

Abstract
Objective
Transient Receptor Potential Melastatin-2 (TRPM2) channel is a non-selective cation channel that mediates influx of Ca2+ and Na+ with relative permeability of PCa:PNa ∼0.6 in response to cellular oxidative stress. As angiogenesis and ischemic neovascularization are both significantly dependent on oxidant signaling, here we investigated the possibile role of VEGF-induced ROS production in activating TRPM2-dependent Ca2+ signaling, and in the mechanism of angiogensis and ischemic neovascularization.
Approach and Results
We observed that VEGF stimulation rapidly induced the association of TRPM2 and c-Src kinase with VE-cadherin forming a signalplex at VE-cadherin junctions in endothelial cells (ECs). Using ECs isolated from TRPM2−/− mice or after siRNA depletion of TRPM2, we demonstrated that TRPM2-activated Ca2+ signaling was required for c-Src kinase-induced phosphorylation of VE-cadherin at Y658 and Y731, the crucial sites involved in VE-cadherin internalization in response to VEGF. VEGF-induced ROS generation activated TRPM2-induced Ca2+ entry whereas the ROS-insensitive TRPM2 mutant (C1008→A) showed impaired Ca2+ entry. ECs depleted of TRPM2 also displayed significantly perturbed migratory phenotype and impaired activation of c-Src in response to VEGF. TRPM2-/- mice reconstituted with wild type myeloid cells demonstrated aberrant angiogenesis and neovascularisation in the hindlimb ischemia model as compared to wild type mice.
Conclusion
VEGF-induced angiogeneis and post-ischemic neovascularisation in mice required ROS generation in ECs and resultant TRPM2 activation. Thus, our findings provide novel insight into the role of TRPM2 in mechanism of angiogenesis and ischemic neovascularisation.
Keywords: TRPM2, Angiogenesis, Hind Limb Ischemia, Endothelial Cells, VE-cadherin
Introduction
The growth of blood vessel induced by VEGF ligation of the receptor VEFGR2 is mediated by an array of parallel and complementary signaling pathways1. In post-natal life angiogenesis and arteriogenesis are the major determinants of blood vessel growth2. Sprouting of new vessels is defined as angiogenesis2 whereas arteriogenesis is defined by enlargement and remodeling of existing collateral arteries and formation of conductance vessels2. Neovascularization in response to persistent ischemia is a fundamental adaptive response encompassing features of both angiogenesis and arteriogenesis2, 3.
Neovascularization in the ischemic milieu is largely driven by the recruitment endothelial cells (ECs) and inflammatory cells and generation of reactive oxygen species (ROS) and growth factors such as VEGF 4, 5. The canonical growth factor VEGF after binding to VEGFR2 induces EC migration, recruitment, and proliferation6. The stability of VE-cadherin homotypic interaction at adherens junctions (AJs) is maintained by its association with cytoplasmic catenin proteins α-, β-, γ-and p120-catenins, which anchor VE-cadherin to the actin cytoskeleton7.
AJ disassembly induced by VEGF signaling is essential in initiating the angiogenesis program 8. In this context, the phosphorylation of Y658 and Y731 in the C-terminal of VE-cadherin prevented VE-cadherin interaction with catenins and was responsible for disassembly of AJs, and thereby facilitated the migration of ECs 9-11. Although VEGF-induced Y658 and Y731 phosphorylation of VE-cadherin occurs as a consequence of generation of ROS12, the signaling mechanisms downstream of ROS signaling are not understood. Studies have shown VEGF-induced ROS generated by activation of NOX2 mediates angiogenesis and neovascularization of ischemic tissue13, 14. In addition to ROS, Ca2+ signaling activated by VEGF is also crucial in the mechanism of EC migration and angiogenesis15, 16. Here we addressed the question whether ROS generation downstream of VEGF signaling activates Ca2+ signaling via the ROS-activated TRPM2 channel, a member of transient receptor potential melastatin family17, and whether TRPM2-activated Ca2+ signaling is a prerequisite for angiogenesis and ischemic neovascularization.
TRPM2 is highly expressed in ECs where it mediates redox-activated Ca2+ entry and participates in increasing endothelial permeability in response to oxidants17. The primary mode of TRPM2 activation is through oxidant-induced generation of adenosine diphosphoribose (ADP-ribose; ADPR)18, which binds to the Nudix box domain (NUDT9-H) in TRPM2 C-terminal19. Oxidants enzymatically convert NAD+ to ADPR through poly (ADP Ribose) Polymerase (PARP)18. We demonstrated that VEGF via the generation of ROS activated TRPM2, resulting in Ca2+ signaling and c-Src kinase activation, and subsequently induced VE-cadherin phosphorylation on Y658 and Y731. We showed, importantly, that ROS activation of TRPM2 in mice was required for mediating angiogenesis and ischemic neovascularization.
Maerials and Methods
Materials and Methods are available in the online-only Data Supplement.
Results
VEGF activates ROS-dependent Ca2+ influx via TRPM2 channel
We first studied whether VEGF-induced ROS production could activate TRPM2 channel expressed in ECs. We observed by DCF-DA fluorescence and the more specific Amplex Red assay 20 that VEGF indeed induced ROS production in ECs (Fig. 1A; Fig S I A) as expected12. In addition, VEGF induced Ca2+ transients in ECs, which were in part inhibited by the general NADPH oxidase blocker apocynin (Fig 1B) or PEG-Catalase (Fig. SI B). The TRPM2 inhibitor 3-aminobenzamide (3ABA), which antagonizes PARP activity responsible for generating the TRPM2 agonist ADPR17, 21, also suppressed Ca2+ transients (Fig.1B). In contrast, expression of the ROS insensitive mutant of TRPM2 C1008→A in ECs reduced VEGF-mediated Ca2+ entry22 (Fig. 1C). VEGF-induced Ca2+ influx was also reduced in ECs from TRPM2-/- mice as compared to ECs from WT mice (Fig. SI C).
