

Abstract
Serum amyloid A (A-SAA/Saa3) was shown before to affect osteoblastic metabolism. Here, using RT-quantitative PCR and/or immunoblotting, we show that expression of mouse Saa3 and human SAA1 and SAA2 positively correlates with increased cellular maturation toward the osteocyte phenotype. Expression is not detected in C3H10T1/2 embryonic fibroblasts but is successively higher in preosteoblastic MC3T3-E1 cells, late osteoblastic MLO-A5 cells, and MLO-Y4 osteocytes, consistent with findings using primary bone cells from newborn mouse calvaria. Recombinant Saa3 protein functionally inhibits osteoblast differentiation as reflected by reductions in the expression of osteoblast markers and decreased mineralization in newborn mouse calvaria. Yet, Saa3 protein enhances osteoclastogenesis in mouse macrophages/monocytes based on the number of multinucleated and tartrate-resistant alkaline phosphatase-positive cells and Calcr mRNA expression. Depletion of Saa3 in MLO osteocytes results in the loss of the mature osteocyte phenotype. Recombinant osteocalcin, which is reciprocally regulated with Saa3 at the osteoblast/osteocyte transition, attenuates Saa3 expression in MLO-Y4 osteocytes. Mechanistically, Saa3 produced by MLO-Y4 osteocytes is integrated into the extracellular matrix of MC3T3-E1 osteoblasts, where it associates with the P2 purinergic receptor P2rx7 to stimulate Mmp13 expression via the P2rx7/MAPK/ERK/activator protein 1 axis. Our data suggest that Saa3 may function as an important coupling factor in bone development and homeostasis.—Thaler, R., Sturmlechner, I., Spitzer, S., Riester, S. M., Rumpler, M., Zwerina, J., Klaushofer, K., van Wijnen, A. J., Varga, F. Acute-phase protein serum amyloid A3 is a novel paracrine coupling factor that controls bone homeostasis.
Keywords: osteoblast, osteoclast, osteocyte, osteogenesis
Many skeletal diseases are due to regulatory defects in bone development or homeostasis. To calibrate bone strength during postnatal development, a complex network of paracrine mechanisms has developed to balance bone deposition and bone resorption (1–3). The activity of bone-forming osteoblasts is balanced by the activity of bone-resorbing osteoclasts and vice versa (4–6). Furthermore, it has become evident that developmental transitions between osteoblasts and osteocytes play a central role in bone metabolism (7). These processes are orchestrated through secretion of a variety of growth factors and cytokines, including receptor activator of NF-κB ligand (RANKL; in humans encoded by the TNFSF11 gene), osteoprotegerin (OPG; in humans encoded by the TNFRSF11B gene), or sclerostin (encoded by the SOST gene). RANKL protein and other proteins are abundantly secreted by different cell types including osteoblasts, and several studies have suggested that RANKL is expressed at even higher levels by osteocytes and controls bone remodeling during postnatal development and/or bone homeostasis in adult mammals (8–12). It acts by binding to the receptor activator of NF-κB (in humans encoded by the TNFRSF11A gene) expressed by osteoclasts and is essential for osteoclast formation, function, and survival. Mature osteoblasts express the RANKL antagonist OPG, which inhibits RANKL-induced osteoclastogenesis (13, 14). Sclerostin is a glycoprotein secreted by osteocytes and exerts antianabolic effects on bone formation (15). Loss-of-function mutations or reduced expression of the SOST gene are linked to the disorder sclerosteosis or to the milder form called van Buchem disease, respectively (16). These pathologies are characterized by bone overgrowth and high bone mass. Because bone development and homeostasis are highly and tightly regulated, the challenge is to gain a better appreciation of the paracrine aspects that control the bone metabolic activities of osteoblasts, osteocytes, and osteoclasts.
Extracellular matrix (ECM) integrity is critical for proper bone strength as well as bone function, and disruption of collagen fibers causes major skeletal defects like osteogenesis imperfecta or lathyrism (17, 18). We have previously shown that inhibition of collagen cross-linking and uncovering of Arg-Gly-Asp (RGD) sequence motifs via disruption of collagen triple-helix formation by homocysteine significantly stimulate expression of the acute-phase protein Serum Amyloid A (A-SAA/Saa3) in osteoblasts. Saa3 affects bone metabolism by modulating the expression of genes involved in inflammation, apoptosis, and bone matrix remodeling like matrix metalloproteinase (MMP) 13 (19). Because our previous study revealed an unexpected bone-related role for A-SAA, we set out to establish what its biologic contribution is to bone cell differentiation and function.
Originally, A-SAA had been characterized as an acute-phase protein of the apoprotein family (20, 21). This family consists of SAA1, SAA2, and SAA4 in humans and Saa1, Saa2, and Saa3 in mice and rabbits (20, 22–25); however, SAA4 does not contribute to acute-phase reactions (22, 26). In humans, the SAA3P gene is referred to as a pseudogene containing an insertion at nucleotide 147 provoking a frameshift and consequently generating a stop codon at position 61. Apart from high levels of A-SAA found in the liver (21, 27, 28), the protein has been found to be expressed in chondrocytes (22, 28, 29), adipocytes (30–32), and monocytes/macrophages (23, 33, 34) where it exerts chemoattractive effects and enhances cell adhesion (35). A-SAA proteins have been shown to be associated with HDLs (36), and elevated A-SAA levels have been correlated with the pathogenesis of chronic diseases such as rheumatoid arthritis (24, 37, 38), Alzheimer's disease (39), cardiovascular diseases (40–42), or insulin resistance (43). In inflamed human synovial tissue and in rabbit synovial fibroblasts, up-regulation of A-SAA induces the expression of several MMPs such as MMP13 (22). Because A-SAA is known to be expressed in both trabecular and cortical human bone (44) and is stimulated by defects in the bone matrix in cell culture (19), we explored the role of this chemoattractive protein in bone metabolism. The principal finding of our work is that A-SAA controls the activity and differentiation of osteoblasts, osteocytes, and osteoclasts and, thus, potentially may represent a novel central regulator of bone remodeling.
MATERIALS AND METHODS
Cell culture
The following murine cell lines were used: C3H10T1/2 embryonic fibroblasts (45, 46) (American Type Culture Collection, Manassas, VA, USA); MC3T3-E1, a clonal preosteoblastic cell line derived from newborn mouse calvaria; the late osteoblast/preosteocyte MLO-A5 and the osteocyte-like MLO-Y4 cell lines; the preosteoclastic, macrophage-like RAW264.7 cell line (American Type Culture Collection); and the human U2-OS osteosarcoma cell line (American Type Culture Collection). All cell lines were cultured in a humidified atmosphere with 5% CO2 at 37°C and were subcultured twice per week using 0.001% Pronase E (Roche, Penzberg, Germany) and 0.02% EDTA in Ca2+- and Mg2+-free PBS before achieving confluence. C3H10T1/2, MC3T3-E1, MLO-A5, MLO-Y4, and U2-OS cells were cultured in α-minimum essential medium (α-MEM; Biochrom, Berlin, Germany) containing 10 μg/ml gentamicin (Sigma-Aldrich, St. Louis, MO, USA). For C3H10T1/2 and MC3T3-E1, culture media were supplemented with 10% heat-inactivated fetal calf serum (FCS; Biochrom); for U2-OS cells, culture media were supplemented with 5% FCS. MLO-A5 and MLO-Y4 cells were cultured on rat tail-derived collagen type I (Roche)–coated dishes (final concentration of 0.15 mg/ml), and culture media were supplemented with 5% FCS (HyClone; GE Healthcare Life Sciences, Logan, UT, USA) and 5% calf serum (HyClone).
Differentiation of MC3T3-E1 cells and MLO-A5 cells was induced with 50 μg/ml ascorbic acid (Sigma-Aldrich) and 5 mM β-glycerophosphate (Sigma-Aldrich) in medium containing 5% FCS (MC3T3-E1 cells) or 10% FCS (MLO-A5 cells). RAW264.7 cells were cultured in DMEM (Invitrogen, Carlsbad, CA, USA) supplemented with 10% fetal bovine serum (HyClone) and 2 mM glutamine (Gibco, Carlsbad, CA, USA). Cells were seeded in culture dishes at a density of 20,000 cells/cm2 and cultured overnight unless stated otherwise. Before cells were treated with compounds, the culture medium was changed. The MAPK/ERK inhibitor PD98059 (PD) was purchased from Sigma-Aldrich and solubilized in DMSO; the P2rx7 inhibitor A438079 (A43) hydrochloride was purchased from Tocris Bioscience (Bristol, United Kingdom) and solubilized in distilled water.
Primary cells and tissues
Primary mouse tissues or cells were obtained according to the regulatory guidelines of the Institutional Animal Care and Use Committee of the Medical University of Vienna. Osteoblastic and early osteocytic cells were isolated from the aseptically dissected long bones of 3- to 4-d-old C57BL/6 mice (Himberg Breeding Facility, Himberg, Austria) by serial digestion with 1 mg/ml collagenase type IA (Sigma-Aldrich) dissolved in α-MEM for 30 min followed by incubation in 4 mM EDTA in PBS for 10 min at 37°C, shaking at 180 rpm. After each sequential digestion, cell suspension was centrifuged, washed with PBS, resuspended in culture medium, and seeded in cell culture dishes.
Human primary osteoblasts from femoral head trabecular explants were derived from patients undergoing hip replacement surgery (obtained with Mayo Clinic institutional board review approval). Outgrowth cultures from minced bone fragments yielded bone marrow stromal cells that were passaged 2–3 times. These cells were seeded at 20,000 cells/cm2 for osteoblast differentiation experiments in osteogenic culture media including 5 mM β-glycerophosphate, 50 μg/ml ascorbic acid, and 10−7 M dexamethasone (Sigma-Aldrich).
Primary mouse osteoclasts were harvested from 8-wk-old C57BL/6 mice that were euthanized, and bone marrow cells were isolated from tibias and femurs under aseptic conditions and cultured in α-MEM containing 10% FCS, 10 mg/ml gentamycin (Sigma-Aldrich), and macrophage colony-stimulating factor (MCSF; 30 ng/ml). After 24 h, nonadherent cells were seeded onto 48-well plates at 1 × 106 cells per well and stimulated with MCSF (30 ng/ml) and RANKL (10 ng/ml; both from R&D Systems, Minneapolis, MN, USA) and recombinant Saa (recSaa) 3 (0.2 µg/ml). Medium was changed every 3 d, and multinuclear osteoclasts were identified by Hoechst dye staining (Sigma-Aldrich) and the presence of ≥3 nuclei after 12 d of stimulation.
Calvaria from 4- to 5-d-old C57BL/6 mice were dissected aseptically. The periosteum was removed from the calvarial bone explants, and then calvarial explants were cultured in 48-well plates in α-MEM containing 10% FCS, 50 μg/ml ascorbic acid, 5 mM β-glycerophosphate, and 10 μg/ml gentamicin. The day after dissection, medium was changed, and a part of the explants was treated with 0.2 µg/ml recSaa3. After 12 d, calvaria were fixed for 1 h in 4% paraformaldehyde (PFA), and mineralization was measured by Alizarin Red S stain (Sigma-Aldrich). For this purpose, Alizarin Red S dye was extracted using 10% cetylpyridinium chloride in 10 mM sodium phosphate (pH 7.0) for 45 min at room temperature. Alizarin Red S absorbance was measured at 562 nm in a multiplate reader (Tecan, Groedig, Austria) and normalized to total protein amount measured by bicinchoninic acid assay (Thermo Fisher Scientific, Waltham, MA, USA).
