium rosenbergii nodavirus capsid protein displaying one copy of influenza A matrix 2 ectodomain; NvC-M2ex3, Macrobrachium rosenbergii nodavirus capsid protein displaying three copies of influenza A matrix 2 ectodomain; NvC-M2ex5, Macrobrachium rosenbergii nodavirus capsid protein displaying five copies of influenza A matrix 2 ectodomain; M, protein markers with their corresponding molecular masses in kDa.
Figure 4.

Transmission electron microscopic analysis of fusion proteins.
Notes: (A) NvC-M2ex1, (B) NvC-M2ex3, and (C) NvC-M2ex5 were negatively stained with uranyl acetate and the micrographs were taken under 100,000× magnification. Spherical VLPs of approximately 30 nm in diameter were observed for all the three fusion proteins.
Abbreviations: NvC-M2ex1, Macrobrachium rosenbergii nodavirus capsid protein displaying one copy of influenza A matrix 2 ectodomain; NvC-M2ex3, Macrobrachium rosenbergii nodavirus capsid protein displaying three copies of influenza A matrix 2 ectodomain; NvC-M2ex5, Macrobrachium rosenbergii nodavirus capsid protein displaying five copies of influenza A matrix 2 ectodomain; VLPs, virus-like particles.
Antigenicity of recombinant proteins fused with different copies of M2e
The antigenicity of M2e epitopes displayed on NvC was analyzed with enzyme-linked immunosorbent assay (ELISA). NvC, which does not have an M2e epitope, served as a negative control, whereas the synthetic M2e peptide served as a positive control. Figure 5 shows that the anti-influenza A M2 antibody reacted strongly with the NvC fused with one copy to five copies of M2e epitopes, but did not react with the NvC protein. This indicates that the M2e epitopes fused with the NvC are antigenic. Interestingly, for protein concentration at 1 μg/mL or lower, interaction of the antibody with M2e displayed on NvC increased in proportion to the copy number of M2e epitopes. The interaction reached a plateau for wells coated with 2 μg/mL of fusion proteins.
Figure 5.
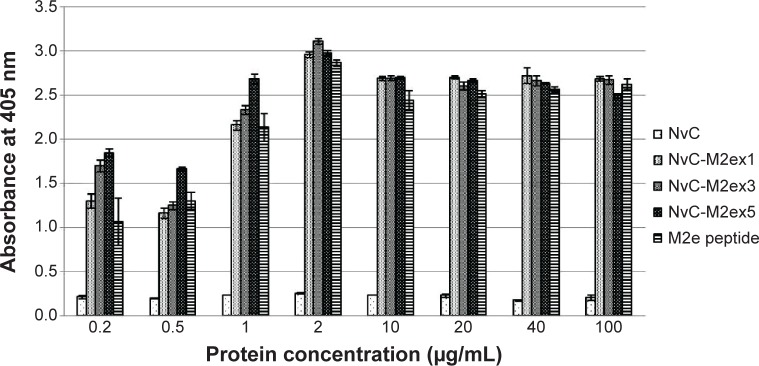
Antigenicity of M2e displayed on NvC.
Notes: Microtiter plate wells were coated with different concentrations of fusion proteins and the synthetic M2e peptide. The wells were blocked with milk diluents and the anti-influenza A M2 antibody was added to react with the coated proteins. NvC-M2ex1, NvC-M2ex3, and NvC-M2ex5 were compared with the synthetic M2e peptide (positive control) and the NvC (negative control).
Abbreviations: M2e, influenza A matrix 2 ectodomain; M2, influenza A matrix 2 protein; NvC, Macrobrachium rosenbergii nodavirus capsid protein; NvC-M2ex1, Macrobrachium rosenbergii nodavirus capsid protein displaying one copy of influenza A matrix 2 ectodomain; NvC-M2ex3, Macrobrachium rosenbergii nodavirus capsid protein displaying three copies of influenza A matrix 2 ectodomain; NvC-M2ex5, Macrobrachium rosenbergii nodavirus capsid protein displaying five copies of influenza A matrix 2 ectodomain.