Figure 1. TRPM2 mediates VEGF-induced Ca2+ entry in endothelial cells.

A) DCFH-DA fluorescence measured in confluent human pulmonary artery ECs stimulated with VEGF. VEGF increased the DCF fluorescence in dose- and time-dependent manner. Experiments were independently reproduced three times in quarduplets.
B) Apocynin (Apo) and 3ABA prevent VEGF-induced Ca2+ entry in ECs. ECs were grown to confluence and were pre-treated with apocynin or 3ABA at indicated concentration 2h before loading ECs with FLIPR-5 Ca2+ sensitive dye. VEGF was added at the arrow (60 s). Although, there was a trend for inhibition of Ca2+ influx at 1mM concentrations of apocynin or 3ABA compared to lower doses, the values were not significant (ns). The experiment was repeated twice in quarduplets. Mean of maximum responses is expressed in the scattered dot plot (with SEM).
C) Ca2+ transients measured using FLIPR-5 Ca2+ sensitive dye following VEGF stimulation of ECs transfected with ADPR-insensitive TRPM2 mutant C1008A or empty vector. VEGF in each case was added at the arrow. Ca2+ ionophore ionomycin was added at the end of experiment for calibration. VEGF-mediated Ca2+ influx was suppressed in cells expressing the TRPM2 mutant (Cys1008 substituted to Ala). Representaive tracing is shown. The experiment was repeated twice with each experiment consisting of 5 or more replicates. Mean of maximum responses is expressed in the scattered dot plot (with SEM).
VEGF induces the association of TRPM2 with c-Src and VE-cadherin
We next addressed possible signaling mechanisms downstream of TRPM2 activation by first determining whether VEGF elicited interaction of c-Src with TRPM2 based on the role of Src activity in promoting disassembly of AJs and hence angiogenesis23. Interestingly, immunoprecipitation studies showed that VEGF stimulation within 3 min induced c-Src binding to TRPM2 (Fig. 2A). In addition, TRPM2 interacted with VE-cadherin, with maximum occurring at 10 min after VEGF stimulation (Fig. 2A). We next used the Duolink proximity ligation assay (PLA)24 to monitor in situ formation of c-Src-TRPM2-VE-cadherin complex. In unstimulated ECs, protein interactions were not seen. However, at 3 min post-VEGF stimulation, we observed TRPM2 and c-Src interaction with VE-cadherin at AJs as well as other sites in ECs (Fig 2B). This interaction was prominent at 10 min and persisted up to 30 min (Fig 2B). No interaction was observed with IgG control antibodies (Fig S II). Thus, both immunoprecipitation and PLA assays showed essentially similar VEGF-induced formation of a tripartite complex consisting of TRPM2 and c-Src at first which was then followed by binding of both to VE-cadherin.
Fig 2. VEGF promotes interaction of TRPM2 and c-Src with VE-cadherin.
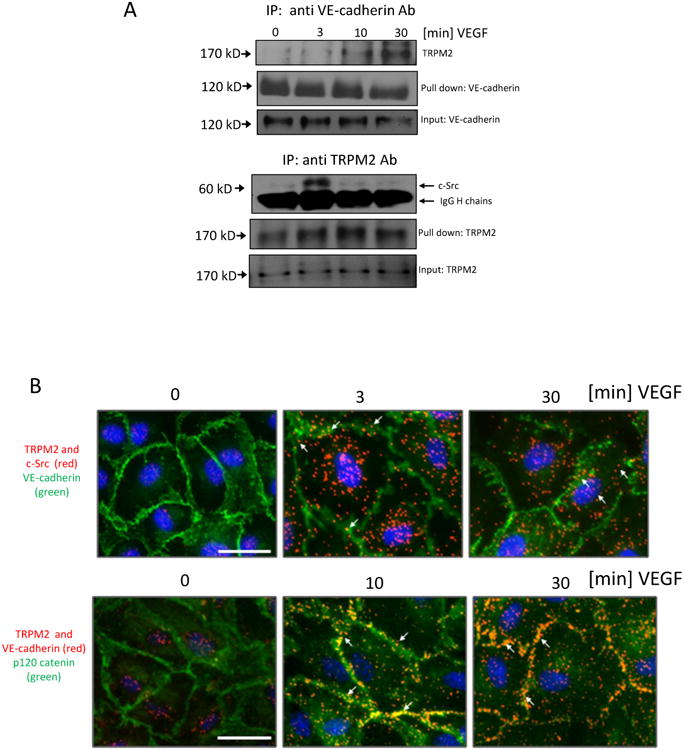
A) Co-immunoprecipitation studies. VE-cadherin was immunoprecipitated from cell lysates with anti-VE-cadherin antibody after VEGF stimulation. TRPM2 was detected using anti-TRPM2 antibody. VEGF induced association of TRPM2 with VE-cadherin with maximum response at 30 min. TRPM2 was also immunoprecipitated with anti-TRPM2 antibody after VEGF stimulation and was immunoblotted using the anti-c-Src antibody. Association of c-Src with TRPM2 was observed within 3 min post-VEGF stimulation. Thick bands denote IgG heavy chains; ∼1/10 of original cell lysate was blotted as input control. The experiment was independently reproduced twice.
B) In situ PLA in ECs treated with VEGF at the indicated times. Primary mouse (c-Src) and rabbit (TRPM2) antibodies were combined with secondary PLA probes (Olink Bioscience). The interaction events are visible as red dots, nuclear staining in blue (DAPI), and endothelial junctions are stained with VE-cadherin (green). In another PLA experiment, interaction between TRPM2 and VE-cadherin was determined using primary rabbit (TRPM2) and goat (VE-cadherin) antibodies combined with secondary with PLA probes. The interaction is visible as red dots and endothelial junctions are stained with p120-catenin (green). Scale bar represent 25μm. The experiment was repeated twice with similar observations.