Reseeding of MLO-Y4 cells on osteoblastic ECM
Interactions of Saa3 were studied with ECMs derived from osteoblasts. ECM preparations were generated by culturing MC3T3-E1 osteoblastic cells in medium containing 50 µg/ml ascorbic acid for 6 d. Living cells were removed using 0.5% sodium deoxycholate in PBS for 20 min at 4°C. The decellularized ECM was thoroughly washed with PBS, and MLO-Y4 cells were seeded onto the ECM at 20,000 cells/cm2, cultured for 3 d. Cells were fixed with 4% PFA for 20 min at room temperature and used for immunostaining as described below.
Cloning, recSAA production, and cell transfections
Murine Saa3 (NM_011315), human SAA1 (NM_000331), and SAA3P (AY209188) cDNA sequences were cloned into the pcDNA3.1 V5-His-TOPO vector (Invitrogen), 3 different lengths of the Mmp13 promoter (CH466522) were cloned from murine genomic DNA into the pGEM-T Easy vector (Promega, Madison, WI, USA), and the activator protein 1 (AP-1) binding site at −47 bp from the transcriptional start site in the Mmp13 promoter was mutated using primers listed in Table 1. All clones were verified by sequencing analyses. The pGEM vectors containing the Mmp13 promoter inserts were then subcloned into the pSEAP2 vector (Clontech, Mountain View, CA, USA).
TABLE 1.
Primers for RT-qPCR, molecular cloning, and ChIP
| Gene |
Primers, 5′–3′ |
Tm (°C) | GenBank accession number | |
|---|---|---|---|---|
| Forward | Reverse | |||
| Primers for RT-qPCR | ||||
| Mouse | ||||
| Alpl | CCAACTCTTTTGTGCCAGAGA | GGCTACATTGGTGTTGAGCTTTT | 62 | NM_007431 |
| Acp5 | CACTCCCACCCTGAGATTTGT | CATCGTCTGCACGGTTCTG | 62 | NM_001102404 |
| Calcr | GAGGTTCCTTCTCGTGAACAG | AGTCAGTGAGATTGGTAGGAGC | 62 | NM_007588 |
| Ctsk | CTCGGCGTTTAATTTGGGAGA | TCGAGAGGGAGGTATTCTGAGT | 62 | NM_007802 |
| Dmp1 | AAAGACCACGACAGTGAGGAT | CATCATCGAACTCAGAACCGTC | 62 | NM_016779 |
| Mepe | AAATATCACGCAGCCTGTAAAGA | GCTGGAATTACGCTTAGAACACT | 62 | NM_053172 |
| Mmp13 | CATTCAGCTATCCTGGCCACCTTC | CAAGTTTGCCAGTCACCTCTAAGC | 62 | CH466522 |
| Saa3 | CTGTTCAGAAGTTCACGGGAC | AGCAGGTCGGAAGTGGTT | 62 | NM_011315 |
| Saa3 TaqMan probe | CAGAGGACTCAAGAGCTGACC | 62 | NM_011315 | |
| Sost | CCTCCTCCTGAGAACAACCA | ACATCTTTGGCGTCATAGGG | 62 | NM_024449 |
| Human | ||||
| BGLAP | GGCGCTACCTGTATCAATGG | GTGGTCAGCCAACTCGTCA | 60 | NM_199173 |
| DMP1 | CTCCGAGTTGGACGATGAGG | TCATGCCTGCACTGTTCATTC | 60 | NM_001079911 |
| MEPE | GGCCAGTGACTGCGATTAAAC | CCTTCGAGTGTGCTTTAGCAT | 60 | NM_001184696 |
| MMP13 | TCCTGATGTGGGTGAATACAATG | GCCATCGTGAAGTCTGGTAAAAT | 60 | NM_002427 |
| RUNX2 | TGGTTACTGTCATGGCGGGTA | TCTCAGATCGTTGAACCTTGCTA | 60 | NM_001015051 |
| SAA1 | TTGGCGAGGCTTTTGATGGGG | AGGTCGGAAGTGATTGGGGT | 60 | NM_000331 |
| SAAL1 | ATGAGGACGTGGCTTTATTTCTC | AGGCAAGTAAGCAACAACCTG | 60 | NM_138421 |
| SAA2 | GCTTCTTTTCGTTCCTTGGCG | GCCGATGTAATTGGCTTCTCTCA | 60 | NM_001127380 |
| SAA3P | ATGAAGCTCTCCTCTGGCATC | TAGTTCCCCCAAGCATGGAAG | 60 | AY209188 |
| SOST | ACTTCAGAGGAGGCAGAAATGG | CAAGGGGGAATCTTATCCAACTTTC | 60 | NM_025237 |
| TaqMan | ||||
| 18S rRNA | Hs00917508_m1a | 60 | ||
| Bglap2 | Mm03413826_mHa | 60 | ||
| Col1a1 | Mm00801666_g1a | 60 | ||
| Runx2 | Mm00501578_m1a | 60 | ||
| Tnfrsf11b | Mm01205928_m1a | 60 | ||
| Tnfsf11 | Mm00441906_m1a | 60 | ||
| Primers for pc3.1 TOPO vector cloning | ||||
| Saa3 | CACCATGAAGCCTTCCATTGCCATCATTC | TCAGTATCTTTTAGGCAGGCCAGCAGGTC | 60 | NM_011315 |
| SAA1 | CACCATGAAGCTTCTCACGGGCCTG | GTATTTCTCAGGCAGGCCAGCAG | 58 | NM_000331 |
| SAA3P | CACCATGAAGCTCTCCTCTGGCATC | TAGTTCCCCCAAGCATGGAAG | 58 | AY209188 |
| Primers for pGEM-T Easy/SEAP2 vector cloning | ||||
| Mmp13 at −1007 bp | CACTAGATGTTAAATAATAATTG | CTCCTTCCCAGGGCAAGCATC | 58 | CH466522 |
| Mmp13 at −501 bp | TGGTTCTTATACATTTCTGAG | 58 | ||
| Mmp13 at −255 bp | CTGCTTCTCCCCACTATATC | 58 | ||
| Mmp13 AP-1 mutant | GTGGTGAAGAATCACTATC | GATAGTGATTCTTCACCAC | 58 | |
| Primers for c-FOS ChIP on murine Mmp13 promoter | ||||
| Mmp13 | AAGTTAACACACACCCCA | CCAGCTCAACAAGAAGAAG | 62 | CH466522 |
Tm, temperature. aSupplied by Applied Biosystems.
Recombinant murine Saa3 (recSaa3), human SAA1 (recSAA1), and human SAA3P (recSAA3P) were produced using the TNT T7 coupled reticulocyte lysate system (Promega) following the manufacturer’s instructions. Recombinant proteins were purified using the MagZ Protein Purification System (Promega).
Transfection and cotransfection experiments were performed with MC3T3-E1 or U2-OS cells seeded in 48- or 6-multiwell plates at 20,000 cells/cm2 using FuGENE Transfection Reagent (Promega) following the manufacturer’s instructions. The day after transfection, medium was changed, and cells were treated with listed inhibitors and/or cultured for the specified time followed by collection of RNA or supernatant. Each promoter activation experiment using SEAP2 assay was performed in quadruplicate and was repeated at least twice.
Saa3 small interfering RNA transfection
For knockdown experiments, MC3T3-E1 cells were seeded at 20,000 cells/cm2 in 6-well plates. After 6 h, cells were transfected with a mixture of 3 different small interfering RNAs (siRNAs) specific for Saa3 (40 pmol each; 5′-AAGATGGGTCCAGTTCATGAA-3′, 5′-AGAAGCTGGTCAAGGGTCTA-3′, and 5′-CTCTGACATGAAGAAAGCTAA-3′; #2606110; Qiagen, Hilden, Germany) or with a scrambled siRNA [siRNA negative (siNeg) control, stealth siNeg control; #12935-112; Invitrogen] using X-tremeGene siRNA transfection reagent (Roche), as described by the supplier. After 3 d, total RNA was isolated as described below and subjected to RT-quantitative PCR (RT-qPCR).
Chromatin immunoprecipitation
MC3T3-E1 cells were transfected with pc3.1-Saa3 or pc3.1 empty control vector and cultured for 3 d. Subsequently, chromatin cross-linking, cell lysis, chromatin sharing, c-FOS immunoprecipitation, and DNA cleanup were performed with the ChampionChIP One-Day kit (Qiagen) following the manufacturer’s instructions. c-FOS binding to Mmp13 promoter was quantified by RT-qPCR [for chromatin immunoprecipitation (ChIP)-Mmp13 promoter primers, see Table 1].
Isolation of RNA and RT-qPCR
Total RNA was extracted using the SV Total RNA Isolation kit (Promega) following the supplier’s instructions. cDNA was synthesized from ∼0.5 μg RNA using the First-Strand cDNA Synthesis kit (Roche) as described by the supplier. The resulting cDNAs were subjected to quantitative PCR amplification with a real-time cycler using the QuantiTect SYBR-Green PCR Kit (Qiagen) for the genes Alpl, Acp5 [tartrate-resistant alkaline phosphatase (TRAP)], Calcr, Ctsk, Dmp1, Mepe, Mmp13, Sost, BGLAP, DMP1, MEPE, MMP13, RUNX2, SAA1, SAAL1, SAA2, SAA3P, and SOST or TaqMan Gene Expression Master Mix (Applied Biosystems, Foster City, CA, USA) for measuring Saa3, Runx2, Bglap2, Col1a1, Tnfsf11, and Tnfrsf11b, and 18S rRNA expression (for primers, see Table 1). RT-qPCRs were performed using standard protocols as shown before (19). Expression was evaluated using the comparative quantification method (47).
Affymetrix GeneChip analysis
Total RNA was isolated using the SV Total RNA Isolation kit. Quality control of the RNAs as well as labeling, hybridization, and scanning of the hybridized arrays were performed by the “Kompetenzzentrum fuer Fluoreszente Bioanalytik” (Regensburg, Germany) using the mouse 430 2.0 chip (Affymetrix, Santa Clara, CA, USA).
Protein isolation and immunoblotting
Whole-cell protein extracts were prepared using SDS sample buffer [2% SDS, 100 mM 2-ME (β-mercaptoethanol), and 125 mM Tris-HCl (pH = 6.8)] and heated at 95°C for 5 min. To obtain supernatant protein extracts, cell medium was collected, and proteins were precipitated with 50% trichloroacetic acid at 4°C for 1 h, concentrated by centrifugation, neutralized with PBS, and dissolved in SDS sample buffer.
Immunoblotting analysis was performed as shown before (19). The following primary antibodies were used: rabbit, anti-mouse Saa3, and rabbit, anti–β-actin (#4967; Cell Signaling, Danvers, MA, USA). Measurements are given as the mean of 3 immunoblots, and representative blots are shown.
