

Abstract
Change of cell shape in vivo plays many roles that are central to life itself, such as embryonic development, inflammation, wound healing, and pathologic processes such as cancer metastasis. Nonetheless, the spatiotemporal mechanisms that control the concerted regulation of cell shape remain understudied. Here, we show that ribosomal S6K, which is normally considered a protein involved in protein translation, is a morphogenic protein. Its presence in cells alters the overall organization of the cell surface and cell circularity [(4π × area)/(perimeter)2] from 0.47 ± 0.06 units in mock-treated cells to 0.09 ± 0.03 units in S6K-overexpressing macrophages causing stellation and arborization of cell shape. This effect was partially reversed in cells expressing a kinase-inactive S6K mutant and was fully reversed in cells silenced with small interference RNA. Equally important is that S6K is itself regulated by phospholipids, specifically phosphatidic acid, whereby 300 nM 1,2-dioleoyl-sn-glycero-3-phosphate (DOPA), but not the control 1,2-dioleoyl-sn-glycero-3-phosphocholine (DOPC), binds directly to S6K and causes an ∼2.9-fold increase in S6K catalytic activity. This was followed by an increase in Filamin A (FLNA) functionality as measured by phospho-FLNA (S2152) expression and by a subsequent elevation of actin nucleation. This reliance of S6K on phosphatidic acid (PA), a curvature-inducing phospholipid, explained the extra-large perimeter of cells that overexpressed S6K. Furthermore, the diversity of the response to S6K in several unrelated cell types (fibroblasts, leukocytes, and invasive cancer cells) that we report here indicates the existence of an underlying common mechanism in mammalian cells. This new signaling set, PA-S6K-FLNA-actin, sheds light for the first time into the morphogenic pathway of cytoskeletal structures that are crucial for adhesion and cell locomotion during inflammation and metastasis.—Henkels, K. M., Mallets, E. R., Dennis, P. B., Gomez-Cambronero, J. S6K is a morphogenic protein with a mechanism involving Filamin-A phosphorylation and phosphatidic acid binding.
Keywords: cytoskeleton signaling, cell morphology, morphogenesis, breast cancer, cell invasion
Maintenance of cell shape is essential for proper cell function, such as wound healing, chemotaxis, and embryonic development. Coordinated reorganization of the cell cytoskeleton occurs during endocytosis and exocytosis (e.g., in synaptic vesicles of the neuronal cell junction), during adhesion and cell chemotaxis (e.g., in leukocytes during inflammation) (1, 2), during the establishment of cell polarity and cell-cell interactions (3) (e.g., gastrointestinal or lens epithelial cells) and also has been observed in invading cells (e.g., cancer metastasis). In the latter, cells can adopt an elongated morphology indicative of a mesenchymal migration mode or a rounded appearance that is displayed as an amoeboid motility that comprises a variety of protrusion types (lamellipodia, filopodia, and blebs) relative to different cell migration modes (4–6).
Stellation or “star shape” is a normal anatomic feature present in astrocytes and neurons, as well as in hepatocytes and pancreatic cells. This plasticity that exists between cell shape and protrusion formation results in cells that can adapt to and modulate aspects of their microenvironment during cell migration. The determinants of the cell shape are provided by the cortical cytoskeleton (7, 8). Many of the cortical proteins in the cytoskeleton (actin, myosin, tubulin, villin, and profilin) are the substrates for a variety of kinases, such as PI3K/Ak strain transforming (AKT) (7–9). However, because PI3K/AKT is the initiator of several cell injury pathways, it is not clear what particular protein member/link is responsible for PI3K-mediated changes in cell shape.
A prominent, downstream member of the PI3K family is S6K that has 2 isoforms, S6K1 and S6K2, and whose activities are increased by phosphorylation on several sites in response to cellular stimulation by mitogens and growth factors. In fact, S6K does not just regulate protein synthesis but may regulate actin polymerization and cytoskeleton integrity (10). S6K and actin have been shown to form a protein-protein interaction through cosedimentation/differential sedimentation assays (10). This interaction is a direct binding event where S6K cross-links with actin filaments. Further, S6K has been shown to localize to the actin arc (9).
The current study defined a new role for S6K in relation to cell shape change, which is the prelude to cell migration. It was found that S6K induced changes in cell morphology that were mediated by phosphorylation of FLNA and S6K was under the regulation of PA, which was needed for the formation of extended membrane protrusions.
MATERIALS AND METHODS
Plasmid DNAs
Full-length, myc-tagged S6K1-wild-type (WT), -T389E, and -kinase-dead (KD) (S6K-T389A) were cloned into pRK5 expression vectors by (11). One-half microliter of each plasmid DNA was transformed separately into 100 μl of Max Efficiency DH5α competent cells (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s protocol. Aliquots (100 μl) of each transformed plasmid was pipetted and evenly spread onto separate sterile Luria broth (LB) + agar plates supplemented with a final concentration of 100 μg/ml ampicillin. Plates were covered, inverted and incubated at 37°C overnight. Three individual colonies from each DNA sample were each grown separately in 5 ml LB supplemented with 100 μg/ml ampicillin and incubated at 37°C with aeration for approximately 6 h. Each plasmid was then produced on a larger scale by seeding an aliquot of each 5 ml liquid culture into separate 500 ml LB cultures supplemented with 100 μg/ml ampicillin. Each large-scale liquid culture was incubated at 37°C with aeration overnight. Three milliliters of each 500 ml culture was used to make 15% glycerol stocks, which were frozen at −70°C to be used as stocks to seed future preparations of these DNA plasmids. The remaining volume of each liquid culture was purified according to the Qiagen Plasmid Maxi-Prep Kit (Invitrogen). The full-length FLNA plasmid (pcDNA3-mycFLNA-WT; Addgene plasmid 8982) was from Dr. John Blenis via Addgene (Cambridge, MA, USA) (12).
Cell migration (chemotaxis) and phagocytosis assays
For S6K inhibitor experiments, untransfected or S6K-transfected RAW264.7 cells were incubated in 0 or 100 nM Ro31-8220 (Sigma-Aldrich, St. Louis, MO, USA) in chemotaxis buffer for 1 h before the start of chemotaxis. Eighteen hours post-transfection, each set of mock or transfected RAW264.7 cells was loosened from the 4 × 35 mm plates using 500 μl cell dissociation solution (Sigma-Aldrich) and combined with the cell culture media in which the cells had been incubated previously. Cell viabilities were determined and cell numbers normalized for all sets. Untransfected or transfected cells were sedimented at 2000 rpm for 5 minutes at room temperature and resuspended in chemotaxis buffer [0.1% bovine serum albumin (BSA) in RAW DMEM] at a concentration of 3 × 106 cells/ml. Approximately 3 × 105 cells were applied to the top of a 24-well, 8 mm pore size transwell insert (Corning, Corning, NY, USA) coated with 10 mg/ml collagen I (Sigma-Aldrich), while 500 μl of either chemotaxis buffer alone or chemotaxis buffer + the appropriate concentration of macrophage inflammatory protein (MIP)-1α (Cell Sciences, Inc., Canton, MA, USA) was added to the bottom well of the transwell plate. Collagen-coated transwells containing migrating cells were incubated in a cell culture incubator at 37°C for approximately 3 hours. The stained filters were removed from the inserts and mounted onto glass microscope slides. Five fields of each filter were photographed at ×20 magnification under bright field light conditions.