TRPM2 activation of c-Src signals VE-cadherin phosphorylation and EC migration
We next studied the function of VEGF-induced binding of TRPM2 and c-Src with VE-cadherin at AJs. Here we observed that c-Src was phosphorylated at the Y-416 active site25 in a TRPM2-dependent manner (Fig. 3A). c-Src activation in turn also induced the phosphorylation of VE-cadherin (Fig. 3B and C). Depletion of either TRPM2 or c-Src suppressed phosphorylation of VE-cadherin at Y-658 and Y-731 (Fig. 3B and C). TRPM2 depletion, however, had no effect on VEGFR2 activation itself based on the finding that phosphorylation of VEGFR2 on the active site Y1175 was not affected (Fig S IV). Results similar to these VEGF-induced responses were also obtained in ECs stimulated with H2O2 (Fig. S III).
Figure 3. TRPM2 is required for VEGF-induced c-Src activation and VE-cadherin phosphorylation on Tyr-658 and Tyr-731.
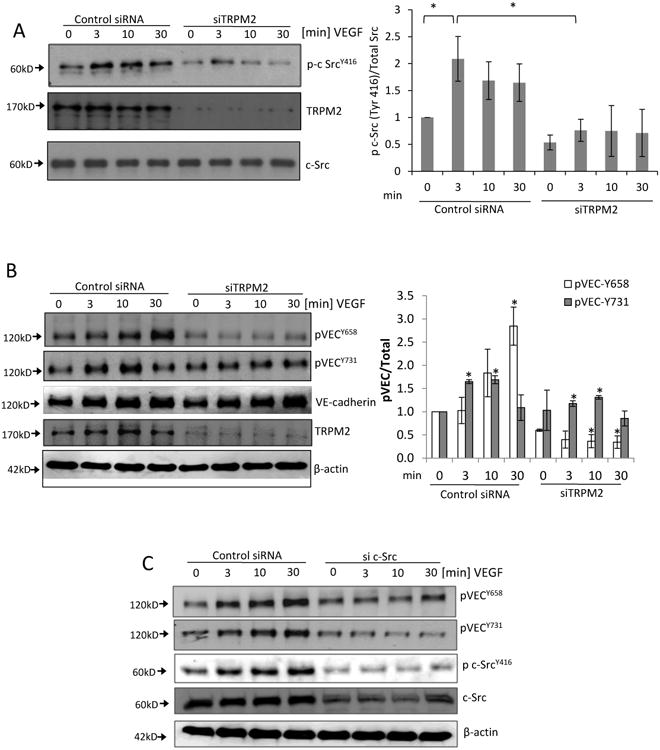
A) Immunoblot showing time-dependent phosphorylation of c-Src at active site Tyr-416 induced by VEGF stimulation of ECs. Maximum phosphorylation was observed at 3 min of VEGF stimulation. Transfection of ECs with TRPM2 siRNA to suppress TRPM2 expression significantly reduced phosphorylation of c-Src at Tyr-416. Mean of n=5 different immunoblots is shown in bar graph (SEM). *p<0.05.
B) Representative Western blots of lysates of ECs transfected with TRPM2 siRNA and then stimulated with VEGF for different times. VEGF induced the phosphorylation of VE-cadherin at Tyr-658 and Tyr-731 peaking at 30 min and 10 min, respectively. Silencing of TRPM2 suppressed VEGF-induced VE-cadherin phosphorylation at both sites. Bar graph shows mean responses of n=3 experiments (SEM) for Tyr-658 and Tyr-731 phosphorylation. *p<0.05 compared to the respective time points in the control group.
C) Immunoblots of human ECs transfected with c-Src siRNA to suppress Src expression, followed by stimulation with VEGF. c-Src knockdown reduced phosphorylation of VE-cadherin on Tyr-658 and Tyr-731. Representative result is shown from two indpependent epxeriments.
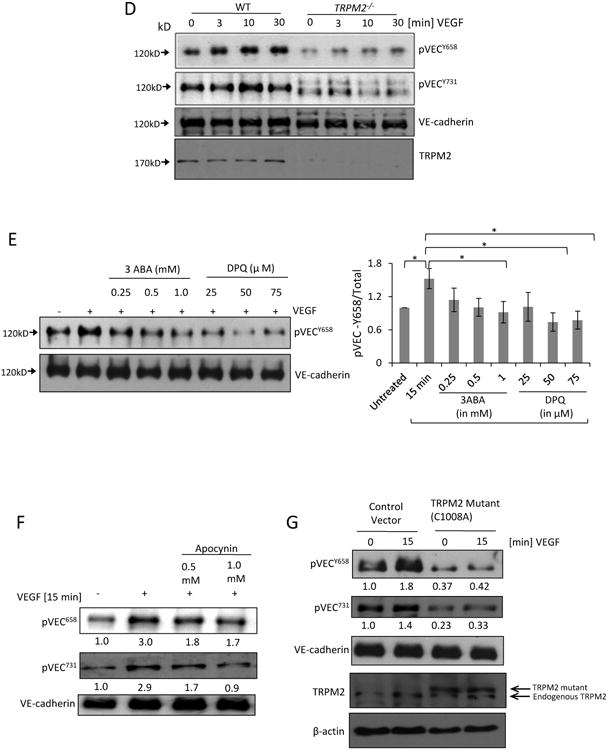
D) Mouse lung vascular ECs obtained from wild type and TRPM2-/- mice were stimulated with VEGF for different times and cell lysate was immunoblotted with anti-phospho VE-cadherin Tyr-658 and Tyr-731 antibodies. Phosphorylation of VE-cadherin was supressed in TRPM2-/ -mouse ECs compared to wild type mouse ECs.
E) Human ECs were treated with 3 aminobenzamide (3ABA) or (3,4-dihydro-5-[4-(1-piperidinyl)butoxyl]-1(2H)-isoquinolinone) (DPQ) at the indicated concentrations, and stimulated with VEGF for 15 min. Both inhibitors suppressed VEGF-induced VE-cadherin phosphorylation with DPQ being more effective. Bar graph shows mean response from 3 different immunoblots (SEM). *p<0.05.