Immunofluorescence microscopy
Samples were fixed and stained as shown before (48). For specific detections, we used the anti-P2rx7 antibody (EB09509; Everest Biotech, Oxfordshire, United Kingdom) diluted 1:300 or the Saa3 antibody in blocking buffer. Analysis was performed using a Leica TCS SP5 confocal laser-scanning microscope (Leica Microsystems, Wetzlar, Germany).
Statistical analysis
Statistical analyses were performed using ANOVA or Student’s t test in Prism 4.03 (GraphPad Software, La Jolla, CA, USA). Values of P ≤ 0.05 were considered significant. Each experiment consisted of at least 3 biologic replicates. For RT-qPCR data, results from technical triplicates were averaged, and the mean value was treated as a single, biologic statistical unit. Results are presented as the mean ± sd.
RESULTS
Mouse Saa3 is a novel osteocyte-related secreted protein
Because denatured collagen matrix strongly stimulates Saa3 expression in osteoblasts (19), we assessed the basal mRNA levels of 3 genes encoding A-SAA in mouse (Saa1, Saa2, and Saa3) in a series of osteoblast and osteocyte-related cell lines and primary cells. Affymetrix GeneChip expression profiling showed that Saa1, Saa2, and Saa3 are all expressed at comparable levels within a 2- to 4-fold range in MC3T3-E1 osteoblasts (Table 2). However, basal levels of Saa3 are substantially higher than Saa1 or Saa2 in MLO-Y4 osteocytes (Table 2). These results are corroborated by RT-qPCR analysis of basal Saa3 mRNA and protein levels in cells representing different stages of the osteocyte differentiation lineage. Saa3 mRNA expression is lowest in C3H10T1/2 embryonic fibroblasts, higher in preosteoblastic MC3T3-E1 cells and MLO-A5 osteoblast/preosteocyte cells, but clearly highest in MLO-Y4 osteocyte-like cells (Fig. 1A). Accordingly, a very similar pattern of protein expression is found in this panel of 4 cell lines in both cell lysates (CL) and the culture supernatant of serum-free conditioned medium (Fig. 1B–D). Importantly, the presence of Saa3 in conditioned medium [Fig. 1B, lower blot (CM); Fig. 1D) suggests that it is a secreted factor in osteocytes, as expected from its chemokine-like activity in other cell types.
TABLE 2.
Saa expression measured by GeneChip analysis
| Gene | MC3T3-E1 (log2 signal) | MLO-Y4 (log2 signal) |
|---|---|---|
| Saa1 | 3.96 | 3.87 |
| Saa2 | 4.46 | 4.38 |
| Saa3 | 5.14 | 10.84 |
Saa3 basal expression is considerably higher in MLO-Y4 cells than in MC3T3-E1 cells; however, there is no difference between Saa1/Saa2 basal expression in the 2 cell lines.
Figure 1.

Mouse Saa3 is highly expressed in osteocytes. Expression pattern of Saa3 on mRNA level (A) and protein level (B–D). Representative Saa3 immunoblots are shown for the cellular (CL, with β-actin as reference, upper blots) and the serum-free conditioned media (CM) fraction (related to media volume, lower blot) (B). Quantification is shown of the cellular lysate fraction (C) and of the conditioned media fraction (D). E) Comparison of Saa3 (green) levels by IF in MLO-Y4 cells and MC3T3-E1. Scale bar, 50 µm. F) Quantification of total Saa3 IF signal found in (E) referenced to total nuclear (blue) or to total F-Actin (red) IF signal. G) Saa3 RT-qPCR analysis of primary mouse osteoblasts [Fraction (Fr) 2–4] or late osteoblasts to early osteocytes (Fr 5–7) obtained by sequential digestion of long bones. H) The osteoblast-to-osteocyte differentiation pattern of the cellular fractions is supported by increasing Tnfsf11 mRNA levels in cell fractions Fr 5–7. 18S rRNA is a reference gene for RT-qPCR analysis; MC3T3-E1 is set to 1, and all other cell lines are referred to as fold change to MC3T3-E1. In (G) and (H), Fr 2 is set to 1; all other cell fractions are referred to as fold change to Fr 2. Values are represented as the mean ± sd; for all experiments n = 3. *P ≤ 0.05; **P ≤ 0.01; ***P ≤ 0.001.
Immunofluorescence (IF) microscopy reveals that the intracellular distribution of Saa3 is distinctly cytoplasmic (Fig. 1E), consistent with its expected localization upon production in the endoplasmatic reticulum, Golgi apparatus, and/or secretory vesicles prior to its secretion. In agreement with our mRNA and protein expression analysis, fluorescence signals for Saa3 appear to be more prominent in MLO-Y4 preosteocytes compared to MC3T3-E1 cells (Fig. 1E, F). To obtain an in vivo correlate for these findings, we analyzed early osteoblast to early osteocyte cell fractions obtained by sequential digestion of long bones from neonatal mice (3–4 d old). In agreement with our cell culture results, RT-qPCR analysis showed that Saa3 expression is highest in late osteoblastic/early osteocytic cells [i.e., pooled fractions 5–7, which represent a mixed population of late osteoblasts and osteocytes (8)] (Fig. 1G). These cells are characterized by expression of the biomarker Tnfsf11/RANKL, which is expressed at elevated levels in mature osteocytes (Fig. 1H). These data suggest collectively that Saa3 is a secreted protein that is prominently expressed in osteocytes.
Mouse Saa3 is phylogenetically related to human SAA1 and SAA2
As a prelude to assess the clinical relevance of human A-SAA proteins in patients, we compared the phylogenetic relationships between the A-SAA encoding genes in mouse and human. It appears that these genes have undergone genetic evolution during vertebrate phylogenetic divergence, which is reflected by distinct duplications and/or mutations that altered the protein coding potential of the A-SAA genes. For example, the human SAA3 gene is reported to be a pseudogene (SAA3P) with an insertion at nucleotide 147 that causes a frameshift generating a stop codon at amino acid 61 (Fig. 2A). However, the phylogenetic dendrogram we generated shows that the murine Saa3 is remarkably similar to the human and murine SAA1/Saa1 and SAA2/Saa2 sequences (Fig. 2B). The gene similarity between the respective human and mouse isoforms is greater than the similarity between putative homologs (Fig. 2B). This observation suggests that the SAA genes were duplicated in a mammalian predecessor and subsequently mutated independently in rodents and mammals.
Figure 2.

Phylogenetic relations between mouse Saa3 and human SAA1 and SAA2. A) Mouse (m) as well as human (h) SAA1/SAA2 sequences show high homology with each other, and mouse Saa3 shows a high similarity to mouse and human SAA1/SAA2 after amino acid 30. In humans, the SAA3 gene is reported to be a pseudogene (SAA3P); a mutation/insertion at base 147 causes a frameshift generating a stop codon at position 61. B) Gene homology tree between human and mouse A-SAA proteins. C) In differentiating primary human osteoblasts, SAA1 and SAA2 mRNA expressions increase drastically after 26–30 d of culture. D) In contrast, SAAL1 expression remains unaffected during the whole differentiation process. E) The differentiation to mature osteoblasts is demonstrated by BGLAP and RUNX2 expressions reaching the maximum at d 12. The drop in BGLAP and RUNX2 expression is accompanied by increased levels of osteocyte markers DMP1, SOST, and MEPE (F). 18S rRNA is a reference gene for RT-qPCR analysis; d 1 is set to 1, and later time points are referred to as fold change relative to d 1. Values are represented as the mean ± sd; for all experiments n = 3.
In contrast to its current database designation, mouse Saa3 is more closely related to human SAA1 and SAA2 than SAA3P. Thus, the possibility arises that mouse Saa3 performs the biologic functions of human SAA1 and/or SAA2 in osteocytes. Indeed, Saa1 and Saa2 are expressed only at low levels in murine bone cells (see Table 2). The corollary of this finding is that expression of human SAA1 increases dramatically upon long-term osteogenic induction (after 26–30 d) in cultures of differentiating primary human osteoblasts (Fig. 2C). SAA2 mRNA expression follows a similar temporal expression trend as SAA1, but its induction is far less pronounced (Fig. 2C). For comparison, the distantly related family member SAAL1 is expressed at constitutive levels during human osteoblast differentiation (Fig. 2D). Expression of the human pseudogene SAA3P was not detected during osteoblastic differentiation as expected by the evolutionary inactivation of this gene.
Because mouse Saa3 is osteocyte related (Fig. 1), we assessed how expression of human SAA1 is modulated in relation to established osteoblast vs. osteocyte markers. This analysis reveals that the up-regulation of SAA1 is initiated after the initial peak of expression of the osteoblast-related genes RUNX2 and BGLAP (Fig. 2E) but occurs concomitantly with induction of the osteocyte-related genes DMP1, SOST, and MEPE (Fig. 2F). These molecular data collectively suggest that human SAA1 and mouse Saa3 are the physiologically relevant and species-specific gene isoforms that produce secreted A-SAA protein in osteocytes.
Osteocyte-specific expression of Saa3 suppresses the osteoblast phenotype
To corroborate cell line data indicating that Saa3 expression is highest in osteocyte-like cells, we examined Saa3 expression in greater detail during osteoblastic to osteocytic differentiation in MLO-A5 cells. Saa3 mRNA levels exhibit a maximal increase in expression during the osteoblast-to-osteocyte transition in MLO-A5 cells when Bglap2 expression begins to decrease. Saa3 expression is elevated from around 22 arbitrary units (d 14) to 30 arbitrary units (d 22). However, the initial induction (and fold change) is highest before d 22 and increases from about 8 arbitrary units at d 11 to 22 arbitrary units at d 14 (Fig. 3A). Because the bone cell–related production of many chemokines and cytokines occurs as part of autologous paracrine mechanisms (49), we examined whether Saa3 expression is biologically linked to progression of osteoblastic or osteocytic differentiation. Therefore, differentiating MC3T3-E1 osteoblastic cells cultured for 10 d in osteogenic media were subsequently cultured for an additional 3 d with purified recSaa3 protein (at 0.2 μg/ml). Administration of Saa3 significantly decreases the mRNA expression of osteoblastic genes like Col1a1, Runx2, Alpl, and Tnfrsf11b (OPG) (Fig. 3B) but does not affect cell viability of MC3T3-E1 cells (Fig. 3C). These results indicate that osteocyte-related production of Saa3 may be part of a paracrine mechanism that suppresses expression of Bglap2, a marker of maturing osteoblasts.
Figure 3.
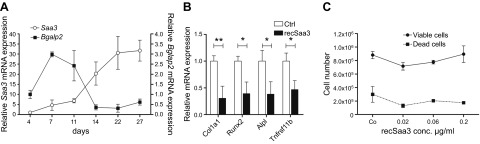
Suppression of the osteoblastic phenotype by Saa3. Saa3 expression increases during osteoblast-to-osteocyte maturation and decreases the expression of genes of the maturing osteoblastic phenotype. A) Saa3 mRNA levels increase slowly but permanently during late osteoblast-to-osteocyte differentiation in MLO-A5 cells. Interestingly, only when Bglap2 decreases between d 11 and 14 do Saa3 mRNA levels reach their maximum. B) Treatment of 10-d-old osteogenic media-matured MC3T3-E1 osteoblasts with recSaa3 for a further 3 d significantly decreases the mRNA expression of the main osteoblast marker genes like Col1a1, Runx2, Alpl, and Tnfrsf11b. Ctrl, control. This treatment does not affect cell proliferation and viability of MC3T3-E1 cells (C). 18S rRNA is a reference gene for RT-qPCR analysis. In (A), d 4, and in (B), untreated controls are set to 1, and time points or treatments are referred to as fold change to d 4 or to control. Values are represented as the mean ± sd; for all experiments n = 3. *P ≤ 0.05; **P ≤ 0.01.
