

Abstract
Objective:
Ataxia-telangiectasia (AT) is a rare, devastating neurodegenerative disease presenting with early-onset ataxia, oculocutaneous telangiectasia, immunodeficiency, radiosensitivity, and proneness to cancer. In a previous phase 2 study, we showed that 6 monthly infusions of autologous erythrocytes loaded with dexamethasone (EryDex; EryDel, Urbino, Italy) were effective in improving neurologic impairment in young patients with AT. The present article reports the results of the extension of this study for an additional 24-month period.
Methods:
After the end of the first trial, 4 patients continued to be treated with monthly EryDex infusions for an additional 24 months, and their clinical outcome was compared with that of 7 age-matched patients who stopped the treatment after the first 6 infusions. The protocol included serial assessment of ataxia (by International Cooperative Ataxia Rating Scale) and adaptive behavior (by Vineland Adaptive Behavior Scales) and clinical and laboratory tests revealing treatment- and steroid-dependent adverse effects, if present.
Results:
Patients in the extended study experienced a continuous neurologic improvement with respect to their pretreatment status, whereas controls showed a progressive neurologic deterioration (according to the natural history of the disease) after the discontinuation of the treatment. The delivery system we adopted proved to be safe and well-tolerated, and none of the side effects usually associated with the chronic administration of corticosteroids were observed during the entire trial.
Conclusions:
These promising preliminary results call for a large-scale controlled study on protracted treatment of patients with AT with dexamethasone-loaded erythrocytes.
Ataxia-telangiectasia (AT) is a rare genetic disease1,2 caused by mutations in the ataxia telangiectasia mutated (ATM) gene, which results in a multisystemic disorder presenting with early-onset ataxia, oculocutaneous teleangiectasias, progressive supranuclear ophthalmoplegia, immunodeficiency, recurrent sinopulmonary infections, radiosensitivity, and proneness to cancer. In the classic form, patients are wheelchair-dependent by the age of 10 years,1 and their life expectancy is approximately 25 years.1,2 No effective disease-modifying therapy is presently available.
Observational studies have suggested that betamethasone may be effective in improving neurologic functions in patients with AT.3,4 A controlled short-term trial confirmed the efficacy of oral betamethasone (0.1 mg/kg),5 but corticosteroid side effects were quickly observed. To overcome these side effects and explore the effects of a more protracted treatment on neurologic outcome, we recently performed a single-arm, open-label, 6-month extended phase 2 trial in which the enrolled patients received a monthly treatment of 50 mL of autologous erythrocytes loaded with 2 vials of 250 mg of dexamethasone phosphate (DSP) by EryDex (EryDel, Urbino, Italy) procedure.6 Similarly to betamethasone, dexamethasone has a high anti-inflammatory potency and lacks mineralcorticoid activity. DSP is converted to dexamethasone by erythrocytes' phosphatases and released into the bloodstream for about 20–30 days.7
After 6 infusions, patients experienced a clinical improvement of about 5 points on the International Cooperative Ataxia Rating Scale (ICARS)8 and a significant improvement in adaptive behavior, assessed by Vineland Adaptive Behavior Scales (VABS),9 without the occurrence of steroid-dependent adverse events. The improvement was more relevant in patients presenting with less severe neurologic impairment and was affected by the efficiency of the DSP loading procedure into erythrocytes.6 We report the results of the extension of this preliminary study for an additional 24-month period.