F-G) Tranfection of TRPM2 mutant C1008A or apocynin (Apo) treatment of ECs suppressed VEGF-induced phosphorylation of VE-cadherin at Tyr-658 and Tyr-731. Normalized band densities are shown at the bottom of each blot.
Next studies made using ECs obtained from TRPM2-/- mice. These studies showed that ECs obtained from TRPM2-/- mice were resistant to VEGF-induced phosphorylation at Y658 and T731, in contrast to wild type ECs (Fig. 3D). In addition, using 2 different PARP inhibitors DPQ and 3ABA17, we observed that both inhibitors interfered with VEGF signaling as evident by markedly reduced VEGF-induced phosphorylation of VE-cadherin (Fig. 3E). These findings together demonstrate the key involvement of TRPM2 in the signaling pathway mediating VE-cadherin phosphorylation downstream of VEGF signaling.
We next addressed whether ROS generation downstream of VEGF signaling was also required for TRPM2 activation. Inhibiting VEGF-induced ROS generation by apocynin or more selectively through siRNA depletion of NOX2 in ECs suppressed VE-cadherin phosphorylation at Y658 and Y731 (Fig. 3F and Supplementary Fig S VA). VEGF exposure of ECs expressing the ADPR-insensitive mutant of TRPM2 C1008→A also suppressed VE-cadherin phosphorylation at Y658 and Y731 (Fig 3G). These findings thus link ROS generation to activation of TRPM2 and thereby to VE-cadherin phosphorylation at Y658 and Y731 sites11. We also observed that blocking intracellular Ca2+ influx by LaCl3 abrogated c-Src activation and VE-cadherin phosphorylation highlighting the essential role of Ca2+ influx in signaling c-Src-induced VE-cadherin phosphorylation (Supplementary Fig S VB).
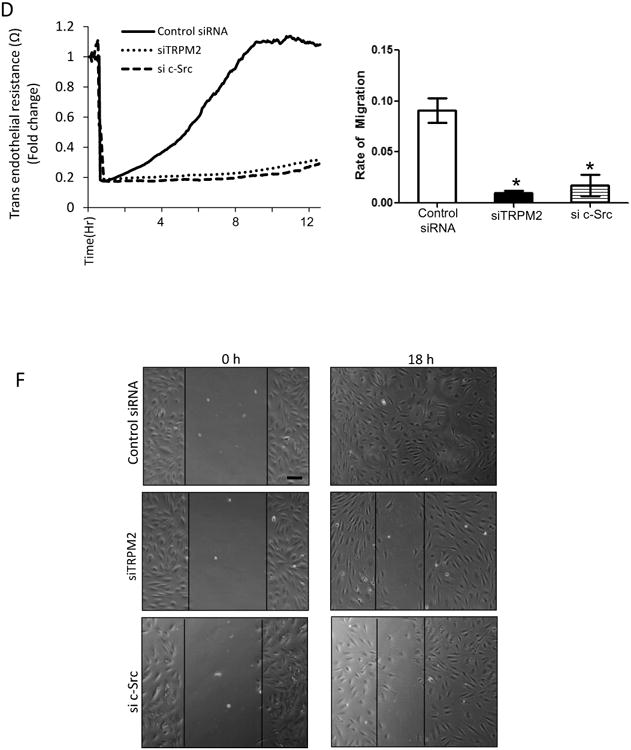
VEGF mediates internalization of VE-cadherin secondary to decreased p120-catenin and β-catenin binding to VE-cadherin, which in turn promotes the migration of ECs26. We observed in control ECs that VEGF significantly reduced the expression of p120-catenin and β-catenin at AJs consistent with internalization of VE-cadherin, whereas the response was significantly attenuated in ECs from TRPM2-/- mice (Fig. 4A, B). These changes were correlated with attenuated decrease in trans-endothelial resistance (TER) in TRPM2-depleted ECs after VEGF stimulation as compared to control ECs (Fig. 4C). VEGF-induced actin stress fiber formation was also reduced in TRPM2-depleted ECs as compared to controls (Fig S VI).
Figure 4. TRPM2 activation mediates VEGF-induced disassembly of adherens junctions and endothelial cell migration.
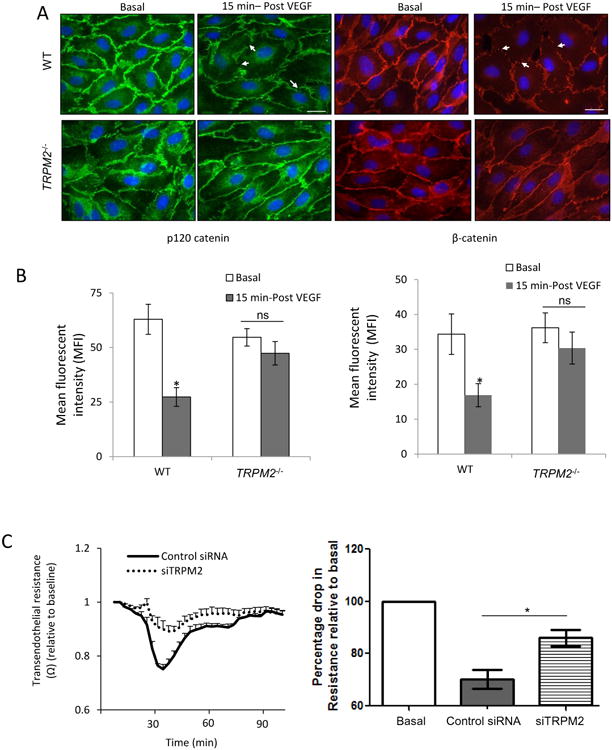
A) Immunofluorescent staining of p120-catenin (green) and β-catenin (red) in mouse lung ECs. VEGF stimulation induced multiple discontinuities in the expression of p120-catenin and β-catenin at the plasma membrabe in wild type ECs (arrows) whereas these changes were reduced in TRPM2-/- mouse ECs and AJs appeared continuous. Scale bar=20μm.