Cell shape/morphology evaluation
Imaging allows quantification of cell size, shape, and texture that are useful in the study of differentiation of stem cells, hematology and oncology. Reducing a cell’s complex shape to a single readout is challenging. We have measured the number of cell protrusions or “arborizations” as described elsewhere (4). Additionally, we have quantified cell form by measuring cell roundness using ImageJ software (13). Cell Circularity can be quantified from 2-dimensional images of the cells by comparing the surface of a cell to its periphery. Cell Circularity = (4π × area)/(perimeter)2 and for a perfect circle is equal to 1, and as the shape becomes more convoluted, the value decreases and is <1.
Immunofluorescence
RAW264.7 cells were transfected using 1.5 μg plasmid DNA and were plated at onto coverslips placed into 35 mm tissue culture dishes. Eighteen hours post-transfection, cells were fixed onto the coverslips using 4% paraformaldehyde for 10 minutes at room temp. Cells were permeabilized using 0.5% Triton X-100 in PBS for 10 min at room temp. Cells were blocked for 4 hours at room temp using 10% fetal calf serum in PBS and 0.1% Triton X-100 (PBS-T). Cells were washed 3× with PBS-T for 5 minutes each wash and then probed overnight at 4°C in a 1:1000 dilution of α-myc-FITC antibody in blocking buffer. Coverslips were again washed 3× with PBS and then incubated in a 1:200 dilution of phalloidin-tetramethylrhodamine isothiocyanate (TRITC) in PBS for 2 hours at room temperature. Slides were kept in the dark until needed and were viewed using a Nikon Upright Eclipse 50i Tissue Culture Microscope, a Plan Fluor 100×/1.30 oil objective and FITC, TRITC, or DAPI fluorescence filters. Photomicrographs were obtained using a Diagnostics Instrument (Sterling Heights, MI, USA) Spot 6 digital camera and MetaVue software.
Kinase assays
The catalytic activity of purified, recombinant S6K protein in the absence or presence of PA was measured in the presence of 8 mM 3-(N-morpholino)propanesulfonic acid (MOPS)/NaOH, pH. 7.0, 0.2 mM EDTA, 10 mM Mg-acetate, 0.1 mM ATP, 1 μCi [γ-32P]ATP and 100 μM S6K/RSK2 peptide substrate 2 at 30°C for 10 minutes. Reactions were stopped by the addition of 5 µl 3% phosphoric acid and then spotted onto P81 Whatman filter paper, which was washed with running water for 5 minutes. Filters were air-dried and cut into individual samples, placed into scintillation vials containing Scintiverse II (Fisher Scientific, Chicago, IL, USA) and quantified using disintegrations per minute per milligram of protein. Alternatively, to measure S6K activity on FLNA directly, purified, recombinant FLNA (Origene, Rockville, MD, USA) or a synthetic peptide substrate representing the primary FLNA phosphorylation site (5′-RRRAPSVANVG-3′) (Bio-Synthesis, Inc., Lewisville, TX, USA) were used in place of the S6K-RSK2 peptide substate.
Actin polymerization assays
The pyrene actin polymerization assay was used (14). RAW264.7/LR5 cells were either transfected with the relevant expression plasmids for 2 days or they were treated with PA for 10 minutes prior to harvesting. The PA was prepared in 0.5% fatty acid–free BSA in PBS. Cells were sonicated in actin lysis buffer (20 mM Tris-HCl, 20 mM NaCl, and 768 nM aprotinin). Ten microliters of cell lysates were added to 85 μl pyrene-labeled actin-containing buffer, which was purchased as a kit (BK003) from Cytoskeleton, Incorporated (Denver, CO, USA). Actin polymerization buffer (10 μl) was added to the reaction for a final total volume of 105 μl. Actin polymerization was measured for 12 minutes at 30-second intervals at the excitation of 350–360 nm with a bandwidth of 20 nm and the emission 401–411 nm with a bandwidth of 10 nm in a TECAN Safire2 (Morrisville, NC, USA) spectrophotometer microplate reader at room temperature. In vitro actin polymerization assay was performed as outlined in the manufacturer’s (Cytoskeleton, Incorporated) instructions except that the protein of interest (Arp3) was incubated with increasing concentrations of PA for 10 min before beginning the assay. The Arp3 recombinant protein was from Novus Biologicals (Littleton, CO, USA).
Protein-lipid binding assays
The method for preparing and detecting protein-lipid binding has previously been described (15). In brief, increasing concentrations of either DOPA or DOPC lipids from Avanti Polar Lipids (Alabaster, AL, USA) were spotted onto a PVDF membrane. DOPA or DOPC were dissolved in a 2.0:1.0:0.8 ratio solution of MeOH:CHCl3:H2O. Appropriate amounts of lipid ranging from 1 to 30 μg were spotted onto the membrane. The membrane was blocked overnight with a 3% fatty acid–free BSA solution. The membrane was then incubated overnight with recombinant S6K protein. After protein incubation, the membrane was incubated overnight with the relevant primary antibody and was then incubated with the corresponding secondary antibody, which was detected using enhanced chemiluminescence.
Coimmunoprecipitation
Immunoprecipitation (IP) experiments were performed with either untransfected or transfected cells (S6K only, FLNA only or both S6K + FLNA together). Cells were harvested and lysed with a special lysis buffer (5 mM HEPES, pH 7.8, 100 μM sodium orthovanadate and 0.1% Triton X-100). Cell lysates for untransfected or transfected cells were treated with 1 μl monoclonal antibody for FLNA or S6K, respectively, along with 10 μl agarose beads (Millipore, Billerica, MA, USA) and incubated at 4°C overnight, which would result in all of the endogenous and overexpressed FLNA or S6K in the cell lysates being immunoprecipitated. After incubation, the immunoprecipitates were washed with LiCl wash buffer (2.1% LiCl, 1.6% Tris-HCl, pH 7.4) and NaCl wash buffer (0.6% NaCl, 0.16% Tris-HCl, 0.03% EDTA, pH 7.4), respectively, and sedimented at 12,000 × g for 1 minute. Immunoprecipitated samples were analyzed by SDS-PAGE using 4–20% gradient gels that were then transferred onto PVDF membranes for subsequent use in Western blot (WB) analyses to detect for the presence of overexpressed myc-tagged S6K binding to FLNA or overexpressed myc-tagged FLNA binding to S6K. Similar co-IP reactions were also performed to detect for the presence of actin in the subsequent WB analyses. In a separated set of duplicate samples that were not immunoprecipitated, equal amounts of protein similar to that of the immunoprecipitated samples was electrophoreses on SDS-gels and used for WB analyses to act as equal protein loading control.
Statistical analysis
Data are presented as means ± sem. The difference between means was assessed by the single factor analysis of variance test. Probability of P < 0.05 indicated a significant difference.