Saa3 is required for retention of the osteocyte phenotype
Because production of Saa3 in osteocytes suppresses the expression of osteoblastic genes, we hypothesized that it may be simultaneously required for progression or maintenance of the osteocyte phenotype. To investigate the role of Saa3 in osteocytes, we depleted Saa3 using a pool of siRNAs in MLO-A5 and MLO-Y4 cells. The loss of Saa3 expression decreases expression of Dmp1 and Mepe, which are prominent ECM mineralization-related osteocyte markers, in MLO-A5 and in MLO-Y4 cells (Fig. 4A, B). Furthermore, in MLO-A5 cells, Saa3 knockdown provokes a decrease in expression of osteocyte-related paracrine factors like Sost, Tnfsf11 (RANKL), and Tnfrsf11b (OPG) (Fig. 4C). Of note, Saa3 depletion did not affect expression of the latter 3 markers in MLO-Y4 cells (Fig. 4D), suggesting that the exact biologic effects of Saa3 may be cell-type dependent. More importantly, the reduced expression of several osteocyte markers upon inhibition of Saa3 expression in 2 different cell types suggests that Saa3 may be involved in progression of osteocyte differentiation.
Figure 4.
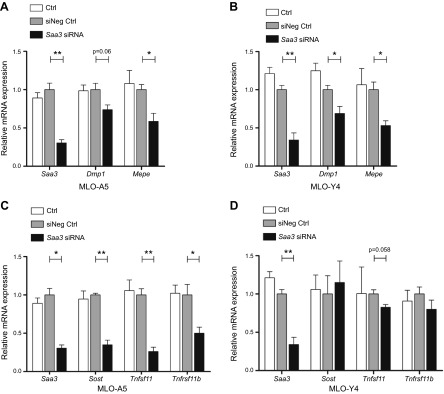
Saa3 retains the osteocyte phenotype in MLO-A5 and MLO-Y4 cells. Knockdown of Saa3 significantly decreases the expression of ECM mineralization-related osteocyte markers like Dmp1 and Mepe in MLO-A5 (A) and in MLO-Y4 (B) cells. C) In MLO-A5 cells, the loss of Saa3 expression (Saa3 expression is as in A) causes down-regulation of secreted factors like Sost, Tnfsf11, and Tnfrsf11b, which is not observed in MLO-Y4 cells (D). 18S rRNA is a reference gene for RT-qPCR analysis. Negative control (siNeg Ctrl) is set to 1, and control (Ctrl) or treated probes are referred to as fold change to siNeg Ctrl. Values are represented as the mean ± sd; for all experiments n = 3. *P ≤ 0.05; **P ≤ 0.01; ***P ≤ 0.001.
Reciprocal feedback regulation of osteoblast-specific Bglap2 and osteocyte-specific Saa3
When cells progress through the osteoblast-to-osteocyte differentiation, we find that Saa3 levels are reciprocally regulated with Bglap2 (osteocalcin) expression (see Figs. 2C, E, and 3A). Because both proteins are secreted factors in bone, we assessed whether reciprocal regulation in their expression reflects a functional coupling. We investigated this hypothesis by monitoring the effects of modulating Saa3 or Bglap2 proteins by administering recombinant proteins or siRNAs in osteoblastic or osteocytic cell types (Fig. 5). Consistent with the proposed osteoblast-suppressive role of Saa3, administration of recSaa3 decreases Bglap2 expression (∼50–70%) (Fig. 5A) in MC3T3-E1 osteoblastic cells (treated at d 10 for 3 d with 0.2 μg/ml recSaa3), whereas siRNA depletion of Saa3 modestly increases Bglap2 mRNA levels (∼30–40%) (Fig. 5B), but not of other osteoblastic factors like Runx2 (data not shown). These results suggest that Saa3 is a paracrine regulator of Bglap2 levels. Furthermore, treatment of MLO-Y4 cells for 3 d with recombinant murine Bglap2 (recBglap2) reduces Saa3 mRNA levels in a concentration-dependent manner (Fig. 5C). We note that we did not analyze other bone-related markers like Sost or Dmp1, which would permit a more detailed understanding of the roles of Saa3 and Bglap2 osteocytogenesis. Although the absence of these data prevents broader biologic interpretations, our results suggest that there is reciprocal negative feedback regulation between Saa3 and Bglap2 during the osteoblast-to-osteocyte transition.
Figure 5.
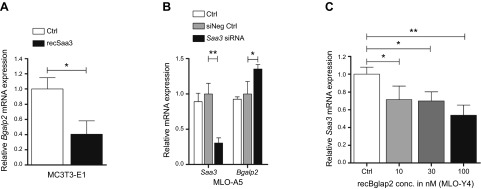
Reciprocal feedback regulation of Bglap2 and Saa3. A) Treatment of 10-d-old MC3T3-E1 matured osteoblasts with recSaa3 for a further 3 d significantly decreases the mRNA expression of the osteoblastic marker Bglap2. B) In MLO-A5 preosteocytes, knockdown of Saa3 expression (Saa3 expression is as in Fig. 4A, C) provokes a significant up-regulation of Bglap2 levels. C) In MLO-Y4 cells, Bglap2 treatment decreases Saa3 expression in a concentration (conc.)-dependent manner. 18S rRNA is a reference gene for RT-qPCR analysis. Control (Ctrl) or negative control (siNeg Ctrl) is set to 1, and control (Ctrl) or treated probes are referred to as fold change to Ctrl or siNeg Ctrl. Values are represented as the mean ± sd; for all experiments n = 3. *P ≤ 0.05; **P ≤ 0.01.
Osteocyte-derived Saa3 binds to osteoblastic ECM
A-SAA interacts with ECM-related glycoproteins (50), raising the possibility that mouse Saa3 may interact with the ECM from osteoblasts. To address this question, we generated nonmineralized ECMs from MC3T3-E1 osteoblasts by culturing cells for 6 d followed by removal of cells using deoxycholate. MLO-Y4 osteocytes were cultured on the resulting scaffolds in normal maintenance medium for 3 d, and deposition of secreted proteins was visualized by confocal IF microscopy (Fig. 6). Apart from removing cells, deoxycholate effectively releases the minimal amounts of Saa3 known to be produced by MC3T3-E1 cells (for example, see Fig. 1E), while retaining the gross integrity of the ECM; IF signals for Saa3 are below the level of detection on the osteoblast ECM (data not shown). Sequential Z sections from the MLO-Y4 cell cultures grown on these ECM preparations clearly show robust intracellular localization of Saa3 in cells residing on top of the osteoblast-derived ECM (Fig. 6A), as expected from previous results (see Fig. 1E). Sequential sections through the osteoblastic ECM show substantial deposition of Saa3 produced by MLO-Y4 osteocytes (Fig. 6B–E represents a Z stack overlay of several ECM sections). This staining is not observed in a negative control in which the ECM was analyzed in the absence of MLO-Y4 cells (data not shown), although the presence of a collagen-rich matrix is evidenced by Picro-Sirius Red staining (Fig. 6F). Thus, Saa3 is effectively deposited in the bone-related ECM produced by osteoblastic cells, consistent with an ECM-associated paracrine function in bone tissue.
Figure 6.
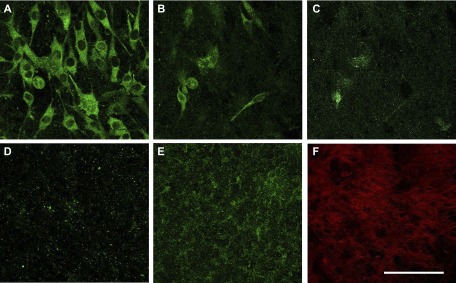
Saa3 interacts with osteoblastic ECM. MLO-Y4–derived Saa3 is deposited in ECM derived from MC3T3-E1 osteoblasts. A) Saa3 (green) in MLO-Y4 cells is attached on the top of the MC3T3-E1-derived, decellularized ECM. B–D) Sequential Z stages of the ECM are shown; green dots represent Saa3 deposition. E) Overlay of the Z stages of the ECM section. F) ECM derived from MC3T3-E1 osteoblasts before MLO-Y4 reseeding as stained by Picro-Sirius Red. Scale bar, 50 µm.
Saa3 stimulates in vitro osteoclastogenesis and ex vivo bone resorption
Secreted proteins generated by bone-producing cells can be released upon bone resorption by osteoclasts, with latent TGF-β representing one classic example (51). We reasoned therefore that deposition of osteocyte-produced Saa3 in the ECM could predicate a role for Saa3 in osteoclastogenesis, similar to the function of osteoblast-produced RANKL in supporting osteoclast maturation. When macrophage-like, preosteoclastic RAW264.7 cells are treated with MCSF and RANKL (M/R) for 3 d, cells begin to differentiate into osteoclast-like cells exhibiting strongly increased expression of classic osteoclast biomarkers, including mRNAs for Acp5 (TRAP) (Fig. 7A), Ctsk (cathepsin K) (Fig. 7B), and Calcr (calcitonin receptor) (Fig. 7C). Remarkably, cotreatment of RAW264.7 cells with M/R and recSaa3 (0.2 μg/ml) additionally increases the expression of these same osteoclast-specific genes. The sole treatment of RAW246.7 cells with recSaa3 revealed a trend of increased mRNA levels of Acp5, Ctsk, and Calcr that did not reach statistical significance (P > 0.05) (Fig. 7A–C). Corroborating these mRNA expression data, histochemical data obtained after treatment of RAW246.7 cells for 10 d revealed that Saa3 stimulates TRAP activity, albeit only in the presence of M/R (Fig. 7D). Hence, Saa3 may synergize with M/R to promote osteoclastogenesis.
Figure 7.