Immunogenicity of fusion proteins in BALB/c mice
BALB/c mice were separated into six groups, containing seven mice each. The mice were injected subcutaneously with HEPES, NvC, NvC-M2ex1, NvC-M2ex3, and NvC-M2ex5. Relative titers of total antibodies specifically against the M2e present in mice serum samples were analyzed by ELISA. Serum samples collected before injection (week 2), after primary injection (week 5), after first booster (week 8), and after second booster (week 9) were analyzed and compared (Figure 6). In general, the antibody titer achieved a maximum level after the first booster (week 8). Interestingly, when the copy number of M2e displayed on NvC increased, total antibodies induced against M2e also increased. This was most obvious after the first injection (week 5), where NvC-M2ex5 induced a significantly higher antibody level when compared with NvC-M2ex3 and NvC-M2ex1, and likewise NvC-M2ex3 to NvC-M2ex1 (P<0.001). Although NvC-M2ex3 induced an antibody titer similar to that of NvC-M2ex5 after the first booster (week 8), it subsided more than NvC-M2ex5 after the second booster (week 9).
Figure 6.

Immunogenicity of the fusion proteins in BALB/c mice.
Notes: Antibodies specifically against M2e in serum samples collected at week 2 (pre-injection), week 5 (3 weeks after first injection), week 8 (3 weeks after first booster injection), and week 9 (1 week after second booster injection) were measured by ELISA. Serum samples from mice were pooled according to the groups injected with HEPES buffer, NvC, NvC-M2ex1, NvC-M2ex3, and NvC-M2ex5. The serum samples were then reacted with synthetic M2e peptide coated onto microtiter plate wells. Alphabets and roman numerals (a, b, c, d; x, y, z; and I, II, III, IV) on top of each bar (group) represent statistical significance (P<0.001) of results within each time point. The asterisks (**) on top of the bar chart show extreme significance (P<0.0001) compared with the group of mice immunized with NvC (control without M2e).
Abbreviations: M2e, influenza A matrix 2 ectodomain; ELISA, enzyme-linked immunosorbent assay; HEPES, 2-[4-(2-hydroxyethyl)piperazin-1-yl]ethanesulfonate; NvC, Macrobrachium rosenbergii nodavirus capsid protein; NvC-M2ex1, Macrobrachium rosenbergii nodavirus capsid protein displaying one copy of influenza A matrix 2 ectodomain; NvC-M2ex3, Macrobrachium rosenbergii nodavirus capsid protein displaying three copies of influenza A matrix 2 ectodomain; NvC-M2ex5, Macrobrachium rosenbergii nodavirus capsid protein displaying five copies of influenza A matrix 2 ectodomain.
Immunophenotyping of mice splenocytes
Immunophenotyping of mice splenocytes was performed after the second booster injection. The cytotoxic T lymphocyte (CTL) to T helper cell ratio was the highest for NvC-M2ex3 and NvC-M2ex5, followed by NvC-M2ex1 (Figure 7). As more copy numbers of M2e were displayed on NvC, a lesser amount of monocytes and macrophages were activated, implying that the displayed M2e may have shielded some epitopes located on the carriers (NvC).
Figure 7.

Immunophenotyping of mice splenocytes.
Notes: Spleens from mice immunized with HEPES, NvC, NvC-M2ex1, NvC-M2ex3, and NvC-M2ex5 were harvested at week 9. The spleens were meshed through cell strainers to obtain single cell suspensions. The splenocytes were then probed with two sets of fluorochrome-labeled antibodies for detection of the (A) CD8+:CD4+ population ratio and (B) F4/80+ population. Roman numerals and alphabets (I, II; x, y, z) on top of each bar (group) represent the statistical significance (P<0.05) of results between different treatment groups.
Abbreviations: HEPES, 2-[4-(2-hydroxyethyl)piperazin-1-yl]ethanesulfonate; NvC, Macrobrachium rosenbergii nodavirus capsid protein; NvC-M2ex1, Macrobrachium rosenbergii nodavirus capsid protein displaying one copy of influenza A matrix 2 ectodomain; NvC-M2ex3, Macrobrachium rosenbergii nodavirus capsid protein displaying three copies of influenza A matrix 2 ectodomain; NvC-M2ex5, Macrobrachium rosenbergii nodavirus capsid protein displaying five copies of influenza A matrix 2 ectodomain; CD8+, cells possessing clusters of differentiation 8 (cytotoxic T lymphocytes); CD4+, cells possessing clusters of differentiation 4 (T helper cells); F4/80+, cells possessing F4/80 cell surface marker (macrophages).