B) Bar graph of mean fluorescent intensities at AJs in wild type and TRPM2-/- ECs show significantly reduced intensity of p120-catenin and β-catenin in WT ECs compared to TRPM2-/- ECs. Mean flluorescent intensity (MFI) was calculated from images taken from 5-6 different fields for each group. The mean response from two indpendent experiments is shown in the bar graph. *p<0.05, ns: non significant.
C) TER measurement of AJ disassembly (with SEM bars) in control and TRPM2-depleted human pulmonary artery ECs. siRNA depletion of TRPM2 suppressed AJ disassembly induced by VEGF. Representative tracings are shown. The mean response after VEGF stimulation is shown in bar graph from two independent experiments each consisting of 4 replicates.
D-E) EC migration measured using TER showing markedly reduced EC migration following VEGF stimulation in siRNA TRPM2-depleted or siRNA c-Src-depleted ECs compared to control ECs. Study was made after uniform injury of EC monolayers made at the arrow. Each trace is representative of 5 or more replicates. The mean response is shown for n=3 for TRPM2 siRNA and n=2 for c-Src siRNA.
F) Wound scratch healing motility assay carried out over 18 h showed that human ECs transfected with TRPM2 siRNA and c-Src siRNA exhibited impairment in wound closure as compared to control siRNA-treated human ECs. Experiment was repeated twice. Bar represent 50 μm.
We next determined the role of TRPM2 in mediating the EC migration using the TER wound scratch assay27. A wound was created in EC monolayers growing on microelectrodes by applying a calibrated electric field27. This resulted in ∼90% decrease in TER (Fig. 4D, E). In controls, EC migration showed full recovery at 8 to 10 hr post-wound. These migration and recovery responses were however significantly impaired after TRPM2 or c-Src depletion in ECs (Fig. 4D and E). The validity of these migration results was confirmed by wound scratch healing assay (Fig. 4F).
TRPM2 is required for angiogenesis and post-ischemic neovascularization
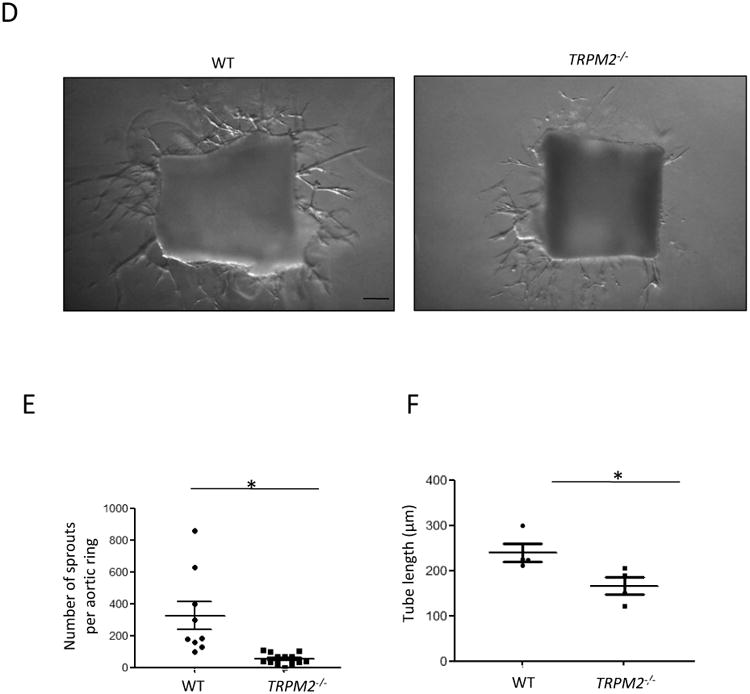
To investigate the role of TRPM2 in the mechanism of angiogenesis in vivo, WT and TRPM2-/- mice were injected subcutaneously with Matrigel plugs supplemented with VEGF (100ng/ml). The plugs obtained from TRPM2-/- mice showed significantly reduced number of CD31+ vessel formation (Fig. 5A, 5B) and angiogenic area compared to WT mice (Fig. 5C). Using the mouse aortic ring assay28, we also observed defective capillary sprouting and reduced capillary lengths from TRPM2-/- mouse rings as compared with WT mice (Fig. 5D-F).
Figure 5. TRPM2 knockout mice (TRPM2-/-) exhibit impaired angiogenesis in the in vivo Matrigel plug assay and ex vivo aortic ring assay.
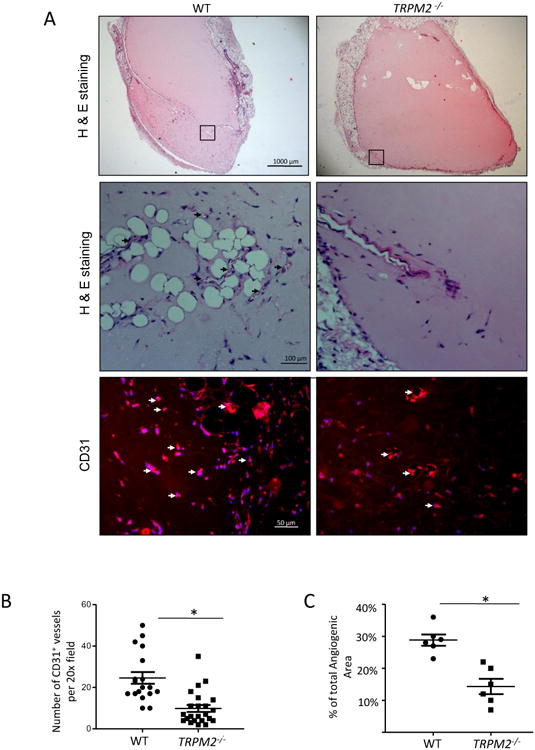
A) Histology of Matrigel plugs by H&E staining and immunofluorescence using anti-CD31 antibody. Micrographs show representative images of Matrigel plugs isolated from WT and TRPM2-/- mice after 10 days of implantation. Control plugs in absence of VEGF supplementation showed no angiogenic response (not shown). Figure shows representative of n=4 Matrigel plug studies per group. Arrows denote neovascularization evident CD31 expression in WT and much reduced response in TRPM2-/- mice.