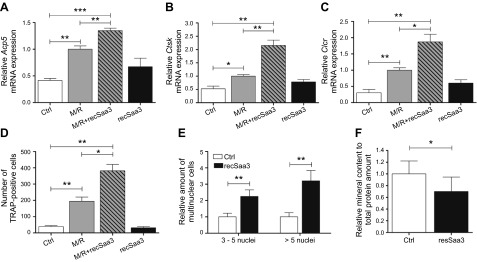
Effect of Saa3 on maturation and activity of osteoclasts. Saa3 up-regulates osteoclast-specific genes, increases osteoclastic fusion, and triggers their bone-resorbing activity. Macrophage-like, preosteoclastic RAW264.7 cells treated with MCSF (30 ng/ml) and RANKL (10 ng/ml) for 3 d significantly up-regulate osteoclast marker genes: Acp5 (A), Ctsk (B), and Calcr (C). Remarkably, cotreatment with M/R and recSaa3 significantly further increases the expression of these osteoclast-specific genes (A–C). Sole treatment with recSaa3 shows a tendency to increase the mRNA levels of Acp5, Ctsk, and Calcr (A–C). D) Treatment of RAW264.7 cells with M/R for 10 d leads to TRAP-positive cells. Cotreatment of these cells with recSaa3 doubles the TRAP-positive osteoclast-like cells. E) Treatment of primary mouse monocytes/macrophages with M/R and with or without recSaa3 for 12 d. Combined treatment significantly increases the number of multinuclear cells, counting 3–5 nuclei per cell and counting over 5 nuclei per cell (E). Calvarial explants from 4- to 5-d-old mice were treated for 12 d with recSaa3, and mineral content was measured with Alizarin Red staining. A significantly decreased mineral content in recSaa3-treated samples is observed, suggesting increased osteoclastic activity in these samples (F). 18S rRNA is a reference gene for RT-qPCR analysis. Values are represented as the mean ± sd. M/R-treated cells are set to 1 in (A)–(C), and control (Ctrl) or treated probes are referred to as fold change to M/R. In (E) and (F), untreated control cells are set to 1, and recSaa3-treated cells/calvaria are referred to as fold change to Ctrl. For all graphs, n = 3 except for (F), n = 10. *P ≤ 0.05; **P ≤ 0.01; ***P ≤ 0.001.
Encouraged by these results, we examined whether Saa3 affects osteoclastic fusion. We treated primary mouse monocytes/macrophages with M/R with or without recSaa3 protein (0.2 μg/ml) for 12 d and determined the relative number of multinuclear cells. Strikingly, recSaa3 significantly increases the number of multinuclear cells with either few (3–5) or many (>5) nuclei per cell (Fig. 7E). Moreover, we tested whether Saa3 can stimulate osteoclastic activity and ex vivo bone resorption in calvarial explants from 4- to 5-d-old mice. As anticipated, treatment of calvarial explant cultures with recSaa3 for 12 d decreases mineral content (Fig. 7F). Taken together, these data collectively suggest that Saa3 produced by osteocytic cells stimulates osteoclast maturation and, consequently, that Saa3 may represent an osteocyte/osteoclast coupling factor for modulating bone homeostasis and/or remodeling.
Saa3 associates with the P2rx7 receptor of osteoblasts and osteoclasts in vitro
Our combined results suggest that Saa3 is produced by osteocytes to control the osteoblast-to-osteocyte transition, while simultaneously promoting osteoclast maturation by a paracrine mechanism. To understand the mechanism by which Saa3 may affect osteoblastogenesis and osteoclastogenesis, we were guided by prior studies with macrophages showing that A-SAA protein activates that purinergic P2RX7 receptor that activates the NLRP3 inflammasome (52). Furthermore, previous studies have shown that the P2rx7 receptor is expressed in osteoblasts and osteoclasts (53). Earlier studies also showed that P2RX7-mediated signaling drives osteoclastic fusion (54) and negatively regulates bone mineralization (55). Therefore, we hypothesized that Saa3 may be an osteoclastogenic coligand for P2rx7. To test this hypothesis, we investigated whether Saa3 colocalizes with P2rx7 in osteoblasts and osteoclasts. IF microscopy using antibodies for Saa3 and P2rx7 in both MC3T3-E1 (Fig. 8A–D) and RAW264.7 (Fig. 8E–H) cells clearly shows similar localization of Saa3 and P2rx7 proteins at the cell surface; the extent of colocalization is most apparent upon digital image magnification (see insets in the lower-left corner of panels in Fig. 8D, H). The in situ proximity of Saa3 with P2rx7 indicates that Saa3 may control the activity of the P2rx7 receptor in osteoblasts and osteoclasts.
Figure 8.
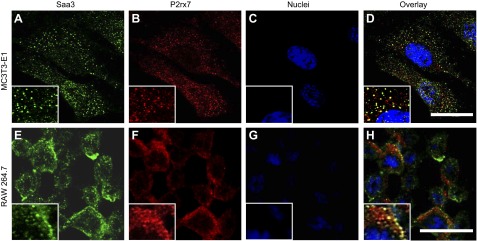
Saa3 colocalizes with P2rx7 receptor. A–D) Coimmunofluorescence of Saa3 (A) and P2rx7 (B) shows colocalization of both proteins (D) in MC3T3-E1 cells. Nuclei are depicted in (C). E–H) A similar pattern is observed in RAW264.7 cells. Saa3 (E) as well as P2rx7 (F) are abundantly expressed in this cell line; overlay of both IF pictures shows the colocalization of both proteins (H). Nuclei are depicted in (G). Insets represent picture magnifications. Scale bars, 25 µm.
The proximity of Saa3 and P2rx7 observed by IF microscopy may suggest that these 2 proteins interact directly through protein/protein interactions. To reinforce our biologic interpretations, we have investigated this possibility using biochemical analysis (e.g., peptide-tag pull-down assays). However, we did not succeed in our extensive attempts to achieve Saa3 precipitation in complex with P2rx7 (unpublished negative observations). Regardless of whether Saa3 interacts directly or through a chain of nearby protein/protein interactions, similar localization by IF microscopy as well as data obtained with the P2rx7 inhibitor A43 (see below) together suggest that Saa3 may function through a P2rx7-dependent mechanism in osteoblasts.
Saa3 stimulates the P2rx7/MAPK/ERK/AP-1 axis to control Mmp13 expression
We next elucidated the molecular mechanism by which Saa3 may control osteoblast activity. In earlier studies, we have shown that Saa3 expression is induced upon ECM disruption and that Saa3 supports repair and/or modeling of the ECM by up-regulating Mmp13 expression in osteoblasts (19). Mmp13 plays a central role in bone turnover because it is a collagenase that degrades the ECM, and its expression is physiologically responsive to parathyroid hormones and TGF-β, as well as MAPK/ERK signaling (56–58) In addition, Mmp13 is required for osteocytic peri-lacunar remodeling (59), as well as stimulating osteoclast differentiation and activation (60). Our new finding that Saa3 colocalizes in situ with the P2rx7 receptor suggests that Saa3 may enhance Mmp13 gene expression via P2rx7-mediated signaling pathways. As expected, forced expression of Saa3 (pc3.1-Saa3) in MC3T3-E1 osteoblasts induces Mmp13 mRNA levels by about 2- to 3-fold (Fig. 9A). Yet, this stimulation is effectively blocked upon inhibition of the P2rx7 receptor by the selective inhibitor A43 (Fig. 9A). Furthermore, treatment with PD, a potent and selective kinase inhibitor of the MAPK/ERK pathway, clearly abrogates induction of Mmp13 mRNA expression by Saa3 in MC3T3-E1 cells (Fig. 9B). These results establish a molecular nexus among Saa3, the P2rx7 receptor, MAPK/ERK signaling, and Mmp13 gene expression.
Figure 9.

Pathway involved in Saa3-mediated Mmp13 up-regulation. In MC3T3-E1 cells, Saa3 up-regulates Mmp13 via P2rx7, MAPK/ERK, and c-FOS/AP-1. A) Forced expression of Saa3 (pc3.1-Saa3) in MC3T3-E1 cells increases Mmp13 expression after 3 d. However, inhibition of the P2rx7 receptor by A43 clearly blocks Mmp13 up-regulation in these cells. Treatment of MC3T3-E1 cells with A43 alone has no effect on Mmp13 expression. B) A very similar effect is seen when MC3T3-E1 cells overexpressing Saa3 are treated with the MAPK/ERK inhibitor PD. PD alone has no effect on Mmp13 expression. C) Schematic representation of Mmp13 promoter region. Large, black arrow shows the first exon, small white bar represents the 3′-UTR, and small open arrows show the start sites of the diverse promoter amplicons cloned into the SEAP2 reporter vector. Sequence between the small black arrows shows the promoter region analyzed to define c-FOS binding by ChIP assay. Small gray box defines selected putative AP-1 transcription binding site, as calculated with Genomatix software. fw, forward; rev, reverse. D) pc3.1-Saa3-dependent stimulation of pSEAP2-Mmp13 promoter fragments in transient transfected MC3T3-E1 cells after 4 d of Saa3 transfection as assessed by alkaline phosphatase reporter gene assay. E) Mutation of the AP-1 binding site at −47 bp upstream from the transcriptional start site in the 0.25 kb Mmp13 promoter construct fully depletes Mmp13 promoter activation by forced Saa3 expression. mut, mutant; WT, wild-type. F) ChIP analysis shows that forced expression of Saa3 for 3 d notably increases c-FOS binding on the selected proximal Mmp13 promoter region in MC3T3-E1 cells. G) Forced expression of human SAA1 (middle white bar) or treatment with human recSAA1 (middle black bar) significantly increases MMP13 mRNA levels in human osteoblast-like U2-OS cells. However, this effect is not seen after forced SAA3P expression (right white bar) or when treating with recSAA3P (right black bar). 18S rRNA is a reference gene for RT-qPCR analysis. Values are represented as the mean ± sd. Control cells (Ctrl) or cells transfected with empty pc3.1 vector (pc3.1 empty) are set to 1, and treated samples are referred to as fold change; for all experiments n = 3. *P ≤ 0.05; **P ≤ 0.01; ***P ≤ 0.001 vs. Co; ++P ≤ 0.01 vs. pc3.1-Saa3.
Mmp13 is regulated by MAPK/ERK signaling via transcription factor AP-1 (56–58). The AP-1 binding site is identified at −47 bp upstream from the transcriptional start site based on Genomatix analysis (Genomatix Software Gesellschaft mit beschränkter Haftung, Munich, Germany) (Fig. 9C). We generated 3 different Mmp13 promoter deletion mutants (1, 0.5, and 0.25 kbp) plus an AP-1 point mutant within (the 0.25 kbp promoter) that were fused to the SEAP2 reporter; the SEAP2 reporter produces a secreted alkaline phosphatase that can be enzymatically monitored in the cell culture medium. Cotransfection experiments were performed with the Mmp13/SEAP2 reporter constructs and a pcDNA3.1-Saa3 expression vector that were introduced into MC3T3-E1 cells for 3 d. As predicted from the location of the AP-1 site at −47 bp, the 0.25 kbp Mmp13 promoter responds robustly to Saa3 coexpression with a 3- to 4-fold increase of the SEAP2 activity (Fig. 9D). Remarkably, Saa3 effects are suppressed by inclusion of additional promoter sequences located upstream of between 0.25 kbp. The latter suggests that normal physiologic activation of the Mmp13 promoter by Saa3 is attenuated by secondary transcriptional pathways that apparently have been dissected by promoter deletion. More importantly, mutation of the AP-1 binding site at −47 bp upstream from the transcriptional start site in the 0.25 kb Mmp13 promoter completely prevents Mmp13 promoter activation by Saa3 (Fig. 9E). Hence, the AP-1 site is the functional endpoint of a Saa3/P2xr7 signal transduction pathway.