RESULTS
S6K-WT overexpression results in increased dendritic protrusions and stellated macrophage morphology
Using a pRK5-N1-myc–tagged vector as the backbone that had WT or mutant p70S6K cloned into it (Fig. 1A–C), we observed that RAW264.7 macrophages and COS-7 cells overexpressing S6K had cell morphologies markedly different from mock-transfected cells (Fig. 1D, E). A significant increase in actin-rich dendritic protrusions visualized using a phalloidin-TRITC label (red fluorescence staining of polymerized actin) occurred following overexpression of S6K-WT in RAW264.7 macrophages (Fig. 1D). In parallel experiments, cells overexpressing myc-tagged S6K-WT were labeled with anti-myc antibodies conjugated to FITC to detect the presence of the overexpressed S6K protein in immunofluorescence images from COS-7 cells. A stellated appearance in COS-7 cells was also evident as a result of S6K-WT overexpression when compared with mock-treated (Fig. 1E), which is similar to that shown for RAW264.7 macrophages. These dendritic protrusions were represented by a ∼3-fold increase in the number of elongated lamellipodia/filopodia (Fig. 1F). Stellation or star shape is a normal anatomic feature present in astrocytes and neurons, as well as in hepatocytes and pancreatic cells. Cells that expressed only endogenous levels of wild-type S6K protein had a more spherical shape that evolved into a more fusiform and finally stellated morphology following overexpression of S6K. Additionally, although S6K overexpression yielded a small increase in the cell area of both types of cells (Fig. 1G, H), there was a considerably more significant increase in the cell perimeter for both the macrophage and fibroblast cell lines used for our study (Fig. 1I, J), which was manifested as a 2- to 3-fold increase compared with mock-treated cells. These data presented in this figure suggest that relative changes in cell morphology are under the control of S6K expression.
Figure 1.

Overexpression of S6K results in increased dendritic protrusions and stellated morphology. A, B) An S6K plasmid (A) transfected into RAW264.7 macrophages produced the expected myc-S6K products detected by PCR peaking at 18–24 hours post-transfection (B). C) A schematic model of the S6K1 architecture with key phosphorylation sites. D–F) S6K overexpression alters cell morphology of RAW264.7 macrophages (D) and COS-7 cells (E) when compared with mock-transfected macrophages. A significant increase in dendritic protrusions occurred following overexpression of S6K-WT, as measured using α-phalloidin-TRITC (specific for polymerized actin) or α-S6K-FITC IgG antibodies that bound to S6K. Micrographs are representative images observed in 6 fields per condition. F) Quantification of dendritic protrusions in cells that overexpress S6K versus mock cells as measured by the relative mean number of protrusions per cell ± sem. G–J) Quantifications of RAW264.7 macrophage (G, H) and COS-7 cell (I, J) cell areas (G, I) and cell perimeters (H, J). MCS, multiple cloning site; ORI, origin of replication; PCMV, cytomegalovirus promoter; PolyA, polyadenylated tail. Data are presented as mean values ± sem from n = 12 different field of cells quantified. *P < 0.05, statistically significant increase, between samples and controls. #P < 0.05, statistically significant decrease, between samples and controls.
Mutation in the active site of S6K results in altered macrophage morphology
Next, we observed the effect of mutant S6K protein expression using macrophages that overexpressed S6K-WT, S6K-T389E, or S6K-T389A (kinase dead, S6K-KD). S6K-WT is the WT S6K protein, S6K-T389E is a mutant S6K that retains kinase activity but is resistant to rapamycin, and S6K-KD is a mutant that is kinase-inactive but still interacts with mammalian target of rapamycin and has some rapamycin-sensitive phosphorylation sites. As indicated in Fig. 2A, B, both macrophages and fibroblast cells transfected with S6K-WT DNA contained numerous filopodia (second panels from top) compared with controls (top panels). This is also shown in cells that overexpressed the S6K-T389E kinase-active mutant that was able to still drive the formation of some filopodia (Fig. 2A, B, second panels from bottom), and the kinase-dead S6K-KD mutant did not (Fig. 2A, B, bottom panels). These effects were also observed in COS-7 cells (Fig. 2B). The S6K-KD mutant is kinase inactive, so it would not demonstrate the enhanced regulated activity that the WT S6K did, and this mutant S6K would probably suppress activation of endogenous S6K through a dominant interfering mechanism.
Figure 2.

S6K mutation reverses cell shape changes. A, B) The specific subcellular localization of overexpressed myc-tagged S6K plasmids in either RAW264.7 macrophages (A) (shown are two representative examples among 6 different fields) or COS-7 fibroblasts (B). Immunofluorescent signals are from c-myc-FITC or c-myc-TRITC IgG antibodies, respectively. Cells transfected with S6K-WT DNA contained numerous filopodia (white arrowheads), which are less evident in S6K-T389E or -KD expressing cells. C) Schematic drawing of cell shape changes typical of macrophage, neuronal or transformed cancer cell morphology. D, E) Cell circularities of RAW264.7 macrophages and COS-7 cells are significantly affected due to S6K overexpression. Cell circularity was calculated as indicated in Materials and Methods. Data are presented as mean values ± sem from n = 12 cells quantified. *P < 0.05, statistically significant increase, between samples and controls. #P < 0.05, statistically significant decrease, between samples and controls.
Additionally, we correlated altered cell morphology as a result of S6K overexpression to altered cell circularity (Fig. 2C), which is a function of both the area and perimeter of the cell. As shown in Fig. 2D, E, and Table 1, when S6K-WT was overexpressed in both RAW264.7 macrophages and COS-7 cells, cell circularity was significantly decreased. Cell circularities were 0.47 ± 0.06 units in mock-treated macrophages compared with 0.088 ± 0.027 units in S6K-overexpressing macrophages and 0.60 ± 0.04 units in mock-treated COS-7 cells compared with 0.10 ± 0.05 units in S6K-overexpressing COS-7 cells. This negative effect was then partially reversed when the other kinase-active S6K-T389E or the kinase-inactive S6K-KD was overexpressed, which resulted in cell circularity that began to approach levels similar to the mock-treated cells. Collectively, the data in Fig. 2 demonstrate that phenotypic changes elicited by S6K overexpression were observed in both macrophages and fibroblast cell lines, suggesting a general, cell-type independent mechanism of action that could be typified in human basal or disease states synonymous with these cell types. This is the first reported observation that S6K induced a dramatic stellation phenotype. Narrow, elongated processes were formed by direct outgrowth from the round cells in all directions. With S6K overexpression, the rounded cell body experienced a dramatic shape change toward stellation or arborization, which is reminiscent of neurons or transformed cells.
TABLE 1.
Mean cell circularities ± sem (relative units)a
| Transfection condition | RAW264.7 Φ | COS-7 cells | MDA-MB-231 |
|---|---|---|---|
| Mock | 0.47 ± 0.06 | 0.61 ± 0.004 | 0.31 ± 0.02 |
| S6K-WT | 0.09 ± 0.03 | 0.10 ± 0.05 | 0.15 ± 0.01 |
| S6K-T389E | 0.20 ± 0.04 | 0.32 ± 0.04 | 0.29 ± 0.02 |
| S6K-KD | 0.32 ± 0.01 | 0.51 ± 0.06 | ND |
ND, not determined.
Data derived from ImageJ software analysis.