METHODS
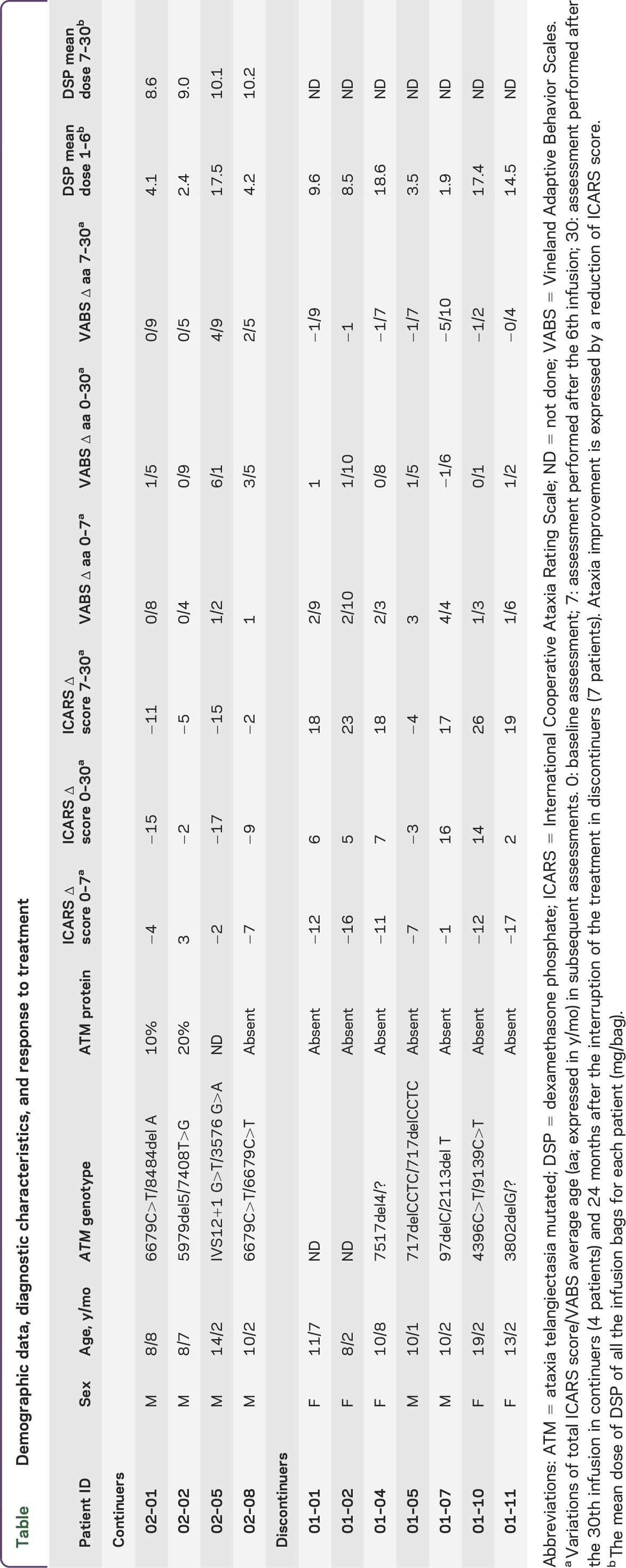
At the end of the first 6-month trial,6 4 male patients (mean age 10.6 years; SD 2.8 years; range 8.7–14.2 years) requested and were authorized by the Ethic Committee to continue EryDex treatment. So far they have been treated monthly for an additional 24-month period. The ICARS and VABS scores of the patients in the extended study before the beginning of the treatment and after 6 and 24 EryDex infusions were compared with those of the 7 patients with AT (2 male, 5 female; mean age 11.8 years; SD 3.5 years; range 8.2–19.2 years) who stopped the treatment after the sixth infusion, as originally planned by the study design.6 In accordance with a drug compassionate use clause, the selection of the continuers was based on the parents' decision (i.e., no external inclusion criterion could be applied), and the controls were selected among discontinuers by trying to best match the clinical characteristics of the continuers (table). During the extension study, the loading procedure was further optimized by obtaining an encapsulated DSP of 9.4 ± 0.8 mg/bag (vs 6.7 ± 6.9 in the previous study6) with the same dose of the medicament. The assessment also included clinical and laboratory tests aimed at detecting treatment- and steroid-dependent adverse effects.
Table.
Demographic data, diagnostic characteristics, and response to treatment
Standard protocol approvals, registrations, and patient consents.
The extension was authorized by the Ethic Committee. Current Controlled Trial registration 2010-022315-19SpA.
RESULTS
The table summarizes the clinical response to the treatment at the end of the first 6 months (11 patients) and after 24 further months in continuers (4 male patients) and discontinuers (7 patients; 2 male, 5 female). The mean DSP incorporation during both the original and extended trials is also shown. At the end of the first 6-month trial, ICARS and VABS scores of continuers and discontinuers were ICARS 52.2 ± 7.1 vs 43 ± 2.9 and VABS 6 ± 2.3 vs 8.2 ± 0.6, respectively.