To corroborate the dependency of Saa3-mediated Mmp13 up-regulation on AP-1, we performed ChIP analysis. Forced expression of Saa3 for 3 d in MC3T3-E1 cells results in a notable increase in c-Fos binding (Fig. 9F) to the −73/+3 region of the proximal Mmp13 promoter that is selectively amplified by our genomic qPCR primers (Fig. 9C). To translate the findings with mouse cells to human cells, we elevated A-SAA protein levels in human osteoblast-like U2-OS osteosarcoma cells by forced expression or administration of human recSAA1 protein (Fig. 9G). Either method significantly increases MMP13 mRNA levels, but this effect is not seen in control experiments with empty expression vector or vectors that express the human pseudogene SAA3P (Fig. 9G). The latter corroborates the functional equivalence of the mouse Saa3 and human SAA1 genes. We conclude that Saa3 enhances Mmp13 gene expression in osteoblasts via a regulatory network that involves P2rx7 and MAPK-ERK, as well as a c-FOS-containing AP-1 complex interacting with the proximal promoter (−73/+3) of the Mmp13 gene.
DISCUSSION
The physiologic interplay between the ECM and osteoblast function is a key question in bone biology. In this study, we show that the secreted acute-phase protein Saa3 has a hitherto unappreciated function in controlling ECM maintenance and bone cell phenotype development. Based on our past and present work, we find that acute induction of Saa3 in osteoblasts contributes to bone ECM repair, whereas Saa3 may also have important roles in osteoblast/osteocyte lineage progression and osteoclastogenesis, presumably through a paracrine mechanism. These principal biologic functions of Saa3 indicate that this protein may have underexplored roles in bone metabolism and skeletal diseases. Importantly, our data suggest that Saa3 may represent a novel coupling factor that controls bone repair and homeostasis.
As an acute-phase protein, A-SAA represents a protein that is expressed as part of an early response to inflammation and perhaps other biologic cues. Apart from its expression in multiple tissues including liver (21, 27, 28), chondrocytes (22, 29), monocytes (25, 33, 34), and adipocytes (30–32), as well as disease states like rheumatoid arthritis (24, 37, 38), Alzheimer's disease (37, 39), cardiovascular diseases (40–42), or insulin resistance (43), we find that A-SAA is expressed in osteoblasts and osteocytes. We observed here that A-SAA gene expression (human SAA1 and mouse Saa3) is robust at distinct developmental stages during osteogenic lineage progression. Due to the lack of a reliable and/or commercially available antibody for Saa3, we were unable to corroborate the cell-type-specific expression of Saa3 in bone tissue by immunohistochemistry. In osteoblasts, the activation of Saa3 due to compromised ECM integrity stimulates expression of Mmp13 (19), a major MMP in bone. Thus, expression of A-SAA proteins in bone could act as a general stress factor, reporting microdamage and other physicochemical disturbances of the bone matrix.
Our data suggest that Saa3 accelerates the conversion of late-stage osteoblasts to an early osteocytic phenotype. Several lines of evidence suggest that Saa3 stimulates but does not suffice to induce osteocytic differentiation. For example, we find that Saa3 is not up-regulated until osteocytes mature from an osteoblastic state (i.e., based on findings with MLO-A5 osteocytes presented in Fig. 3A). Furthermore, MC3T3-E1 osteoblasts do not have the potential to differentiate into osteocytes, and Saa3 is not expressed at appreciable levels during MC3T3-E1 differentiation unless cells are treated with homocysteine (19), which does not convert osteoblasts into osteocytes. Therefore, we envision that Saa3 is an enhancer of the osteocytic program, and not an inducer.
Our results also show that MLO-Y4 and MLO-A5 differ in their biologic responses to depletion of Saa3. Although the exact cause for this cell-type-specific difference remains to be determined, distinctions in the biologic properties of the 2 cell types could account for the effects of Saa3. For example, MLO-Y4 cells represent a more mature osteocytic cell type than MLO-A5 cells. However, whereas MLO-Y4 cells can grow to high density, they do not deposit much ECM and do not mineralize efficiently in our experience. Furthermore, MLO-Y4 cells maintain a confluent proliferative state in which single cells start to detach at high density followed by replacement through cell division. In contrast, the MLO-A5 cells grow robustly and at confluence produce a visible ECM upon which they initiate differentiation and mineralization. Because our own data show that Saa3 is deposited within the ECM produced by mature osteoblasts, we postulate that the biologic effects of Saa3 may be proportional to the amount of ECM that is produced by the 2 cell types.
Clinical translation of findings on mouse Saa3 to human bone biology and skeletal disease is hampered by the lack of an easily identifiable human homolog due to Saa/SAA gene evolution in the mammalian lineage. The human as well as the murine A-SAA gene family includes 5 gene loci (SAA1/Saa1, SAA2/Saa2, SAA3P/Saa3, SAA4/Saa4, and SAAL1/Saal1). In both organisms, the gene clusters are organized in the same way indicating a common evolutionary origin of the entire SAA gene cluster (61–63). Our data indicate that the mouse Saa3 and human SAA1 genes have evolved into functionally analogous genes. Furthermore, the C-terminal parts of the human SAA1 and mouse Saa3 protein exhibit very high sequence identity with only few, mostly conservative changes of amino acids. Thus, the C-terminal region of human SAA1/SAA2 may have assumed the biologic functions of mouse Saa3, consistent with previous suggestions regarding the evolution of A-SAA protein functions (64). We note that 2 other A-SAA genes, SAAL1 and SAA4, differ significantly from the other members of the A-SAA gene family (30, 62). However, SAA4 is only marginally expressed, and SAAL1 is a constitutive protein that is ubiquitously expressed. High-throughput RNA sequence expression data across >250 human musculoskeletal tissues and mesenchymal cell types show that SAA1 is preferentially expressed in osteogenic cells (Mayo Clinic Musculoskeletal Genomics Atlas, unpublished data), consistent with our interpretation that human SAA1 is the functional equivalent of mouse Saa3 and thus guiding future avenues for clinical translation.
Although our study indicates that SAA3 is important for bone cell maturation, 10- to 26-wk-old mice lacking global Saa3 expression show no obvious phenotypical differences under standard laboratory conditions. However, when exposed to a high sucrose and cholesterol diet, Saa3−/− mice gain less weight compared to wild-type littermates. Notably, female but not male Saa3−/− mice have less diet-induced adipose tissue inflammation and macrophage content, as well as improved plasma cholesterol, triglycerides, and lipoprotein profiles, thus potentially revealing gender-specific functions of Saa3 (65). The published results for Saa3−/− mice do not exclude the possibility that Saa3 may have a more prominent function under pathogenic conditions or in aging females (e.g., osteoporosis due to estrogen loss) and that loss of Saa3 could create a bone phenotype in old, stressed, and/or ovariectomized Saa3−/− mice.
Our results establish a novel connection between Saa3 and the purinergic P2rx7 receptor in regulating the osteocyte/osteoclast axis. P2 purinergic receptors increasingly gain attention in bone biology because they are known to play major roles not only in osteoblast cell functions but also in MSC differentiation, osteoclast activity and fusion, bone mineralization, and osteocyte-related mechano-transduction (53, 54, 66–68). This receptor drives osteoclast fusion by increasing extracellular adenosine concentrations (54), negatively regulates bone mineralization (55), and activates the NLRP3 inflammasome in macrophages by A-SAA binding (52). Interestingly, osteocytes were shown to respond to in situ mechanical loading by Ca2+ oscillations via P2 purinergic receptors (66). In this study, we observed that Saa3-dependent up-regulation of Mmp13 is dependent on the P2rx7/MAPK/ERK/AP-1 pathway. This finding indicates that Saa3-related paracrine effects are physiologically linked to P2rx7-regulated bone metabolism.
Previous gene expression studies from our group examined the functional consequences of bone ECM damage due to alterations in the architectural organization of collagen, which is known to affect bone strength, perturb bone homeostasis, and contribute to skeletal disease. This prior work revealed that Saa3 mRNA is induced in osteoblastic cells upon ECM insult using collagen-damaging agents (e.g., compounds that affect collagen cross-linking and/or triple-helix formation). The resulting damage in the ECM exposes RGD sequence motifs that trigger a PTK2/FAK-PXN-CTNNB1-RELA pathway and stimulate Saa3 gene expression (19). In this study, we extend these findings by showing that osteocyte-secreted Saa3 is deposited on the collagen matrix and that Saa3 is capable of activating a P2rx7/MAPK/ERK/AP-1 signaling pathway in osteoblasts that activates Mmp13 gene expression. Mmp13 was shown to be required for osteocytic perilacunar remodeling and may maintain bone fracture resistance (59). Mmp14, which is also involved in remodeling in osteocytic lacunae (69), is not regulated after Saa3 depletion in MLO-Y4 cells or after forced expression in MC3T3-E1 cells (data not shown). Thus, the ECM damage-related induction of Saa3 in osteoblasts supports a cellular function related to ECM repair.
Beyond its induction in response to collagen damage in osteoblasts to support ECM repair, we also established that Saa3 is maximally expressed at basal levels in osteocytes. In addition, Saa3 is capable of inhibiting both osteoblast differentiation, as reflected by its ability to suppress osteoblast-related genes (e.g., Bglap2), as well as stimulating osteoclastogenesis (e.g., formation of multinucleated TRAP-positive cells). Conceptually, these findings are consistent with a physiologic model in which Saa3 has a local biologic function in osteocytes, where its deposition in the bone ECM creates a form of latent Saa3 (e.g., analogous to latent TGF-β) (51, 70). This latent Saa3 may support bone resorption and/or remodeling by blocking new osteoblast function and enhancing osteoclastogenesis in adults. Equally possible, Saa3 may function by accelerating osteocyte differentiation, while enhancing osteoclast-mediated replacement of hypertrophic cartilage by new bone during earlier stages of postnatal development. Taken together, we propose that Saa3 may have multiple different biologic functions associated with ECM repair, bone remodeling, bone resorption, and skeletal development.
Acknowledgments
The authors thank their colleagues at Mayo Clinic for support and stimulating discussions. They are especially grateful to Florian Haider and David Neidhart (LBIO, Vienna, Austria), Eric Lewallen, Amel Dudakovic, Emily Camilleri, Jennifer Westendorf, and David Deyle (Mayo Clinic) for stimulating discussions. They also thank Dr. Masayoshi Kumegawa (Meikai University, Urayasu, Chiba, Japan) and Dr. Lynda Bonewald (University of Missouri-Kansas City, Kansas, MI, USA) for generously providing cell lines and Dr. Philipp Scherer (University of Texas, Southwestern, Dallas, TX, USA) for providing a mouse Saa3 antibody. This study was supported by the Fonds zur Foerderung der Wissenschaftlichen Forschung (The Austrian Science Fund) Project P24370-B19, the Wiener Gebietskrankenkasse (Social Health Insurance Vienna), and the Allgemeine Unfallversicherungsanstalt (Austrian Social Insurance for Occupational Risks). Additional support was provided by U.S. National Institutes of Health Grant R01-AR049069 (to A.J.v.W.), as well as intramural grants from the Center for Regenerative Medicine at Mayo Clinic and a Patrick J. Kelly Research Fellowship award (to S.M.R.).