Morphologic changes in macrophages occurred independent of a negative effect on S6K transfection efficiency and cell viability
To eliminate the possibility that the observed changes in cell morphology were not as a result of a negative effect on transfection efficiency or cell viability, we transfected RAW264.7 macrophages with similar levels of the different S6K plasmids, both WT and mutant, for 36 hours. As shown in Fig. 3A, overexpression of the different S6K proteins was achieved compared with endogenous levels of S6K in the mock sample (top panel), which validated these expression vectors for subsequent experiments dealing with changes in cell morphology. Next, we stained live, S6K-transfected cells with crystal violet, as only viable, live cells will take up this dye, which allowed us to determine that no significant difference existed in the cell viability (as measured using crystal violet assay) of the S6K-overexpressing cells, which was between 70% and 80%, compared with mock-transfected cells, which had a cell viability of ∼80% (Fig. 3B, right panels). Immediately following crystal violet staining, the same samples were then fixed with paraformaldehyde and used for immunoblotting using α-myc-TRITC and DAPI to detect overexpressed S6K and to stain nuclei. We observed that transfection/electroporation efficiencies were between 85% and 90% for all transfected cells at 36 hours post-transfection (Fig. 3B, left panels). Figure 3C is the quantification of the microscopic images shown in Fig. 3B. These data indicate that the S6K WT and mutant proteins were similarly overexpressed and had similar transfection efficiencies/cell viabilities. Additionally, the morphologic changes we observed in Fig. 1 were as a result of some other mechanism not associated with a negative effect on cell transfection/viability. We propose that these cell shape changes occurred as a result of overexpression of S6K-WT in macrophages (Fig. 3D) and epithelial (Fig. 3E) cells, whereby the changes in cell shape from 0 to 48 hours post-transfection evidenced using bright-field microscopy were a continual and active process of outgrowth of the cells when compared with the mock-treated cells that had fewer dendritic outgrowths.
Figure 3.

S6K overexpression does not affect transfection efficiency or cell viability. A–C) RAW 264.7 cells were transfected with 1.5 μg plasmid DNA for 18 h. A) Cell lysates were prepared from S6K transfected cells and used for WB analysis. Effect of overexpression of S6K plasmids is shown. Chemiluminescent detection of the overexpressed 70 kDa myc-tagged S6K protein (upper panel) is denoted on the right by an arrow. Actin is presented as the equal protein loading control (lower panel). Results presented are typical of 3 different assays. B) Cells were incubated with 0.1% crystal violet in 2% EtOH, fixed with paraformaldehyde and then used for immunoblotting using myc-TRITC to detect overexpressed S6K and DAPI to detect nuclei staining. Only viable, live cells will take up crystal violet and result in intense purple staining within the cell. C) Quantification of transfection efficiency and cell viability shown in (B). Data are presented in terms of mean percent transfection efficiency [(red-stained cells only/all cells) ∣ 100%] ± sem or mean percent cell viability [(crystal violet-stained cells only/all cells) ∣ 100%] ± sem. *P < 0.05, statistically significant increase, between samples and controls. D, E) Time lapse change in cell morphology as a result of S6K overexpression. RAW 264.7 (D) or COS-7 (E) cells were transfected with 1.5 μg plasmid DNA for up to 48 hours and bright-field photomicrographs were imaged at 0, 6, 12, 24, 36, and 48 hours post-transfection.
S6K mediates cell circularity
By silencing expression of either S6K1 or S6K2 in macrophages using 300 nM siRNA specific for either of the 2 different S6K isoforms (Fig. 4A, inset), cells became more rounded in the presence of siS6K1 or siS6K2 (Fig. 4A) when compared with the less rounded appearance of the mock-treated cells, which also reiterated that S6K was important to cell shape changes. As a control, we also measured MIP-1α–mediated RAW264.7 cell chemotaxis with the same experimental conditions and found cell migration was also inhibited by silencing S6K (Fig. 4E). Additionally, we found that macrophages were more rounded in the presence of the S6K small-molecule inhibitor Ro31-8220 (10 nM) compared with cells that overexpressed S6K-WT alone (Fig. 4B). This increase in cell circularity due to Ro31-8220 treatment also resulted in significantly reduced cell migration in the absence or presence of S6K-WT overexpression (Fig. 4F). Control chemotaxis experiments using optimal conditions are shown in Fig. 4C, D. Data presented in this figure established a link between both S6K-mediated cell migration and S6K-mediated macrophage cell shape change.
Figure 4.

S6K-mediated cell shape changes contribute to cell migration. A, B) RAW264.7 macrophages were transfected with 300 nM siRNAs specific for either S6K1 or S6K2 (A) or 2 µg S6K-WT plasmid for 3 days using electroporation and then mock-treated or treated with 10 nM of the S6K-specific inhibitor Ro31-8220 for 20 minutes (B); subsequent cell lysates were used for WB (insets, respectively). A) Bright-field photomicrographs of RAW264.7 macrophages were imaged at 72 hours post-transfection and are presented as duplicate fields that indicate cells became more circular in appearance following silencing of S6K. C, D) Optimization of MIP-1α–mediated chemotaxis as a function of time (C) and MIP-1α concentration (D) in the absence or presence of 10 nM of the S6K-specific inhibitor Ro31-8220. Results are presented as the mean number of cell migrated compared with the control ± sem of triplicate samples. E, F) Samples in (A) and (B) were also used for MIP-1α–mediated chemotaxis in response to 10 nM MIP-1α for 1 h. *P < 0.05, statistically significant increase, between samples and controls. #P < 0.05, statistically significant decrease, between samples and controls.
Certain physiologic processes are dependent on kinase-active S6K expression: chemotaxis, phagocytosis, and cell invasion
Next, we determined whether cell shape changes driven by mutation of S6K shown in Fig. 2 resulted in measurable cytologic changes in macrophage biology, specifically chemotaxis and phagocytosis. Transfection of S6K-WT or -T389E plasmid significantly stimulated RAW264.7 cell chemotaxis, whereas no such increase was observed for the S6K-KD mutant (Fig. 5A). Additionally, when the S6K small-molecule inhibitor Ro31-8220 was present in the cell migration reaction at a concentration of 10 nM, macrophage chemotaxis was inhibited approximately to that of basal levels in the cells that overexpressed WT S6K or the S6K-KD mutant, and the S6K inhibitor Ro31-8220 had no effect on MIP-1α–independent or –dependent chemotaxis of S6K-T389E overexpressing macrophages. As a second physiologic readout of macrophage functionality, we studied phagocytosis. Very similar to our chemotaxis results, S6K overexpression in macrophages increased zymosan-stimulated phagocytosis compared with the mock control (Fig. 5B), which was significantly decreased by 10 nM Ro31-8220 treatment. For the most part, although the chemotaxis and phagocytosis are two different systems of physiologic readout of the cells, chemotaxis and phagocytosis do not necessarily have similar pathways involved in both processes, which could be at play in our cells following transfection/overexpression of S6K-WT and its mutants.
Figure 5.
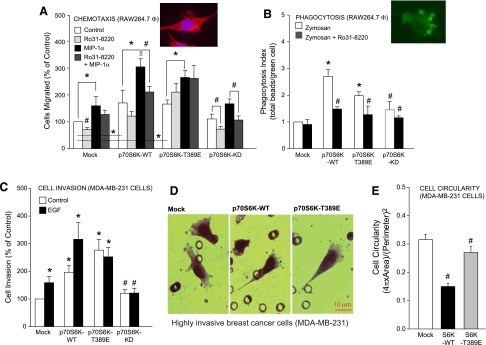
Physiologic relevance of S6K to chemotaxis, phagocytosis, and cell invasion. RAW264.7 macrophages (A, B) or MDA-MB-231 breast cancer cells (C, D) were transfected with 2 µg of the relevant S6K plasmid and 2 days post-transfection cells were used for MIP-1α–mediated chemotaxis (A), zymosan-mediated phagocytosis (B), or EGF-mediated cell invasion of matrigel embedded PET membranes (C, D). A) Chemotaxis results are presented as the mean effect from triplicate sets of assays compared with control ± sem. B) Phagocytosis index is expressed in terms of the total number of zymosan beads phagocytosed by the cells/total number of zymosan-containing cells per field ± sem. Triplicate assays were performed. C) Cell invasion is expressed in terms of the mean percent of invading cells (%) compared with the control ± sem. D) Hematoxylin-stained MDA-MB-231 breast cancer cells are more stellated following overexpression of kinase-active S6K (-WT or -T389E, middle and right panels) compared with mock-treated cells (left panel) following cell invasion through matrigel matrix and the PET membrane. E) Mean cell circularity measurements of samples shown in ± sem (D). *P < 0.05, statistically significant increase, between samples and controls. #P < 0.05, statistically significant decrease, between samples and controls.