After 30 months, the patients in the extended study experienced an improvement in ICARS score of 10.7 points (SD 6.75; range 2–17) with respect to the score obtained at basal clinical evaluation. Conversely, the ICARS score of the 7 patients who discontinued the treatment worsened by 6.7 points (SD 6.5; range +3–16) (Mann-Whitney U = 1.00; z = 2.45; p = 0.013803). Moreover, all continuers and only 1 of 7 discontinuers improved their ICARS score (Fisher exact test, p = 0.0152). Correspondingly, VABS average age improved by 2.10 years (SD 2.4 years; range 9 months–6.1 years) and by 0.9 years (SD 1 year; range −1.6 to +1.10 years) in continuers and discontinuers, respectively (Mann-Whitney U = 5.50; z = 1.60; p = 0.10). Moreover, ICARS scores at the end of the first 6-month trial and after 30 months improved by 8.2 points (SD 5.8; range 2–17) in the continuers and worsened by 17.8 points (SD 6.9, range −4–26) in the discontinuers (Mann-Whitney U = 0.00; z = 2.64; p = 0.008). Ataxia improved in 4 of 4 continuers and in 1 of 7 discontinuers (Fisher exact test, p = 0.015) (figure). Concomitant variations in VABS among continuers and discontinuers were 2.6 years (SD 2.3 years; range 0.5–4.9 years) and −1.7 years (SD 11 months; range −4 months +2.9 years), respectively (Mann-Whitney U = 0.00; z = 2.64; p = 0.008).
Figure. Neurologic impairment variation and duration of the treatment in patients with ataxia-telangiectasia.
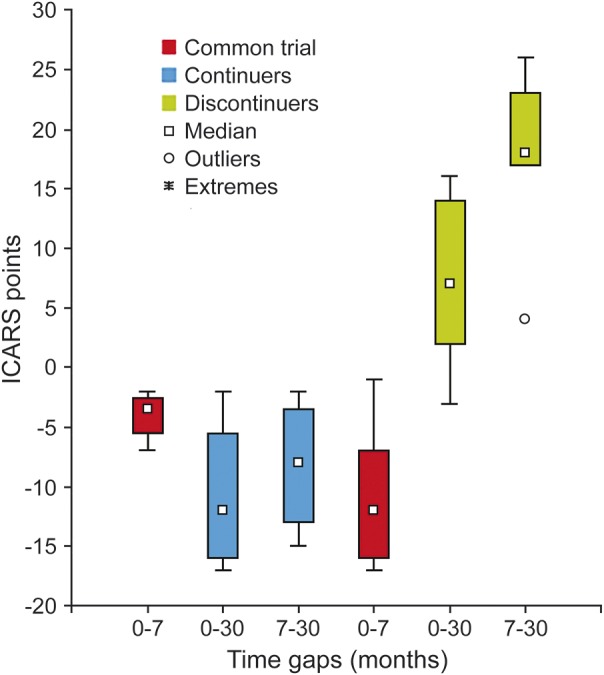
The trend of International Cooperative Ataxia Rating Scale (ICARS) score variations (with SD) in 4 patients with ataxia-telangiectasia who were treated for 30 months with monthly infusions of autologous dexamethasone phosphate–loaded erythrocytes compared with the ICARS scores of 7 patients who discontinued the treatment after the first 6 monthly infusions. 0 (baseline), 7 (after the first 6 infusions), and 30 (after 30 months) represent the scheduled time points for the subsequent assessments. In continuers, 30 is also the total number of infusions before the last evaluation. In red is the common trial (0–7) for all 11 patients, in blue are the ICARS score variations in 4 continuers (0–30; 7–30), and in green are the ICARS score variations in 7 discontinuers (0–30; 7–30). The increase of the score reflects the worsening of ataxia.
No steroid-dependent adverse reactions were observed during the entire treatment period. Standard laboratory tests, physical examination, vital signs, and ECG were all normal. Additional laboratory parameters such as cholesterol (total, high-density lipoprotein, and low-density lipoprotein), glycosylated hemoglobin (HbA1c), and blood and urinary cortisol were all within the normal range. CD4+ lymphocyte count remained stable during the treatment period but below the normal range for 3 of the 4 patients. Blood α-fetoprotein levels increased in all patients, regardless of the treatment they underwent.