B) Scatter dot graph showing mean number of newly formed CD31+ vessels in Matrigel plugs in WT and TRPM2-/- mice. CD31+ vessel number was significanlty less in plugs isolated TRPM2-/- mice than WT (shown as SEM). *p<0.005
C) Percentage angiogenic area in Matrigel plugs isolated from WT and TRPM2-/- mice at day# 10 (n=4) of plug implantation. TRPM2-/- mice showed significantly lower angiogenic area compared to WT mice. Shown as SEM. *p<0.05
D-F) Aortic rings from TRPM2-/- mice exhibited impaired vessel sprouting and tube elongation in the ex vivo aortic ring assay. Aortas were obtained from WT and TRPM2-/- mice and aortic rings in 1mm diam were cultured in Matrigel supplemented with VEGF (100ng/ml) for 7 days. Capillary sprouts and average tube length were measured in 5 aortic rings from group under microscopy. The data is represented as a scatter plot (with SEM). Bars represent 100 μm. The experiment was repeated twice. *p<0.05.
To extend these studies to in vivo ischemia model, we used the mouse model of hindlimb ischemia4, 5, 13, 14. The lethally irradiated TRPM2-/- mice were reconsituted with WT bone marrow cells5 to determine the role of EC- as opposed myeloid cell-expressed TRPM2 in mediating post-ischemia neovascularization. The control group consisted of WT mice similarly irradiated and transplanted with WT bone marrow cells. Laser Doppler blood flow measurements in ischemic and non-ischemic limbs after femoral artery ligation demonstrated that hind limb blood flow was markedly reduced after femoral artery ligation compared to non-ischemic control (day 0), and importantly blood flow recovered to 60% of basal level by day 28 in WT mice (Fig. 6A). However, blood flow recovery was severely compromised in TRPM2-/- mice (Fig. 6A). In the bone marrow transplantation experiments, the effectiveness in TRPM2-/- mice was confirmed by determining TRPM2 protein expression in bone marrow cells isolated from chimeric TRPM2-/- mice (Fig. 6B).
Figure 6. Impaired post-ischemic neovascularization in TRPM2−/− mice.
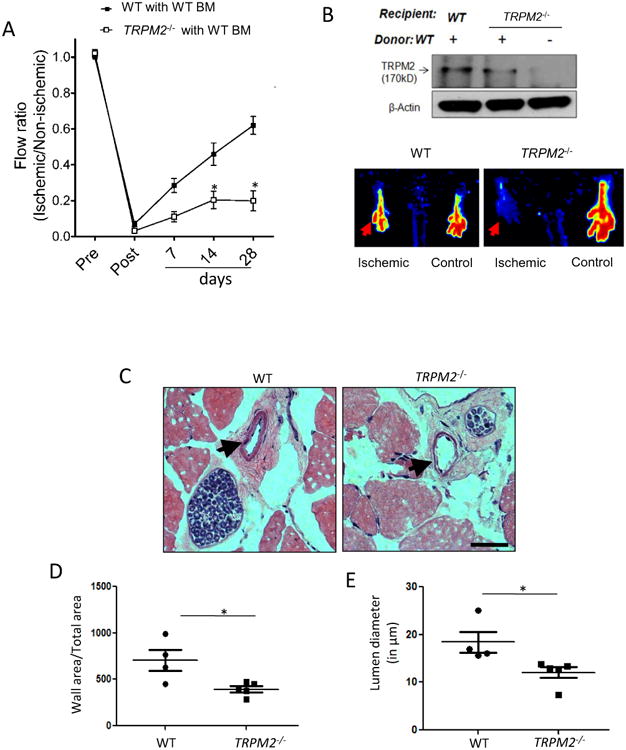
A) Blood flow recovery after hindlimb ischemia in WT and TRPM2−/− mice shown as ratio of laser Doppler-measured perfusion values (SEM) of ischemic (left) and non-ischemic (right) legs (WT, n = 4; TRPM2−/−, n = 5). Right panel show representative perfusion images at day# 28 measured by laser Doppler.
B) Effects of bone marrow transplantation of WT myeloid cells into WT and TRPM2−/− recipients. Analysis of bone marrow myeloid cells showed effective reconstitution of TRPM2 in TRPM2−/− mice.
C-E) Hematoxylin and eosin (H&E) staining of mouse adductor muscle. Arrowhead shows collateral vessels in the semi-membranosus muscle. The wall area is calculated from measurements of luminal and perivascular tracings. The lumen diameter was calculated for vessels in the range 5-30μm diameter size. Data are shown as scatter plots (with SEM). Bar represents 50 μm.