In a further effort to see whether the effects of S6K on cell shape would extrapolate beyond those observed in macrophages, we changed cell types and looked at cell invasion using a cancer cell line. We found that endothelial growth factor (EGF)–mediated cell invasion and EGF-mediated formation of dendritic protrusions in human breast cancer cells (MDA-MB-231) was more pronounced in cells that overexpressed S6K compared with mock-transfected control cells (Fig. 5C and D, respectively). We also noticed that cells that migrated through both the matrigel matrix and the pores in the polyethylene terephthalate (PET) membrane were more fusiform in shape in S6K-WT or S6K-T389E overexpressing cancer cells compared with the more rounded appearance of cells in the mock-transfected samples. Additionally, we also observed a significant decrease in cell circularity of the S6K-overexpressing MDA-MB-231 cancer cells compared with mock-transfected controls (Fig. 5E), which is similar to data obtained from RAW264.7 macrophages and COS-7 epithelial cells in Fig. 2. Collectively, data from S6K-transfected RAW264.7 and MDA-MB-231 cells suggested that chemoattractant-induced chemotaxis, phagocytosis, and cell invasion occurred as a result of a S6 kinase-mediated process. Thus, the effect of S6K overexpression on cell morphology was observed in multiple cell lines from diverse lineages, representative of many different normal or disease states.
PA interacts with S6K to form a functional, lipid-protein complex that aids in actin polymerization
Earlier reports have documented the positive effect of PA on S6K (16–18), which links the catalytic product of the phospholipase D (PLD) reaction or PLC/diacylglycerol kinase (DAGK) in tandem to the S6K pathway. Therefore, we concentrated next on the effects of lipids, particularly PA, on S6K. As shown in Fig. 6A, B, S6K bound to increasing amounts of PA in the form of DOPA when DOPA was first bound to a PVDF membrane and then incubated in the presence of purified, recombinant S6K protein. Following incubation with primary antibodies specific to the S6K protein, the protein-lipid (S6K-PA) interaction was observed using enhanced chemiluminescence and autoradiography. Of note, no binding occurred between S6K and phosphatidylcholine in the form of DOPC.
Figure 6.
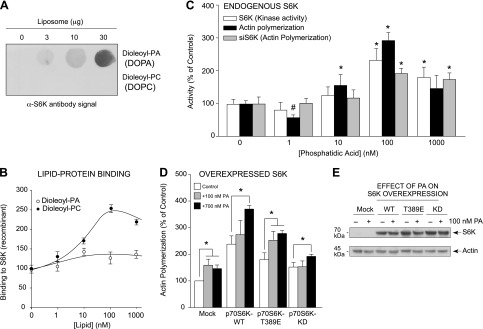
PA and S6K bind together and form a lipid-protein complex that aids in actin polymerization. A, B) In vitro assay of PA:S6K lipid-protein binding. Increasing concentrations of either DOPA or DOPC lipids were spotted onto the PVDF membrane that was incubated first with purified, recombinant S6K protein and then α-S6K antibody. A) The resulting lipid-protein interaction was detected using enhanced chemiluminescence and autoradiography that was then densitometrically quantified in (B). C) Measurements in parallel of actin polymerization and S6K kinase activity from PA-treated samples, similar to (B), using purified, recombinant S6K (kinase activity and actin polymerization) or cell lysates from macrophages that were silenced with 300 nM siRNA specific for S6K and subsequently treated with increasing PA for actin polymerization assays. D) RAW264.7 macrophages were transfected with 2 µg of the relevant S6K plasmid. Two days post-transfection, cells were incubated in the absence or presence of 300 nM PA for 20 minutes, and then lysates were prepared that were used for the pyrene-based actin polymerization assays. Overexpression of kinase-active S6K resulted in increased actin polymerization, while kinase-inactive S6K had no additional effect on actin polymerization. E) WB protein loading controls from samples used in (D) that show overexpression of S6K proteins during 300 nM PA treatment. *P < 0.05, statistically significant increase, between samples and controls. #P < 0.05, statistically significant decrease, between samples and controls.
Another way the interaction between S6K and PA was also evidenced was as a positive effect on both S6K activity and actin polymerization when in vitro based assays were used (Fig. 6C). We measured the ability of purified, recombinant, full-length, low basal activity S6K to phosphorylate a synthetic substrate peptide in the presence of PA that also affected the polymerization of monomeric actin. Optimal binding of PA to S6K occurred at ∼100 nM DOPA (Fig. 6B), which was also the optimal concentration of PA in the S6K activity and actin polymerization assays (Fig. 6C). These results suggest that a direct binding event between S6K and PA occurred in the cell that positively enhanced actin polymerization and dendritic protrusions.
We have shown in Fig. 1 that overexpressing S6K-WT yielded an increase in filopodia that could be visualized with TRITC-phalloidin. To confirm these data, we also measured actin polymerization in vitro (Fig. 6D). Using a pyrene-based assay, actin polymerization increased ∼2.5-fold in the case of WT S6K overexpression and about half that amount in S6K-T389E overexpressing extracts, whereas the kinase-inactive mutant S6K-KD demonstrated in vitro actin polymerization comparable to that of the mock-transfected cells. The addition of 100 nM PA to the reaction further increased actin polymerization in the case of the mock or kinase-active S6K samples (S6K-WT and S6K-T389E), which was not the case for the kinase-inactive S6K (S6K-KD). Taking into consideration the data shown in Fig. 6C, D, there is a dependence of the cells with endogenous levels of S6K for PA during acting polymerization that is even more pronounced when S6K is overexpressed and in abundance. We hypothesize that S6K overexpression resulted in S6K flooding the cells and this condition required more PA for actin polymerization to occur, which yielded a positive effect on actin polymerization only at 700 nM PA (Fig. 6D). Figure 6E is the WB-positive controls of S6K overexpression relative to actin loading controls. Although this in vitro PA/S6K binding could have consequences in vivo, these data agree with observations made in Fig. 2 and also indicate the importance of S6K activity in macrophages to signaling events that affect the level of actin polymerization and cell protrusion formation.