Cytokine concentration in peripheral blood of immunized mice
The concentrations of IFN-γ and IL-10 in mice inoculated with the fusion proteins were analyzed with a sandwich ELISA. NvC-M2ex3 strongly induced the production of IFN-γ (Table 2), which corresponds to the CTL ratio in spleen. However, NvC-M2ex1 and NvC-M2ex5 have no significant effect on production of IFN-γ. All the fusion proteins containing M2e (NvC-M2ex1, NvC-M2ex3, and NvC-M2ex5) suppressed IL-10 secretion with a significance level of P<0.05, suggesting a type 1 T helper immune response (Table 2).
Table 2.
Cytokine concentrations in pooled serum samples
| Group | Cytokine concentrations (pg/mL)
|
|
|---|---|---|
| IFN-γ | IL-10 | |
| HEPES | 498.312±84.619a | 421.099±0y |
| NvC | 620.540±131.630a | 432.181±11.082y |
| NvC-M2ex1 | 611.138±103.423a | 365.692±33.245x |
| NvC-M2ex3 | 1541.948±131.630b | 365.692±33.245x |
| NvC-M2ex5 | 432.498±94.0212a | 354.610±22.163x |
Notes: a,b and x,yindicate statistical significance (P,0.05) in between a and b or x and y.
Abbreviations: IFN-γ, interferon gamma; IL-10, interleukin-10; HEPES, group of mice injected with buffer; NvC, group of mice injected with Macrobrachium rosenbergii nodavirus capsid protein; NvC-M2ex1, group of mice injected with Macrobrachium rosenbergii nodavirus capsid protein displaying one copy of influenza A matrix 2 ectodomain; NvC-M2ex3, group of mice injected with Macrobrachium rosenbergii nodavirus capsid protein displaying three copies of influenza A matrix 2 ectodomain; NvC-M2ex5, group of mice injected with Macrobrachium rosenbergii nodavirus capsid protein displaying five copies of influenza A matrix 2 ectodomain.
Discussion
Currently available influenza vaccines are derived from inactivated viruses propagated in embryonated chicken eggs. The effectiveness of these vaccines depends heavily on the H and N antigens of influenza viruses, which are always susceptible to mutations. Influenza A M2e was found to be highly conserved regardless of the high mutation rates of the H and N glycoproteins. In addition, due to the vital role of M2e in viral replication, it is of great interest to develop a universal vaccine based upon M2e, which is believed to be protective regardless of viral strain.16–18
M2e is present in very low amounts on the influenza virus, so is unable to induce an immune response specifically to this epitope. Although M2e in virus-infected cells is abundant,19 it cannot induce production of specific antibodies against M2e.20,21 This is likely due to shielding of M2e by other highly glycosylated proteins, preventing its contact with immune cells.22 Thus, influenza virus infection induced poor M2e antibody responses.11 To overcome this, multiple copies of M2e were displayed on the surface of NvC nanoparticles.
The present study demonstrates that NvC is capable of carrying at least 158 amino acid residues at its C-terminal end without impairing particle formation, making it a potential carrier for subunit vaccines. De Filette et al22 linked up to three copies of M2e epitopes to the N-terminal end of the hepatitis B core antigen, which self-assembled into nanoparticles. In our study, up to five copies of M2e were successfully fused to the C-terminal end of NvC without affecting nanoparticle formation.
Three copies of M2e displayed on NvC nanoparticles induced significantly higher anti-M2e antibody production (P<0.001) compared with NvC-M2ex1 harboring only one copy of M2e. This finding is in good agreement with that of De Filette et al22 who demonstrated that the protective efficacy increased with the copy number of M2e from one, two, to three copies. Interestingly, we found that NvC-M2ex5 induced an even stronger antibody response than NvC-M2ex3, which was particularly significant (P<0.001) after the primary injection. However after the first booster injection, both NvC-M2ex3 and NvC-M2ex5 achieved a maximum antibody titer. Thus, antibody production did not increase further after the second booster injection.