DISCUSSION
Long-term corticosteroid administration is effective in slowing the progression of Duchenne muscular dystrophy.10 We recently reported a statistically significant improvement in ataxic symptoms with DSP-loaded red cells in a cohort of young patients with AT.6 Although this new treatment did not have the usual side effects of chronic steroid treatment, it was apparently less effective then oral betamethasone treatment, which resulted in a median improvement of 13 points on the ICARS after a month of therapy.5 Present data are more encouraging, showing that after 30 months of treatment the patients experienced an improvement of 10.7 points in mean ICARS score and 3 years in VABS average age with respect to their basal condition. Conversely, the ICARS score of the discontinuers at 30 months had worsened by 6.7 points and the VABS average age had improved just 0.9 years. The gap of 26 ICARS points between continuers and discontinuers at 30 months with respect to the score obtained by the 2 groups after the first 6 months of treatment is an important finding.
Albeit preliminary, the present results are important: 2.5 years is a time span long enough to disclose the progressive neurologic impairment that characterizes the natural clinical history of AT.1,2 The fact that the discontinuers experienced a decline of their neurologic conditions once steroids were interrupted would suggest a symptomatic effect of the treatment rather than a plastic change involving the basic pathogenetic mechanism of the disease. Otherwise, the persistent clinical improvement we observed so far in the continuers conflicts with this hypothesis.
Several methodologic limitations (lack of blinding and placebo in the treatment assignation, small size of the sample, lack of external clinical criteria for patient enrollment) affect our study, which was based on an empiric post hoc observation. The lack of AT-dedicated rating scales is a further intrinsic methodologic limitation. Nevertheless, in the face of such a dramatic, untreatable disease, any clinical observation suggesting a potential treatment deserves to be considered.
Whatever the biological effect of steroids in AT, present data support the view that a protracted treatment may delay and perhaps bias the natural course of the disease. Moreover, the delivery system we adopted was confirmed to be safe and well-tolerated, since after this extended period of time no side effects could be detected. A large-scale controlled study on protracted steroid treatment by DSP loaded into erythrocytes in patients with AT is mandatory in order to verify these preliminary data.
ACKNOWLEDGMENT
The authors thank Drs. L. Buccone, C. Pignata, F. Specchia, and A. Trizzino for referring patients; the associations (Amici di Valentina, Noi per Lorenzo, AISA, AIP) that assisted the families in traveling and staying in Rome or Brescia; all the hospital personnel (doctors, nurses, physiotherapists, technicians, etc.); and the sponsor EryDel for providing economic support and the EryDex system.
GLOSSARY
- AT
ataxia-telangiectasia
- ATM
ataxia telangiectasia mutated
- DSP
dexamethasone phosphate
- ICARS
International Cooperative Ataxia Rating Scale
- VABS
Vineland Adaptive Behavior Scales
AUTHOR CONTRIBUTIONS
All authors have made a substantial contribution to qualify for authorship. Each author listed on the manuscript has seen and approved the submission of this version of the manuscript and takes full responsibility for the manuscript. Vincenzo Leuzzi: drafting/revising the manuscript, analysis and interpretation of clinical and biochemical data, acquisition of data. Roberto Micheli: drafting/revising the manuscript, analysis and interpretation of clinical and biochemical data, acquisition of data. Daniela D'Agnano: drafting/revising the manuscript, analysis and interpretation of clinical and biochemical data, acquisition of data. Anna Molinaro: drafting/revising the manuscript, analysis and interpretation of clinical and biochemical data, analysis and interpretation of patients' neuromotor patterns. Tullia Venturi: drafting/revising the manuscript, analysis and interpretation of clinical and biochemical data, analysis and interpretation of patients' neuromotor patterns. Alessandro Plebani: drafting/revising the manuscript, analysis and interpretation of clinical and biochemical data. Annarosa Soresina: drafting/revising the manuscript, analysis and interpretation of clinical and biochemical data. Mirella Marini: drafting/revising the manuscript, analysis and interpretation of clinical and biochemical data. Pierino Ferremi Leali: drafting/revising the manuscript, analysis and interpretation of clinical and biochemical data. Isabella Quinti: drafting/revising the manuscript, analysis and interpretation of clinical and biochemical data. Maria C. Pietrogrande: drafting/revising the manuscript, analysis and interpretation of clinical and biochemical data. Andrea Finocchi: drafting/revising the manuscript, analysis and interpretation of clinical and biochemical data. Elisa Fazzi: drafting/revising the manuscript, analysis and interpretation of clinical and biochemical data, analysis and interpretation of patients' neuromotor patterns. Luciana Chessa: drafting/revising the manuscript, analysis and interpretation of clinical and biochemical data, molecular analysis interpretation. Mauro Magnani: drafting/revising the manuscript, analysis and interpretation of clinical and biochemical data, acquisition of data.