S6K and FLNA form a protein-protein complex that contributes to increased actin polymerization
Next, we investigated the mechanism by which S6K would affect cell shape and increase actin polymerization. We found that S6K used FLNA as a phosphorylation substrate (Fig. 7), which increased actin polymerization (Fig. 8). FLNA is a high molecular weight protein central to actin functionality that aligns along actin filaments at F-actin junctions and is known to be involved in cell shape changes (19, 20). Using the BLAST database (http://blast.st-va.ncbi.nlm.nih.gov/Blast.cgi), we found that there was considerable homology of the FLNA amino acid sequence to that of the ribosomal S6 protein, which is a well-characterized downstream protein substrate of S6K. We found that S2152 of FLNA had significant sequence alignment with S236 of S6 (Fig. 7A). This alignment between S6 and FLNA is also consistent with consensus sequences for both AKT [RXR(X2)S/T] and p70S6K [R(X2)SX] that typifies potential phosphorylation sites on downstream targets that AKT and S6K each favor (21, 22). Additionally, the BLAST search yielded a second area of homology between 2 regions on FLNA and one region on S6, which is signified by a consensus sequence of VK(X6)PG. These two areas of homology on FLNA are located within the 17th and 24th repeat β-pleated sheet units (VKYNEQHVPG and VKWGDEHIPG, respectively) that contain alternating runs of hydrophilic and hydrophobic residues, while the corresponding homologous region on S6 resides (VIVKKGEKDIPG) in its second exon (23, 24).
Figure 7.

In vitro phosphorylation of filamin-A by S6K. A) BLAST database search results of amino acid sequence homology between various S6K phosphorylated protein targets: S6K autophosphorylation site (S421), ribosomal S6 phosphorylation site (S236), and potential phosphorylation sites on LFNA (S2152, S2157, and S2163). B) In-gel phosphorylation assay of FLNA by S6K. Varying concentrations of FLNA (0–300 ng) was used in vitro with 50 µg recombinant S6K and 1 µCi [32P]ATP. Proteins were electrophoresed on SDS-polyacrylamide gels and transferred onto PVDF membranes that were then used for autoradiography to detect 32P incorporation. A long (4 hours) exposure (A) was needed to observe FLNA phosphorylation by S6K, whereas a short exposure (30 minutes) was needed to observe auto-phosphorylation of S6K (B). C) Densitometric quantification of the 280 kDa phospho-band from the in-gel phosphorylation assay similar to the one presented in (B). D) In vitro kinase assays in which 32P was incorporated onto FLNA by S6K relative to similar S6K reactions in the presence of the downstream RSK2 synthetic peptide substrate only (no FLNA) that were measured using scintillation counting. The red line and open circles represent the results of S6K using FLNA as a substrate and the corresponding phosphorylation. The green line and closed circles represent a positive control using the standard peptide substrate of S6K (RSK2) used only as a readout of phosphorylation. E, F) S6K catalytic assays were performed using purified, recombinant S6K and measured the incorporation of [γ-32P]ATP in the presence of increasing concentrations of various synthetic peptide substrates representing potential FLNA serine residue phosphorylation sites and constant S6K (E) or increasing concentrations of S6K with constant 100 µM FLNA peptide substrates (F).
Figure 8.

S6K and FLNA form a protein-protein complex that contributes to increased actin polymerization. A) Schematic cartoon representing the myc tags and both S6K and FLNA protein domains used for co-IP reactions. B) Co-immunoprecipitation assays of protein-protein interaction between FLNA and S6K. RAW264.7 cell lysates that overexpressed mock, S6K, FLNA, or both S6K + FLNA were prepared and immunoprecipitated with rabbit antibodies specific to FLNA (top panel) or S6K (second panel from top) that were then used for SDS-PAGE and subsequent WB and detection using rabbit α-myc antibodies specific for myc-tagged S6K (top panel) or myc-tagged FLNA (second panel from top). Interactions of actin with S6K and FLNA are shown in the panels second and third from the bottom, respectively, indicating that actin is also complexed with the FLNA-S6K heterodimer. Protein loading controls are presented in the lowest panel (WB α-actin). C, D) S6K-WT overexpression increased phosphorylation of FLNA. RAW264.7 macrophages were transfected with increasing concentrations of S6K-WT (0–2 µg) plasmid DNA and 2 d post-transfection cell lysates were prepared that were then used for SDS-PAGE and WB analyses. C) Total S6K, phospho-FLNA, total FLNA, and the actin protein loading controls are shown in the panels from top to bottom, respectively. All samples were run in triplicate. D) Densitometric quantification of samples shown in (C). *P < 0.05, statistically significant, increases between samples and controls. E) Actin polymerization of cell lysates from RAW264.7 cells that were mock-treated (open circles) or overexpressed with 2 µg S6K-WT+2 µg FLNA-WT (red circles) or with 2 µg S6K-KD+2 µg FLNA-WT (black circles). Lag and growth phases of actin polymerization are increased following co-overexpression of kinase-active S6K-WT and FLNA-WT. F) Actin polymerization of 500 ng purified FLNA phosphorylated by 150 ng S6K or S6K alone in a kinase reaction that used cold-ATP in place of radiolabeled [γ-32P]ATP, which was then utilized for the actin polymerization reaction. Significant delay in the lag phase of actin polymerization was evident in response to phospho-FLNA.
Based on the data indicating sequence homology of a serine residue on FLNA with a known serine residue on S6 that is known to be phosphorylated by S6K, we used purified, recombinant S6K and FLNA in an in vitro kinase reaction that incorporated [γ-32P]ATP onto FLNA via S6K action and performed SDS-PAGE and WB transfer onto PVDF. The subsequent PVDF membrane was used for autoradiography, whereby phosphorylation of FLNA by S6K was first detected (Fig. 7B, upper panel), as was autophosphorylated S6K (lower panel). The relative densitometry of phosphorylated FLNA compared with S6K phosphorylation in this reaction is shown in Fig. 7C. We also compared S6K-mediated phosphorylation of full-length FLNA to S6K-mediated phosphorylation of a well-known downstream synthetic peptide substrate, RSK2, in a filter-binding assay and found that optimal phosphorylation of FLNA by S6K occurred between 50 and 100 ng FLNA (Fig. 7D). Knowing that S6K phosphorylation of FLNA could be detected using a filter binding assay, we next utilized synthetic peptide substrates that comprised the amino acid sequence of FLNA S2147-G2157 (Fig. 7A) in similar kinase reactions, whereby a S6K synthetic peptide substrate was used as the positive control (Fig. 7E, F, insets). As shown in the larger panels for Fig. 7E, F, the FLNA substrate was phosphorylated by S6K when either the peptide substrate or the enzyme were varied, respectively.
Taking into consideration that S6K phosphorylated FLNA, we next determined whether this protein-protein interaction could also be detected using coimmunoprecipation. By use of COS-7 lysates that were mock-treated or overexpressed S6K, FLNA or both S6K + FLNA, we immunoprecipitated either FLNA or S6K from the reaction using antibodies specific to either protein and then used these samples for SDS-PAGE and WB. Following transfer to PVDF membranes, blots were probed for the presence of the opposite overexpressed myc-tagged protein (either IP α-FLNA, then WB α-myc for S6K or IP α-S6K, then WB α-myc for FLNA) (cartoon representation of each protein is shown in Fig. 8A). The top panel in Fig. 8B used an FLNA antibody to immunoprecipitate FLNA from cell lysates that were either mock-transfected or transfected with S6K, FLNA, or both S6K + FLNA. When we probed the resulting WB after SDS-PAGE of the immunoprecipitates with an anti-myc antibody specific for S6K, we detected the presence of overexpressed S6K in the samples that contained only transfected S6K or both co-overexpressed S6K + FLNA. If FLNA was complexed in the cell with myc-tagged S6K, then a band at 70 kDa (which is the molecular weight for S6K) should be shown in these 2 samples, which is what we observed. When we performed the inverse detection (i.e., immunoprecipitated samples using an anti-S6K antibody that were subjected to SDS-PAGE and subsequently were probed on the resulting WB using an antibody specific to the overexpressed myc-tagged FLNA), we detected the presence of FLNA at its molecular weight (which was ∼280 kDa). As shown in Fig. 8B, S6K was pulled down by FLNA (top panel) and FLNA was pulled down by S6K (second panel from top). Protein loading controls are shown at the bottom. These results show that although all of the endogenous and all of the recombinant FLNA or S6K was immunoprecipitated in the cell lysates, we were able to detect interaction of these populations of protein with only the recombinant/overexpressed protein partner. Additionally, this interaction was more favorably detected when all of the FLNA in the cells (both endogenous and overexpressed) was first immunoprecipitated and then detected on the WB using myc antibodies specific for the overexpressed myc-S6K.