Glossary
- A43
A438079
- AP-1
activator protein 1
- A-SAA
serum amyloid A
- Calcr
calcitonin receptor
- ChIP
chromatin immunoprecipitation
- CL
cell lysate
- Ctsk
cathepsin K
- ECM
extracellular matrix
- FCS
fetal calf serum
- IF
immunofluorescence
- MCSF
macrophage colony-stimulating factor
- MEM
minimum essential medium
- MMP
matrix metalloproteinase
- M/R
macrophage colony-stimulating factor and receptor activator of NF-κB ligand
- OPG
osteoprotegerin
- PD
PD98059
- PFA
paraformaldehyde
- RANKL
receptor activator of NF-κB ligand
- RGD
Arg-Gly-Asp
- recBGLAP2
recombinant BGLAP2
- recSAA
recombinant SAA
- RT-qPCR
RT-quantitative PCR
- Saa
serum amyloid A
- siNeg
small interfering RNA negative
- siRNA
small interfering RNA control
- TRAP
tartrate-resistant alkaline phosphatase
REFERENCES
- 1.Li X., Cao X. (2006) BMP signaling and skeletogenesis. Ann. N. Y. Acad. Sci. 1068, 26–40 [DOI] [PubMed] [Google Scholar]
- 2.Hadjidakis D. J., Androulakis I. I. (2006) Bone remodeling. Ann. N. Y. Acad. Sci. 1092, 385–396 [DOI] [PubMed] [Google Scholar]
- 3.Kong Y. Y., Yoshida H., Sarosi I., Tan H. L., Timms E., Capparelli C., Morony S., Oliveira-dos-Santos A. J., Van G., Itie A., Khoo W., Wakeham A., Dunstan C. R., Lacey D. L., Mak T. W., Boyle W. J., Penninger J. M. (1999) OPGL is a key regulator of osteoclastogenesis, lymphocyte development and lymph-node organogenesis. Nature 397, 315–323 [DOI] [PubMed] [Google Scholar]
- 4.Del Fattore A., Teti A., Rucci N. (2012) Bone cells and the mechanisms of bone remodelling. Front Biosci. (Elite Ed.) 4, 2302–2321 [DOI] [PubMed] [Google Scholar]
- 5.Matsuoka K., Park K. A., Ito M., Ikeda K., Takeshita S. (2014) Osteoclast-derived complement component 3a stimulates osteoblast differentiation. J. Bone Miner. Res. 29, 1522–1530 [DOI] [PubMed] [Google Scholar]
- 6.Ryu J., Kim H. J., Chang E. J., Huang H., Banno Y., Kim H. H. (2006) Sphingosine 1-phosphate as a regulator of osteoclast differentiation and osteoclast-osteoblast coupling. EMBO J. 25, 5840–5851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dallas S. L., Bonewald L. F. (2010) Dynamics of the transition from osteoblast to osteocyte. Ann. N. Y. Acad. Sci. 1192, 437–443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nakashima T., Hayashi M., Fukunaga T., Kurata K., Oh-Hora M., Feng J. Q., Bonewald L. F., Kodama T., Wutz A., Wagner E. F., Penninger J. M., Takayanagi H. (2011) Evidence for osteocyte regulation of bone homeostasis through RANKL expression. Nat. Med. 17, 1231–1234 [DOI] [PubMed] [Google Scholar]
- 9.Bellido T. (2014) Osteocyte-driven bone remodeling. Calcif. Tissue Int. 94, 25–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Komori T. (2014) Mouse models for the evaluation of osteocyte functions. J. Bone Metab. 21, 55–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Price J. S., Sugiyama T., Galea G. L., Meakin L. B., Sunters A., Lanyon L. E. (2011) Role of endocrine and paracrine factors in the adaptation of bone to mechanical loading. Curr. Osteoporos. Rep. 9, 76–82 [DOI] [PubMed] [Google Scholar]
- 12.Aubin J. E., Bonnelye E. (2000) Osteoprotegerin and its ligand: a new paradigm for regulation of osteoclastogenesis and bone resorption. Osteoporos. Int. 11, 905–913 [DOI] [PubMed] [Google Scholar]
- 13.Lacey D. L., Timms E., Tan H. L., Kelley M. J., Dunstan C. R., Burgess T., Elliott R., Colombero A., Elliott G., Scully S., Hsu H., Sullivan J., Hawkins N., Davy E., Capparelli C., Eli A., Qian Y. X., Kaufman S., Sarosi I., Shalhoub V., Senaldi G., Guo J., Delaney J., Boyle W. J. (1998) Osteoprotegerin ligand is a cytokine that regulates osteoclast differentiation and activation. Cell 93, 165–176 [DOI] [PubMed] [Google Scholar]
- 14.Simonet W. S., Lacey D. L., Dunstan C. R., Kelley M., Chang M. S., Lüthy R., Nguyen H. Q., Wooden S., Bennett L., Boone T., Shimamoto G., DeRose M., Elliott R., Colombero A., Tan H. L., Trail G., Sullivan J., Davy E., Bucay N., Renshaw-Gegg L., Hughes T. M., Hill D., Pattison W., Campbell P., Sander S., Van G., Tarpley J., Derby P., Lee R., Boyle W. J. (1997) Osteoprotegerin: a novel secreted protein involved in the regulation of bone density. Cell 89, 309–319 [DOI] [PubMed] [Google Scholar]
- 15.Winkler D. G., Sutherland M. K., Geoghegan J. C., Yu C., Hayes T., Skonier J. E., Shpektor D., Jonas M., Kovacevich B. R., Staehling-Hampton K., Appleby M., Brunkow M. E., Latham J. A. (2003) Osteocyte control of bone formation via sclerostin, a novel BMP antagonist. EMBO J. 22, 6267–6276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Van Bezooijen R. L., ten Dijke P., Papapoulos S. E., Löwik C. W. (2005) SOST/sclerostin, an osteocyte-derived negative regulator of bone formation. Cytokine Growth Factor Rev. 16, 319–327 [DOI] [PubMed] [Google Scholar]
- 17.Paschalis E. P., Tatakis D. N., Robins S., Fratzl P., Manjubala I., Zoehrer R., Gamsjaeger S., Buchinger B., Roschger A., Phipps R., Boskey A. L., Dall’Ara E., Varga P., Zysset P., Klaushofer K., Roschger P. (2011) Lathyrism-induced alterations in collagen cross-links influence the mechanical properties of bone material without affecting the mineral. Bone 49, 1232–1241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Thaler R., Spitzer S., Rumpler M., Fratzl-Zelman N., Klaushofer K., Paschalis E. P., Varga F. (2010) Differential effects of homocysteine and beta aminopropionitrile on preosteoblastic MC3T3-E1 cells. Bone 46, 703–709 [DOI] [PubMed] [Google Scholar]
- 19.Thaler R., Zwerina J., Rumpler M., Spitzer S., Gamsjaeger S., Paschalis E. P., Klaushofer K., Varga F. (2013) Homocysteine induces serum amyloid A3 in osteoblasts via unlocking RGD-motifs in collagen. FASEB J. 27, 446–463 [DOI] [PubMed] [Google Scholar]
- 20.Uhlar C. M., Whitehead A. S. (1999) Serum amyloid A, the major vertebrate acute-phase reactant. Eur. J. Biochem. 265, 501–523 [DOI] [PubMed] [Google Scholar]
- 21.Meek R. L., Eriksen N., Benditt E. P. (1992) Murine serum amyloid A3 is a high density apolipoprotein and is secreted by macrophages. Proc. Natl. Acad. Sci. USA 89, 7949–7952 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vallon R., Freuler F., Desta-Tsedu N., Robeva A., Dawson J., Wenner P., Engelhardt P., Boes L., Schnyder J., Tschopp C., Urfer R., Baumann G. (2001) Serum amyloid A (apoSAA) expression is up-regulated in rheumatoid arthritis and induces transcription of matrix metalloproteinases. J. Immunol. 166, 2801–2807 [DOI] [PubMed] [Google Scholar]
- 23.Malle E., De Beer F. C. (1996) Human serum amyloid A (SAA) protein: a prominent acute-phase reactant for clinical practice. Eur. J. Clin. Invest. 26, 427–435 [DOI] [PubMed] [Google Scholar]
- 24.Kumon Y., Loose L. D., Birbara C. A., Sipe J. D. (1997) Rheumatoid arthritis exhibits reduced acute phase and enhanced constitutive serum amyloid A protein in synovial fluid relative to serum. A comparison with C-reactive protein. J. Rheumatol. 24, 14–19 [PubMed] [Google Scholar]
- 25.Meek R. L., Benditt E. P. (1986) Amyloid A gene family expression in different mouse tissues. J. Exp. Med. 164, 2006–2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Thorn C. F., Lu Z. Y., Whitehead A. S. (2003) Tissue-specific regulation of the human acute-phase serum amyloid A genes, SAA1 and SAA2, by glucocorticoids in hepatic and epithelial cells. Eur. J. Immunol. 33, 2630–2639 [DOI] [PubMed] [Google Scholar]
- 27.Rygg M., Nordstoga K., Husby G., Marhaug G. (1993) Expression of serum amyloid A genes in mink during induction of inflammation and amyloidosis. Biochim. Biophys. Acta 1216, 402–408 [DOI] [PubMed] [Google Scholar]
- 28.Subramanian S., Han C. Y., Chiba T., McMillen T. S., Wang S. A., Haw A. III, Kirk E. A., O’Brien K. D., Chait A. (2008) Dietary cholesterol worsens adipose tissue macrophage accumulation and atherosclerosis in obese LDL receptor-deficient mice. Arterioscler. Thromb. Vasc. Biol. 28, 685–691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zerega B., Pagano A., Pianezzi A., Ulivi V., Camardella L., Cancedda R., Cancedda F. D. (2004) Expression of serum amyloid A in chondrocytes and myoblasts differentiation and inflammation: possible role in cholesterol homeostasis. Matrix Biol. 23, 35–46 [DOI] [PubMed] [Google Scholar]
- 30.Chiba T., Han C. Y., Vaisar T., Shimokado K., Kargi A., Chen M. H., Wang S., McDonald T. O., O’Brien K. D., Heinecke J. W., Chait A. (2009) Serum amyloid A3 does not contribute to circulating SAA levels. J. Lipid Res. 50, 1353–1362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Reigstad C. S., Lundén G. O., Felin J., Bäckhed F. (2009) Regulation of serum amyloid A3 (SAA3) in mouse colonic epithelium and adipose tissue by the intestinal microbiota. PLoS One 4, e5842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sommer G., Weise S., Kralisch S., Scherer P. E., Lössner U., Blüher M., Stumvoll M., Fasshauer M. (2008) The adipokine SAA3 is induced by interleukin-1beta in mouse adipocytes. J. Cell. Biochem. 104, 2241–2247 [DOI] [PubMed] [Google Scholar]
- 33.Maier W., Altwegg L. A., Corti R., Gay S., Hersberger M., Maly F. E., Sütsch G., Roffi M., Neidhart M., Eberli F. R., Tanner F. C., Gobbi S., von Eckardstein A., Lüscher T. F. (2005) Inflammatory markers at the site of ruptured plaque in acute myocardial infarction: locally increased interleukin-6 and serum amyloid A but decreased C-reactive protein. Circulation 111, 1355–1361 [DOI] [PubMed] [Google Scholar]
- 34.Yamada T., Wada A., Itoh K., Igari J. (2000) Serum amyloid A secretion from monocytic leukaemia cell line THP-1 and cultured human peripheral monocytes. Scand. J. Immunol. 52, 7–12 [DOI] [PubMed] [Google Scholar]