STUDY FUNDING
EryDel provided economic support and the EryDex System.
DISCLOSURE
V. Leuzzi, R. Micheli, D. D'Agnano, A. Molinaro, T. Venturi, A. Plebani, A. Soresina, M. Marini, and P. F. Leali report no disclosures. I. Quinti is on the scientific advisory board for CSL Behring, is on the editorial board for Frontiers in Immunology, and received research support from Jeffrey Modell Foundation. M.C. Pietrogrande reports no disclosures. A. Finocchi received research support from Italian Ministry of Health. E. Fazzi and L. Chessa report no disclosures. M. Magnani is the CSO of EryDel SpA, holds a patent for encapsulation of agents within erythrocytes, received research support from Fondazione Cassa di Risparmio di Fano, and holds stock in EryDel SpA. Go to Neurology.org/nn for full disclosure forms.
REFERENCES
- 1.Sedgwick O, Boder E. Ataxia-teleangiectasia. In: Vinken PJ, Bruyn GW, editors. Handbook of Clinical Neurology. Vol 14 Amsterdam: Elsevier; 1972:267–239. [Google Scholar]
- 2.Ataxia-Teleangiectasia; AT, #208900. Online Mendelian Inheritance in Man (OMIM), Center for Medical Genetics, John Hopkins University, Baltimore, MD, and the National Center for Biotechnology Information, National Library of Medicine, Bethesda, MD. Available at: http://omim.org/entry/208900. Accessed October 24, 2012.
- 3.Buoni S, Zannolli R, Sorrentino L, Fois A. Betamethasone and improvement of neurological symptoms in ataxia telangiectasia. Arch Neurol 2006;63:1469–1482. [DOI] [PubMed] [Google Scholar]
- 4.Broccoletti T, Del Giudice E, Cirillo E, et al. Efficacy of very-low-dose betamethasone on neurological symptoms in ataxia-telangiectasia. Eur J Neurol 2011;18:564–570. [DOI] [PubMed] [Google Scholar]
- 5.Zannolli R, Buoni S, Betti G, et al. A randomized trial of oral betamethasone to reduce ataxia symptoms in ataxia telangiectasia. Mov Disord 2012;27:1312–1316. [DOI] [PubMed] [Google Scholar]
- 6.Chessa L, Leuzzi V, Plebani A, et al. Intra-erythrocyte infusion of dexamethasone reduces neurological symptoms in ataxia teleangiectasia patients: results of a phase 2 trial. Orphanet J Rare Dis 2014;9:5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Magnani M, Rossi L, D'ascenzo M, Panzani I, Bigi L, Zanella A. Erythrocyte engineering for drug delivery and targeting. Biotechnol Appl Biochem 1998;28:1–6. [PubMed] [Google Scholar]
- 8.Trouillas P, Takayanagi T, Hallett M, et al. International Cooperative Ataxia Rating Scale for pharmacological assessment of the cerebellar syndrome. The Ataxia Neuropharmacology Committee of the World Federation of Neurology. J Neurol Sci 1997;145:205–211. [DOI] [PubMed] [Google Scholar]
- 9.Van Duijn G, Dijkxhoorn Y, Noens I, Scholte E, van Berckelaer-Onnes I. Vineland Screener 0–12 years research version (NL). Constructing a screening instrument to assess adaptive behaviour. Int J Methods Psychiatr Res 2009;18:110–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Moxley RT, III, Ashwal S, Pandya S, et al. ; Quality Standards Subcommittee of the American Academy of Neurology, Practice Committee of the Child Neurology Society. Practice parameter: corticosteroid treatment of Duchenne dystrophy: report of the Quality Standards Subcommittee of the American Academy of Neurology and the Practice Committee of the Child Neurology Society. Neurology 2005;64:13–20. [DOI] [PubMed] [Google Scholar]