Additionally, as it has previously been established that S6K and actin interact and colocalize in the cell (10), we were also able to show that FLNA and S6K were bound to actin when either FLNA or S6K was immunoprecipitated and then actin was detected in the WBs (Fig. 8B, second and third panels from the bottom). Interestingly, we also found that overexpression of S6K was sufficient to positively impact FLNA activity. As shown in Fig. 8C, D, increasing levels of phospho-FLNA (which can also be considered as activated FLNA) were detected in COS-7 cell lysates that concomitantly overexpressed increasing amounts of S6K-WT. The relative densitometry of phospho-FLNA is graphically shown in Fig. 8D compared with the actin loading control. It has been shown that if FLNA is phosphorylated, then FLNA is not cleaved and subsequently cannot translocate to the nucleus (25). The severity of cancer metastasis correlates with the cytoplasmic accumulation of FLNA and targeting the phosphorylation/cleavage of FLNA to nuclear localizations yields negative effects on metastasis (25).
The interaction of S6K with FLNA that we document herein was also evidence of a positive effect on both the lag and growth phases of actin polymerization in cell lysates that overexpressed both S6K-WT + FLNA-WT (Fig. 8E, red circles) compared with mock-treated (Fig. 8E, open circles). Additionally, cell lysates that overexpressed kinase-inactive S6K (S6K-KD) along with FLNA-WT (Fig. 8E, black circles) also had actin polymerization levels similar to mock-treated samples (Fig. 8E, open circles). We also determined that phosphorylated FLNA (via S6K action) was inhibitory to the lag phase of actin polymerization and had no effect on the growth and steady-state phases of actin polymerization (Fig. 8F). Data from Fig. 8 indicate the importance of S6K overexpression to FLNA phosphorylation/activation, which contributes to altered morphology reminiscent of chronic or pathologic disease states.
DISCUSSION
Morphogenesis involves the formation of new cytoskeleton or its remodeling that is accompanied by the appearance of new cellular surface structures (filopodia, villi, spikes, and other protrusions) on the cell surface. We report here that S6K is an intrinsic determinant for cell shape change and morphogenesis. Stellation/arborization or “star shape” is a normal anatomic feature present in astrocytes and neurons, as well as in hepatocytes and pancreatic cells. Narrow, elongated processes are formed by direct outgrowth from the round cells in all directions. We have found that S6K induces a stellation phenotype in a variety of normal, round-shaped cultured cells. However, this aborization is also more pronounced in tumor cells.
Although this morphologic change we report herein was first observed in macrophages that naturally adopt stellated morphologies on their own, the same changes were also found in remarkably different cells types, specifically in fibroblasts and cancer cells. The effects of S6K on cell shape recapitulate the process of malignant transformation. The diversity of the cell’s response to S6K in several, unrelated cell types indicates the existence of a common, underlying mechanism that involves actin dynamics. The present study also defined the mechanism involved in shape changes, demonstrating that PA and FLNA are targets for S6K. The fact that S6K binds to PA is an interesting parallel to AKT and the protein kinase C family, all protein kinase A, G, and C family members, which demonstrate lipid binding (26).
Cell shape changes occur in vivo during both inflammatory processes and cancer progression. As shown in the model presented in Fig. 9, cell shape changes of spherical-shaped, normal cells (a) occurred as a result of overexpression of kinase-active S6K. S6K and PA, the product of the catalytic reaction from PLD, PLC and/or DAGK (b), bound together and formed a lipid-protein complex (c) that allowed for S6K to bind to FLNA (d) and contributed to accumulation of phosphorylated FLNA in vivo (e), which increased polymerization of actin monomers in vitro (f). The final outcome of S6K overexpression yielded significant changes in cell morphology that resembled stellated macrophages (g) or fusiform cancer cells (h). Actin-associated proteins localized in the actin arc maintain S6K at this location, which might also regulate actin polymerization and cytoskeleton integrity.
Figure 9.

Model representing the in vivo S6K-mediated phosphorylation of FLNA, which contributes to S6K-dependent cell shape changes. Cell shape changes occur in vivo during both inflammatory processes and cancer progression and the present study supports an active role of S6K. Overexpression of S6K led to large cell shape changes of spherical-shaped, normal cells (a) that occurred as a result of overexpression of kinase-active S6K. S6K and PA, the product of the catalytic reaction from PLD, PLC, and/or DAGK (b), bound together and formed a lipid-protein complex (c) that allowed for S6K to bind to FLNA (d) and contributed to accumulation of phosphorylated FLNA by S6K both in vitro and in vivo (e), which increased polymerization of actin monomers in vitro (f). The final outcome of S6K overexpression yielded significant changes in cell morphology that resembled stellated macrophages (g) or fusiform cancer cells (h).
It makes sense that PA is implicated in actin polymerization and cell shape change. This is because PA is a central metabolic molecule that can gives rise to phospholipids (phosphatidylcholine and phosphatidylserine, etc.), which are needed for the formation of new surface structures. Moreover, PA is a membrane curvature-inducing phospholipid. In fact, our results indicate that although there is little to no change in cell area after S6K overexpression, the cell perimeter and ultimately the cell circularity dramatically increase. The sprouting surface structures that cover the cell surface in the new topology need to accommodate extra plasma membrane.
We have shown here that S6K is linked directly to the mechanism of actin polymerization through an interaction with PA that plays a key role in determining initial changes in cellular morphology during chemotaxis, phagocytosis, and cell invasion. These observations established an essential role for S6K in cell shape changes that illustrate a novel signaling pathway that links receptor stimulation at the cell membrane with actin dynamics in fundamental cellular processes. Having identified PA-S6K-FLNA-actin as key to this mechanism raises the possibility that targeted suppression of this pathway can effectively inhibit unwanted migration during inflammatory processes or cancer.
Collectively, data presented herein suggest that S6K-mediated morphologic changes correlate with changes observed with expression of signaling elements involved in inflammatory-related (migration and phagocytosis) and cancer-related (invasion) pathways. Although this study was initiated in macrophages that naturally adopt stellated morphologies, the same changes were also found in remarkably different cells types. The effects of S6K on cell shape also recapitulate malignant transformation. The diversity of the response to S6K in several, unrelated cell types, favors the existence of a common, underlying mechanism.
Acknowledgments
The authors acknowledge Dr. Francisco Alvarez (Emory University) for helpful discussions on cell morphology and Marium Husain and Francis Speranza for technical contribution on the transfection efficiency/cell viability assay and actin polymerization and phagocytosis experiments, respectively. This work was supported by the U.S. National Institutes of Health National Heart, Lung, and Blood Institute Grant HL056653-14, and the American Heart Association Grant 13GRNT17230097.