The mechanisms of protection based on M2e are not yet fully understood. However, protection against viral infection was shown to be transferable by serum containing antibodies specifically against M2e, demonstrating the importance of these specific antibodies.23 El Bakkouri et al24 and Ibañez et al17 demonstrated that M2e conferred protection against influenza virus infection through antibody-dependent cytotoxicity and antibody-mediated phagocytosis in Fcγ receptor knockout mice. Dong et al25 demonstrated that impairment of CTL production resulted in increased susceptibility to influenza virus infection. It has also been shown that passive transfer of alveolar macrophages and antibodies gave protection to otherwise unprotected mice due to the lack of Fcγ receptors.24 Our findings showed an increased in CTL but not macrophages when M2e epitopes were displayed on NvC nanoparticles. In fact, the concentration of macrophages reduced as more copies of M2e were displayed on the nanoparticles, suggesting that M2e itself is incapable of stimulating proliferation of macrophages.
In this study, NvC-M2ex3 was shown to induce very high secretion of IFN-γ. Baumgarth and Kelso26 demonstrated that IFN-γ is required for a high level of production of specific immunoglobulin G2a, which is correlated with protection against influenza virus infection.17,27,28 IFN-γ has also been shown to control viral replication at an early stage of infection and reduce the severity of influenza.29–31 To our surprise, NvC-M2ex5 did not result in an increased level of IFN-γ in mouse serum despite an increase in CTL, indicating that cytotoxicity does not always correlate with IFN-γ secretion.32–35 In contrast with IFN-γ production, administration of NvC-M2ex1, NvC-M2ex3, and NvC-M2ex5 resulted in a significant decrease in IL-10, suggesting a type 1 T helper immune response.
Conclusion
In summary, one to five copies of M2e were fused to the C-terminal end of NvC protein and transmission electron microscopic analysis revealed that all the chimeric proteins self-assemble into nanosized VLPs. These nanoparticles displaying different copy numbers of M2e were shown to elicit humoral-mediated and cell-mediated immune responses. This study demonstrates the potential of MrNV capsid protein as a nanocarrier for influenza A virus epitopes. Recommendations for future studies include immunization with different adjuvants and routes of vaccine administration, as well as challenge with various strains of influenza virus.
Acknowledgments
This study was supported by the Ministry of Higher Education and the Ministry of Science, Technology and Innovation of Malaysia.
Footnotes
Disclosure
The authors report no conflicts of interest in this study.
References
- 1.Dawood FS, Iuliano AD, Reed C, et al. Estimated global mortality associated with the first 12 months of 2009 pandemic influenza A H1N1 virus circulation: a modelling study. Lancet Infect Dis. 2012;12(9):687–695. doi: 10.1016/S1473-3099(12)70121-4. [DOI] [PubMed] [Google Scholar]
- 2.De Filette M, Ramne A, Birkett A, et al. The universal influenza vaccine M2e-HBc administered intranasally in combination with the adjuvant CTA1-DD provides complete protection. Vaccine. 2006;24(5):544–551. doi: 10.1016/j.vaccine.2005.08.061. [DOI] [PubMed] [Google Scholar]
- 3.Cox NJ, Subbarao K. Influenza. Lancet. 1999;354(9186):1277–1282. doi: 10.1016/S0140-6736(99)01241-6. [DOI] [PubMed] [Google Scholar]
- 4.Reed C, Meltzer MI, Finelli L, Fiore A. Public health impact of including two lineages of influenza B in a quadrivalent seasonal influenza vaccine. Vaccine. 2012;30(11):1993–1998. doi: 10.1016/j.vaccine.2011.12.098. [DOI] [PubMed] [Google Scholar]