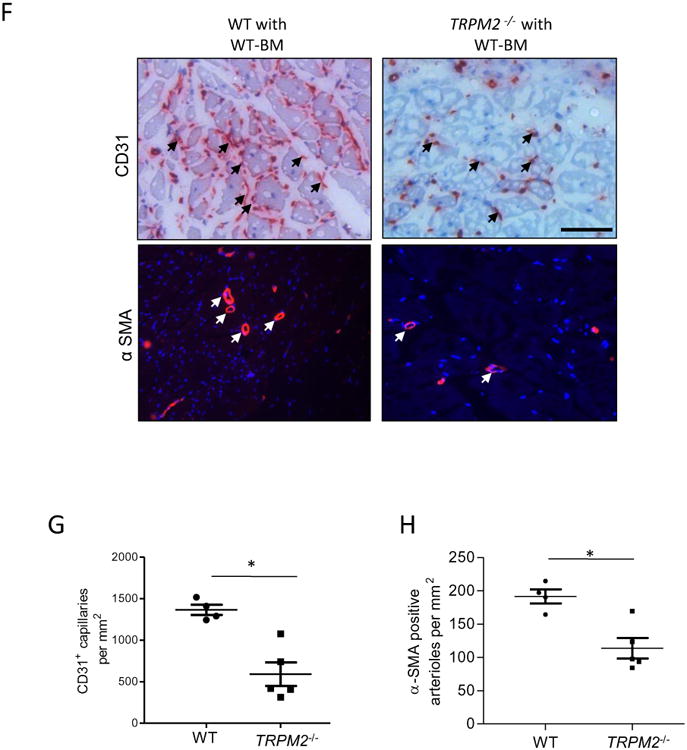
F-H) Histological analysis of mouse gastrocnemius (GC) muscle showing CD31 and α-SMA immunostaining obtained at day#28 after femoral artery ligation. Staining (red) by both histological and immunofluorescenee show CD31+ capillaries and α-SMA+ arterioles; DAPI stained nuclei are blue. Scale bar represents 50 μm. Scatter plots (with SEM) shows quantification of the number of CD31+ and α-SMA+ structures per mm2 area (*p<0.05).
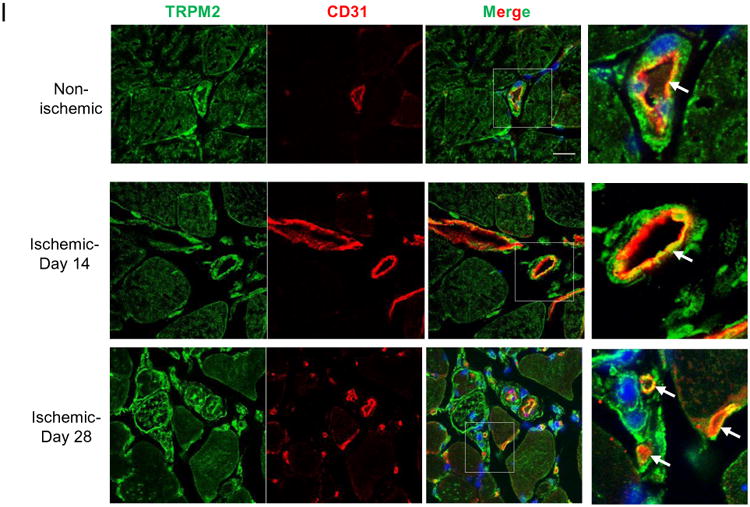
I) Immunofluorescent staining of GC muscle at days #14 and #28 of post-ischemic injury. Expression of TRPM2 was localized in endothelium of vessels. Ischemia induced the expression of TRPM2 in endothelial cells of newly formed vessels at day#14 and #28 post-induction of ischemic injury. Scale bar represent 20μm.
We additionally analyzed alterations in arteriogenesis in the adductor muscle of WT and TRPM2-/- mice. Here we measured wall thickness and lumen diameter of arterial vessels. Both the lumen diameter and the wall thickness of vessels were significantly lower in TRPM2-/- mice than WT mice after ischemic injury (Fig 6 C-E). In addition, depletion of TRPM2 in ECs supressed VEGF-induced ERK phsophorylation (Fig S VIIA) that is implicated in mediating VEGF-induced arteriogenesis29 (Fig S VIIA).
In addition, ischemic angiogenesis was evaluated in the gastrocnemius (GC) muscle of WT and chimeric TRPM2-/- mice. Capillaries visualized by staining with anti-CD31 antibody were counted in ischemic regions (Fig. 6 F-H). Mean number of CD31+ capillaries after ischemia in WT mice was 1336±90 per mm2 compared to 591±141 in TRPM2-/- mice (Fig. 6G). There were also significantly fewer α-SMA+ arterioles in TRPM2-/- mice compared to WT mice (WT 189±29 vs TRPM2-/- 119±23) (Fig.6G, H). The expression of TRPM2 was localised in the endothelium and vascular smooth muscle cells of arteries in GC muscle (Fig 6I and S7B) and in macrophages using Mac-3, a marker of macrophages (Fig S VIIB). Interestingly, the newly formed capillaries at day 28 of post-ischemic injury showed strong expression of TRPM2 suggesting possible role of TRPM2 in driving post-ischemic flow recovery (Fig 6 I).
Discussion
Neovascularisation is an adaptive response in ischemic vascular disorders30 that is coordinated by two distinct processes: angiogenesis involving proliferation of ECs and extension of the capillary network and arteriogenesis involving outgrowth of collateral arteries and de novo arterialization 2, 31. Both are driven by VEGF but require distinct signaling pathways2. Here we demonstrated the novel role of ROS-activated TRPM2 channel in activating c-Src kinase, which thereby mediated VEGF-induced post-ischemic neovascularisation through both induction of angiogenesis and arteriogenesis. The following key observations support this conclusion. First, we observed that TRPM2 deficiency severely impaired VEGF-induced EC migration, angiogenesis and arteriogenesis resulting in defective ischemic neovascularization in the standardized mouse hind limb ischemia model. Second, VEGF signalling induced the association of c-Src and TRPM2 with VE-cadherin at AJs, forming a tripartite complex. Third, c-Src activation occurring downstream of TRPM2-activated Ca2+ signaling was responsible for phosphorylating VE-cadherin at Y658 and Y731, the specific “phospho-switches” responsible for VE-cadherin internalization and EC migration12, 11, 26, and resulting in neovascularization32.
Although our focus was on TRPM2, we cannot rule the possiblity that additional ROS-sensitive trp channels expressed in ECs, such as TRPC3 and TRPC433, 34 may also be involved in mediating neovascularization. However, of these channels TRPM2 is specialaized based on the ROS-dependent mechanism of activation involving generation of the messenger adenosine diphospho ribose (ADP-ribose a.k.a ADPR)19. We observed that ADP-ribose-induced activation of TRPM2 increased endothelial permeability secondary to Ca2+ influx17. The present results provide compelling evidence that TRPM2 channel activation is also required for EC migration and the formation of new blood vessels.