Glossary
- AKT
Ak strain transforming
- BSA
bovine serum albumin
- DAGK
diacylglycerol kinase
- DOPA
1,2-dioleoyl-sn-glycero-3-phosphate
- DOPC
1,2-dioleoyl-sn-glycero-3-phosphocholine
- EGF
endothelial growth factor
- FCS
fetal calf serum
- I.P.
immunoprecipitation
- FLNA
Filamin-A
- KD
kinase-dead
- LB
Luria broth
- MIP
macrophage inflammatory
- p70S6K
p70S6 kinase
- PA
phosphatidic acid
- PET
polyethylene terephthalate
- PLD
phospholipase D
- RSK2
p90 ribosomal S6 kinase 2
- PBS-T
PBS and 0.1% Triton X-100
- siRNA
small interference RNA
- TRITC
tetramethylrhodamine isothiocyanate
- WB
Western blot
- WT
wild-type
REFERENCES
- 1.Bussolino F., Ziche M., Wang J. M., Alessi D., Morbidelli L., Cremona O., Bosia A., Marchisio P. C., Mantovani A. (1991) In vitro and in vivo activation of endothelial cells by colony-stimulating factors. J. Clin. Invest. 87, 986–995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cretel E., Pierres A., Benoliel A. M., Bongrand P. (2008) How cells feel their environment: a focus on early dynamic events. Cell. Mol. Bioeng. 1, 5–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tweedy L., Meier B., Stephan J., Heinrich D., Endres R. G. (2013) Distinct cell shapes determine accurate chemotaxis. Sci. Rep. 3, 2606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bergert M., Chandradoss S. D., Desai R. A., Paluch E. (2012) Cell mechanics control rapid transitions between blebs and lamellipodia during migration. Proc. Natl. Acad. Sci. USA 109, 14434–14439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Friedl P., Wolf K. (2010) Plasticity of cell migration: a multiscale tuning model. J. Cell Biol. 188, 11–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ridley A. J. (2011) Life at the leading edge. Cell 145, 1012–1022 [DOI] [PubMed] [Google Scholar]
- 7.Lee A., Fischer R. S., Fowler V. M. (2000) Stabilization and remodeling of the membrane skeleton during lens fiber cell differentiation and maturation. Dev. Dyn. 217, 257–270 [DOI] [PubMed] [Google Scholar]
- 8.Wang E., Cross R. K., Choppin P. W. (1979) Involvement of microtubules and 10-nm filaments in the movement and positioning of nuclei in syncytia. J. Cell Biol. 83, 320–337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Berven L. A., Willard F. S., Crouch M. F. (2004) Role of the p70(S6K) pathway in regulating the actin cytoskeleton and cell migration. Exp. Cell Res. 296, 183–195 [DOI] [PubMed] [Google Scholar]
- 10.Ip C. K., Cheung A. N., Ngan H. Y., Wong A. S. (2011) p70 S6 kinase in the control of actin cytoskeleton dynamics and directed migration of ovarian cancer cells. Oncogene 30, 2420–2432 [DOI] [PubMed] [Google Scholar]
- 11.Pearson R. B., Dennis P. B., Han J. W., Williamson N. A., Kozma S. C., Wettenhall R. E., Thomas G. (1995) The principal target of rapamycin-induced p70s6k inactivation is a novel phosphorylation site within a conserved hydrophobic domain. EMBO J. 14, 5279–5287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Woo M. S., Ohta Y., Rabinovitz I., Stossel T. P., Blenis J. (2004) Ribosomal S6 kinase (RSK) regulates phosphorylation of filamin A on an important regulatory site. Mol. Cell. Biol. 24, 3025–3035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wessels D., Voss E., Von Bergen N., Burns R., Stites J., Soll D. R. (1998) A computer-assisted system for reconstructing and interpreting the dynamic three-dimensional relationships of the outer surface, nucleus and pseudopods of crawling cells. Cell Motil. Cytoskeleton 41, 225–246 [DOI] [PubMed] [Google Scholar]
- 14.Doolittle L. K., Rosen M. K., Padrick S. B. (2013) Measurement and analysis of in vitro actin polymerization. Methods Mol. Biol. 1046, 273–293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dowler S., Kular G., Alessi D. R. (2002) Protein lipid overlay assay. Sci. STKE 2002, pl6. [DOI] [PubMed] [Google Scholar]
- 16.Frondorf K., Henkels K. M., Frohman M. A., Gomez-Cambronero J. (2010) Phosphatidic acid is a leukocyte chemoattractant that acts through S6 kinase signaling. J. Biol. Chem. 285, 15837–15847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lehman N., Ledford B., Di Fulvio M., Frondorf K., McPhail L. C., Gomez-Cambronero J. (2007) Phospholipase D2-derived phosphatidic acid binds to and activates ribosomal p70 S6 kinase independently of mTOR. FASEB J. 21, 1075–1087 [DOI] [PubMed] [Google Scholar]
- 18.Toschi A., Lee E., Xu L., Garcia A., Gadir N., Foster D. A. (2009) Regulation of mTORC1 and mTORC2 complex assembly by phosphatidic acid: competition with rapamycin. Mol. Cell. Biol. 29, 1411–1420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sato M., Nagano T. (2005) Involvement of filamin A and filamin A-interacting protein (FILIP) in controlling the start and cell shape of radially migrating cortical neurons. Anat. Sci. Int. 80, 19–29 [DOI] [PubMed] [Google Scholar]
- 20.Nakamura F., Osborn T. M., Hartemink C. A., Hartwig J. H., Stossel T. P. (2007) Structural basis of filamin A functions. J. Cell Biol. 179, 1011–1025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Montminy M. (1997) Transcriptional regulation by cyclic AMP. Annu. Rev. Biochem. 66, 807–822 [DOI] [PubMed] [Google Scholar]
- 22.Flotow H., Thomas G. (1992) Substrate recognition determinants of the mitogen-activated 70K S6 kinase from rat liver. J. Biol. Chem. 267, 3074–3078 [PubMed] [Google Scholar]
- 23.Gorlin J. B., Yamin R., Egan S., Stewart M., Stossel T. P., Kwiatkowski D. J., Hartwig J. H. (1990) Human endothelial actin-binding protein (ABP-280, nonmuscle filamin): a molecular leaf spring. J. Cell Biol. 111, 1089–1105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pata I., Hoth S., Kruppa J., Metspalu A. (1992) The human ribosomal protein S6 gene: isolation, primary structure and location in chromosome 9. Gene 121, 387–392 [DOI] [PubMed] [Google Scholar]
- 25.Bedolla R. G., Wang Y., Asuncion A., Chamie K., Siddiqui S., Mudryj M. M., Prihoda T. J., Siddiqui J., Chinnaiyan A. M., Mehra R., de Vere White R. W., Ghosh P. M. (2009) Nuclear versus cytoplasmic localization of filamin A in prostate cancer: immunohistochemical correlation with metastases. Clin. Cancer Res. 15, 788–796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhang C., Wendel A. A., Keogh M. R., Harris T. E., Chen J., Coleman R. A. (2012) Glycerolipid signals alter mTOR complex 2 (mTORC2) to diminish insulin signaling. Proc. Natl. Acad. Sci. USA 109, 1667–1672 [DOI] [PMC free article] [PubMed] [Google Scholar]