- 5.Fang R, Min Jou W, Huylebroeck D, Devos R, Fiers W. Complete structure of A/duck/Ukraine/63 influenza hemagglutinin gene: animal virus as progenitor of human H3 Hong Kong 1968 influenza hemagglutinin. Cell. 1981;25(2):315–323. doi: 10.1016/0092-8674(81)90049-0. [DOI] [PubMed] [Google Scholar]
- 6.Lamb RA, Lai CJ, Choppin PW. Sequences of mRNAs derived from genome RNA segment 7 of influenza virus: colinear and interrupted mRNAs code for overlapping proteins. Proc Natl Acad Sci U S A. 1981;78(7):4170–4174. doi: 10.1073/pnas.78.7.4170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rossman JS, Lamb RA. Influenza virus assembly and budding. Virology. 2011;411(2):229–236. doi: 10.1016/j.virol.2010.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schnell JR, Chou JJ. Structure and mechanism of the M2 proton channel of influenza A virus. Nature. 2008;451(7178):591–595. doi: 10.1038/nature06531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Stouffer AL, Acharya R, Salom D, et al. Structural basis for the function and inhibition of an influenza virus proton channel. Nature. 2008;451(7178):596–599. doi: 10.1038/nature06528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fiers W, De Filette M, El Bakkouri K, et al. M2e-based universal influenza A vaccine. Vaccine. 2009;27(45):6280–6283. doi: 10.1016/j.vaccine.2009.07.007. [DOI] [PubMed] [Google Scholar]
- 11.Feng J, Zhang M, Mozdzanowska K, et al. Influenza A virus infection engenders a poor antibody response against the ectodomain of matrix protein 2. Virol J. 2006;3:102. doi: 10.1186/1743-422X-3-102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Goh ZH, Tan SG, Bhassu S, Tan WS. Virus-like particles of Macrobrachium rosenbergii nodavirus produced in bacteria. J Virol Methods. 2011;175(1):74–79. doi: 10.1016/j.jviromet.2011.04.021. [DOI] [PubMed] [Google Scholar]
- 13.Ho CW, Chew TK, Ling TC, et al. Efficient mechanical cell disruption of Escherichia coli by an ultrasonicator and recovery of intracellular hepatitis B core antigen. Process Biochem. 2006;41(8):1829–1834. [Google Scholar]
- 14.Yap WB, Tey BT, Alitheen NB, Tan WS. Purification of the His-tagged hepatitis B core antigen from unclarified bacterial homogenate using immobilized metal affinity-expanded bed adsorption chromatography. J Chromatogr A. 2010;1217(21):3473–3480. doi: 10.1016/j.chroma.2010.03.012. [DOI] [PubMed] [Google Scholar]
- 15.Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72(1–2):248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 16.Gao X, Wang W, Li Y, et al. Enhanced Influenza VLP vaccines comprising matrix-2 ectodomain and nucleoprotein epitopes protects mice from lethal challenge. Antiviral Res. 2013;98(1):4–11. doi: 10.1016/j.antiviral.2013.01.010. [DOI] [PubMed] [Google Scholar]
- 17.Ibañez LI, Roose K, De Filette M, et al. M2e-displaying virus-like particles with associated RNA promote T helper 1 type adaptive immunity against influenza A. PLoS One. 2013;8(3):e59081. doi: 10.1371/journal.pone.0059081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Price GE, Soboleski MR, Lo CY, et al. Single-dose mucosal immunization with a candidate universal influenza vaccine provides rapid protection from virulent H5N1, H3N2 and H1N1 viruses. PLoS One. 2010;5(10):e13162. doi: 10.1371/journal.pone.0013162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lamb RA, Zebedee SL, Richardson CD. Influenza virus M2 protein is an integral membrane protein expressed on the infected-cell surface. Cell. 1985;40(3):627–633. doi: 10.1016/0092-8674(85)90211-9. [DOI] [PubMed] [Google Scholar]
- 20.Black RA, Rota PA, Gorodkova N, Klenk HD, Kendal AP. Antibody response to the M2 protein of influenza A virus expressed in insect cells. J Gen Virol. 1993;74(Pt 1):143–146. doi: 10.1099/0022-1317-74-1-143. [DOI] [PubMed] [Google Scholar]