- 35.Han C. Y., Subramanian S., Chan C. K., Omer M., Chiba T., Wight T. N., Chait A. (2007) Adipocyte-derived serum amyloid A3 and hyaluronan play a role in monocyte recruitment and adhesion. Diabetes 56, 2260–2273 [DOI] [PubMed] [Google Scholar]
- 36.Westermark G. T., Sletten K., Grubb A., Westermark P. (1990) AA-amyloidosis. Tissue component-specific association of various protein AA subspecies and evidence of a fourth SAA gene product. Am. J. Pathol. 137, 377–383 [PMC free article] [PubMed] [Google Scholar]
- 37.Cunnane G. (2001) Amyloid precursors and amyloidosis in inflammatory arthritis. Curr. Opin. Rheumatol. 13, 67–73 [DOI] [PubMed] [Google Scholar]
- 38.Connolly M., Veale D. J., Fearon U. (2011) Acute serum amyloid A regulates cytoskeletal rearrangement, cell matrix interactions and promotes cell migration in rheumatoid arthritis. Ann. Rheum. Dis. 70, 1296–1303 [DOI] [PubMed] [Google Scholar]
- 39.Chung T. F., Sipe J. D., McKee A., Fine R. E., Schreiber B. M., Liang J. S., Johnson R. J. (2000) Serum amyloid A in Alzheimer’s disease brain is predominantly localized to myelin sheaths and axonal membrane. Amyloid 7, 105–110 [DOI] [PubMed] [Google Scholar]
- 40.Katayama T., Nakashima H., Honda Y., Suzuki S., Yamamoto T., Iwasaki Y., Yano K. (2007) The relationship between acute phase serum amyloid A (SAA) protein concentrations and left ventricular systolic function in acute myocardial infarction patients treated with primary coronary angioplasty. Int. Heart J. 48, 45–55 [DOI] [PubMed] [Google Scholar]
- 41.Johnson B. D., Kip K. E., Marroquin O. C., Ridker P. M., Kelsey S. F., Shaw L. J., Pepine C. J., Sharaf B., Bairey Merz C. N., Sopko G., Olson M. B., Reis S. E.; National Heart, Lung, and Blood Institute (2004) Serum amyloid A as a predictor of coronary artery disease and cardiovascular outcome in women: the National Heart, Lung, and Blood Institute-Sponsored Women’s Ischemia Syndrome Evaluation (WISE). Circulation 109, 726–732 [DOI] [PubMed] [Google Scholar]
- 42.Lewis K. E., Kirk E. A., McDonald T. O., Wang S., Wight T. N., O’Brien K. D., Chait A. (2004) Increase in serum amyloid a evoked by dietary cholesterol is associated with increased atherosclerosis in mice. Circulation 110, 540–545 [DOI] [PubMed] [Google Scholar]
- 43.Scheja L., Heese B., Zitzer H., Michael M. D., Siesky A. M., Pospisil H., Beisiegel U., Seedorf K. (2008) Acute-phase serum amyloid A as a marker of insulin resistance in mice. Exp. Diabetes Res. 2008, 230837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kovacevic A., Hammer A., Stadelmeyer E., Windischhofer W., Sundl M., Ray A., Schweighofer N., Friedl G., Windhager R., Sattler W., Malle E. (2008) Expression of serum amyloid A transcripts in human bone tissues, differentiated osteoblast-like stem cells and human osteosarcoma cell lines. J. Cell. Biochem. 103, 994–1004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Morris A. G. (1981) Neoplastic transformation of mouse fibroblasts by murine sarcoma virus: a multi-step process. J. Gen. Virol. 53, 39–45 [DOI] [PubMed] [Google Scholar]
- 46.Walker P. R., Boynton A. L., Whitfield J. F. (1977) The inhibition by colchicine of the initiation of DNA synthesis by hepatocytes in regenerating rat liver and by cultivated WI-38 and C3H10T1/2 cells. J. Cell. Physiol. 93, 89–97 [DOI] [PubMed] [Google Scholar]
- 47.Pfaffl M. W. (2001) A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 29, e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Thaler R., Rumpler M., Spitzer S., Klaushofer K., Varga F. (2011) Mospd1, a new player in mesenchymal versus epidermal cell differentiation. J. Cell. Physiol. 226, 2505–2515 [DOI] [PubMed] [Google Scholar]
- 49.Canalis E. (2009) Growth factor control of bone mass. J. Cell. Biochem. 108, 769–777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Preciado-Patt L., Hershkoviz R., Fridkin M., Lider O. (1996) Serum amyloid A binds specific extracellular matrix glycoproteins and induces the adhesion of resting CD4+ T cells. J. Immunol. 156, 1189–1195 [PubMed] [Google Scholar]
- 51.Dallas S. L., Rosser J. L., Mundy G. R., Bonewald L. F. (2002) Proteolysis of latent transforming growth factor-beta (TGF-beta )-binding protein-1 by osteoclasts. A cellular mechanism for release of TGF-beta from bone matrix. J. Biol. Chem. 277, 21352–21360 [DOI] [PubMed] [Google Scholar]
- 52.Niemi K., Teirilä L., Lappalainen J., Rajamäki K., Baumann M. H., Öörni K., Wolff H., Kovanen P. T., Matikainen S., Eklund K. K. (2011) Serum amyloid A activates the NLRP3 inflammasome via P2X7 receptor and a cathepsin B-sensitive pathway. J. Immunol. 186, 6119–6128 [DOI] [PubMed] [Google Scholar]
- 53.Wesselius A., Bours M. J., Agrawal A., Gartland A., Dagnelie P. C., Schwarz P., Jorgensen N. R. (2011) Role of purinergic receptor polymorphisms in human bone. Front Biosci (Landmark Ed) 16, 2572–2585(Elite Ed) [DOI] [PubMed] [Google Scholar]
- 54.Pellegatti P., Falzoni S., Donvito G., Lemaire I., Di Virgilio F. (2011) P2X7 receptor drives osteoclast fusion by increasing the extracellular adenosine concentration. FASEB J. 25, 1264–1274 [DOI] [PubMed] [Google Scholar]
- 55.Syberg S., Schwarz P., Petersen S., Steinberg T. H., Jensen J. E., Teilmann J., Jørgensen N. R. (2012) Association between P2X7 Receptor Polymorphisms and Bone Status in Mice. J. Osteoporos. 2012, 637986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Im H. J., Pacione C., Chubinskaya S., Van Wijnen A. J., Sun Y., Loeser R. F. (2003) Inhibitory effects of insulin-like growth factor-1 and osteogenic protein-1 on fibronectin fragment- and interleukin-1beta-stimulated matrix metalloproteinase-13 expression in human chondrocytes. J. Biol. Chem. 278, 25386–25394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Selvamurugan N., Chou W. Y., Pearman A. T., Pulumati M. R., Partridge N. C. (1998) Parathyroid hormone regulates the rat collagenase-3 promoter in osteoblastic cells through the cooperative interaction of the activator protein-1 site and the runt domain binding sequence. J. Biol. Chem. 273, 10647–10657 [DOI] [PubMed] [Google Scholar]
- 58.Boumah C. E., Selvamurugan N., Partridge N. C. (2005) Transcription in the osteoblast: regulatory mechanisms utilized by parathyroid hormone and transforming growth factor-beta. Prog. Nucleic Acid Res. Mol. Biol. 80, 287–321 [DOI] [PubMed] [Google Scholar]
- 59.Tang S. Y., Herber R. P., Ho S. P., Alliston T. (2012) Matrix metalloproteinase-13 is required for osteocytic perilacunar remodeling and maintains bone fracture resistance. J. Bone Miner. Res. 27, 1936–1950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Pivetta E., Scapolan M., Pecolo M., Wassermann B., Abu-Rumeileh I., Balestreri L., Borsatti E., Tripodo C., Colombatti A., Spessotto P. (2011) MMP-13 stimulates osteoclast differentiation and activation in tumour breast bone metastases. Breast Cancer Res. 13, R105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Betts J. C., Edbrooke M. R., Thakker R. V., Woo P. (1991) The human acute-phase serum amyloid A gene family: structure, evolution and expression in hepatoma cells. Scand. J. Immunol. 34, 471–482 [DOI] [PubMed] [Google Scholar]
- 62.Sipe J. (1999) Revised nomenclature for serum amyloid A (SAA). Nomenclature Committee of the International Society of Amyloidosis. Part 2. Amyloid 6, 67–70 [DOI] [PubMed] [Google Scholar]
- 63.Uhlar C. M., Burgess C. J., Sharp P. M., Whitehead A. S. (1994) Evolution of the serum amyloid A (SAA) protein superfamily. Genomics 19, 228–235 [DOI] [PubMed] [Google Scholar]
- 64.Olsen H. G., Skovgaard K., Nielsen O. L., Leifsson P. S., Jensen H. E., Iburg T., Heegaard P. M. (2013) Organization and biology of the porcine serum amyloid A (SAA) gene cluster: isoform specific responses to bacterial infection. PLoS One 8, e76695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Den Hartigh L. J., Wang S., Goodspeed L., Ding Y., Averill M., Subramanian S., Wietecha T., O’Brien K. D., Chait A. (2014) Deletion of serum amyloid A3 improves high fat high sucrose diet-induced adipose tissue inflammation and hyperlipidemia in female mice. PLoS One 9, e108564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Jing D., Baik A. D., Lu X. L., Zhou B., Lai X., Wang L., Luo E., Guo X. E. (2014) In situ intracellular calcium oscillations in osteocytes in intact mouse long bones under dynamic mechanical loading. FASEB J. 28, 1582–1592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Sun D., Junger W. G., Yuan C., Zhang W., Bao Y., Qin D., Wang C., Tan L., Qi B., Zhu D., Zhang X., Yu T. (2013) Shockwaves induce osteogenic differentiation of human mesenchymal stem cells through ATP release and activation of P2X7 receptors. Stem Cells 31, 1170–1180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Armstrong S., Pereverzev A., Dixon S. J., Sims S. M. (2009) Activation of P2X7 receptors causes isoform-specific translocation of protein kinase C in osteoclasts. J. Cell Sci. 122, 136–144 [DOI] [PubMed] [Google Scholar]
- 69.Holmbeck K., Bianco P., Pidoux I., Inoue S., Billinghurst R. C., Wu W., Chrysovergis K., Yamada S., Birkedal-Hansen H., Poole A. R. (2005) The metalloproteinase MT1-MMP is required for normal development and maintenance of osteocyte processes in bone. J. Cell Sci. 118, 147–156 [DOI] [PubMed] [Google Scholar]
- 70.Karsdal M. A., Hjorth P., Henriksen K., Kirkegaard T., Nielsen K. L., Lou H., Delaissé J. M., Foged N. T. (2003) Transforming growth factor-beta controls human osteoclastogenesis through the p38 MAPK and regulation of RANK expression. J. Biol. Chem. 278, 44975–44987 [DOI] [PubMed] [Google Scholar]