- 21.Liu W, Li H, Chen YH. N terminus of M2 protein could induce antibodies with inhibitory activity against influenza virus replication. FEMS Immunol Med Microbiol. 2003;35(2):141–146. doi: 10.1016/S0928-8244(03)00009-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.De Filette M, Min Jou W, Birkett A, et al. Universal influenza A vaccine: optimization of M2-based constructs. Virology. 2005;337(1):149–161. doi: 10.1016/j.virol.2005.04.004. [DOI] [PubMed] [Google Scholar]
- 23.Neirynck S, Deroo T, Saelens X, Vanlandschoot P, Jou WM, Fiers W. A universal influenza A vaccine based on the extracellular domain of the M2 protein. Nat Med. 1999;5(10):1157–1163. doi: 10.1038/13484. [DOI] [PubMed] [Google Scholar]
- 24.El Bakkouri K, Descamps F, De Filette M, et al. Universal vaccine based on ectodomain of matrix protein 2 of influenza A: Fc receptors and alveolar macrophages mediate protection. J Immunol. 2011;186(2):1022–1031. doi: 10.4049/jimmunol.0902147. [DOI] [PubMed] [Google Scholar]
- 25.Dong L, Mori I, Hossain MJ, Kimura Y. The senescence-accelerated mouse shows aging-related defects in cellular but not humoral immunity against influenza virus infection. J Infect Dis. 2000;182(2):391–396. doi: 10.1086/315727. [DOI] [PubMed] [Google Scholar]
- 26.Baumgarth N, Kelso A. In vivo blockade of gamma interferon affects the influenza virus-induced humoral and the local cellular immune response in lung tissue. J Virol. 1996;70(7):4411–4418. doi: 10.1128/jvi.70.7.4411-4418.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fiers W, De Filette M, Birkett A, Neirynck S, Min Jou W. A “universal” human influenza A vaccine. Virus Res. 2004;103(1–2):173–176. doi: 10.1016/j.virusres.2004.02.030. [DOI] [PubMed] [Google Scholar]
- 28.Hashemi H, Pouyanfard S, Bandehpour M, et al. Immunization with M2e-displaying T7 bacteriophage nanoparticles protects against influenza A virus challenge. PLoS One. 2012;7(9):e45765. doi: 10.1371/journal.pone.0045765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.He XS, Draghi M, Mahmood K, et al. T cell-dependent production of IFN-gamma by NK cells in response to influenza A virus. J Clin Invest. 2004;114(12):1812–1819. doi: 10.1172/JCI22797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Liu B, Mori I, Hossain MJ, Dong L, Takeda K, Kimura Y. Interleukin-18 improves the early defence system against influenza virus infection by augmenting natural killer cell-mediated cytotoxicity. J Gen Virol. 2004;85(Pt 2):423–428. doi: 10.1099/vir.0.19596-0. [DOI] [PubMed] [Google Scholar]
- 31.Weiss ID, Wald O, Wald H, et al. IFN-gamma treatment at early stages of influenza virus infection protects mice from death in a NK cell-dependent manner. J Interferon Cytokine Res. 2010;30(6):439–449. doi: 10.1089/jir.2009.0084. [DOI] [PubMed] [Google Scholar]
- 32.Bordignon V, Cordiali-Fei P, Rinaldi M, et al. Evaluation of antigen specific recognition and cell mediated cytotoxicity by a modified lysispot assay in a rat colon carcinoma model. J Exp Clin Cancer Res. 2012;31:9. doi: 10.1186/1756-9966-31-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Karulin AY, Hesse MD, Tary-Lehmann M, Lehmann PV. Single-cytokine-producing CD4 memory cells predominate in type 1 and type 2 immunity. J Immunol. 2000;164(4):1862–1872. doi: 10.4049/jimmunol.164.4.1862. [DOI] [PubMed] [Google Scholar]
- 34.Lim DG, Bieganowska Bourcier K, Freeman GJ, Hafler DA. Examination of CD8+ T cell function in humans using MHC class I tetramers: similar cytotoxicity but variable proliferation and cytokine production among different clonal CD8+ T cells specific to a single viral epitope. J Immunol. 2000;165(11):6214–6220. doi: 10.4049/jimmunol.165.11.6214. [DOI] [PubMed] [Google Scholar]
- 35.Slifka MK, Rodriguez F, Whitton JL. Rapid on/off cycling of cytokine production by virus-specific CD8+ T cells. Nature. 1999;401(6748):76–79. doi: 10.1038/43454. [DOI] [PubMed] [Google Scholar]