We observed that VEGF signaling mediated the formation of a tripartite complex consisting of TRPM2 and c-Src associating with VE-cadherin, which was required for c-Src phosphorylation of VE-cadherin at Y658 and Y731. Formation of this complex was a prerequisite for activation of TRPM2-dependent angiogenesis and neovascularization. This mechanism fits well with the proposed scaffold function of VE-cadherin in aggregating essential signaling constitutents such as Gα13 that regulate assembly and disassembly of adherens junctions35, 36. Thus, VE-cadherin in addition to its role as an adhesive protein mediating formation of a confluent EC monolayer also has the ability to assemble TRPM2 and c-Src AJs that are essential for induction of the angiogenesis and arteriogenesis programs.
Phosphorylation of VE-cadherin at Y-658 proximal to the binding site for p120-catenin prevented p120-catenin binding to VE-cadherin and enabled the internalization of VE-cadherin and subsequent disassembly of AJs 26,37. In addition, phosphorylation of Y-731 prevented the binding of β-catenin to VE-cadheirn also promoting the loss of VE-cadherin junctional integrity38. We observed that VEGF-induced phosphorylation of VE-cadherin was suppressed at both Y-658 and Y-731 by TRPM2 depletion and in ECs isolated from TRPM2-/- mice. In addition, internalization of p120-catenin and β-catenin from AJs was reduced in TRPM2-/- ECs compared to wild type ECs consistent with the VE-cadherin phosphorylation results. A similiar VE-cadherin tyrosine phosphorylation was observed in ECs associated with rapid angiogenesis in ovaries and uterus and ECs from tumor vessels9, 39. Phospho-defective Y658 VE-cadherin mutant was preferentially localized at AJs and these ECs also failed to migrate properly11, 26, 38. In another study, mice having EC-specific deletion of phosphotyrosine phosphatase 1b (PTP1b), which functions to stabilise VE-cadherin junctions, showed enhanced EC migration, capillary sprouting, and accelerated blood flow recovery after femoral artery ligation40, 41. Thus, our results showing that VE-cadherin phosphorylation at Y658 and Y731 was c-Src- dependent and downstream of TRPM2-activation are consistent with the concept that VE-cadherin phosphoylation at these sites is a prerequisite for EC migration and thereby new vessel growth.
We observed that depletion of TRPM2 prevented the phosphorylation of c-Src at the Y-416 active site, indicating that TRPM2 was required for c-Src activation. A likely mechanism c-Src activation in this case is through ROS-mediated inactivation of tyrosine phosphatase SHP-2 that occurs secondary to oxidation of cysteine residue in SHP-2 catalytic domain 42,43. We also observed that ECs expressing ADPR-insensitive mutant of TRPM2 (C1008→A) or pharmacological inhibition of TRPM2 with 3 ABA suppressed Ca2+ entry as well as VE-cadherin phosphorylation in response to VEGF. Although Ca2+ influx was only partially reduced in these experiments, it is possible that a subthreshold level of Ca2+ influx was insufficient to phosphorylate VE-cadherin. The concentrations of inhibitors used for 3- ABA or apocynin was based on previous observation and had no cytotoxic effects44, 45, although with any inhibitor studies we cannot rule out certain non-specific effects. While our findings point to the key role of TRPM2-mediated Ca2+ entry in mediating c-Src activation, it remains unclear how TRPM2-induced Ca2+ influx activates c-Src at the level of VE-cadherin junctions. One possiblity is that TRPM2-mediated Ca2+ entry faciltiates the inactivation of phosphatases such as SHP-2 and PTP1B through enhanced oxidants generation 46, 47.
EC-derived H2O2 was required for neovascularization in response to ischemia4. Mice deficient in NOX2 also exhibited significantly delayed blood flow recovery and capillary formation13. EC-specific NOX4 overexpressing transgenic mice showed enhanced ischemia-induced angiogenesis 48. The present findings reinforce the essential role of ROS generation by ECs in signaling angiogenesis and ischemic neovascularisation; however, our findings interpose the requisite role of ROS activation of TRPM2 as a fundamental mechanism of ROS signaling-mediated angiogenesis and ischemic neovascularisation.
Ligation of the femoral artery in mice induced ischemic angiogenesis as well as arteriogenesis of collateral arteries3. Angiogenesis in this model has a limited capacity to increase perfusion of ischemic tissue49. The major part of reperfusion after ischemic injury is mediated by arteriogeneis49. TRPM2-/- mice exhibited marked defects in both angiogenesis and arteriogenesis as indicated by reduced formation of CD31+ capillaries and α-SMA+ arterial vessels of gastronemius muscle as well as reduced collateral vessel formation in the hind limb. Thus, TRPM2 activated signaling appears to be important for both angiogenesis and arteriogenesis. An impairment in ERK activation has been implicated in arterial morphogenesis29. In this regard in the present study, the observed supressed ERK activation after TRPM2 knockdown may underlie the arteriogenic defect seen in TRPM2-/- mice. We also observed enhanced expression of TRPM2 in small capillaries at day 14 and 28 post-onset of ischemia raising the intriguing possiblity that upregulation of TRPM2 in itself is an adaptive response to prolonged ischemia.
The recruitment of inflammatory myeloid cells is important for post-ischemic angiogenesis and neovascularization5. As TRPM2 was shown to negatively regulate ROS generation in myeloid cells and thereby to modulate inflammation50, it is possible that this function of TRPM2 influenced the formation of vessels following ischemia. To rule out the role of heightened activation of TRPM2-/- inflammatory myeloid cells in the mechanism of angiogenesis and neovascularization, we carried out studies by reconstituting the TRPM2-/- mice with WT myeloid cells.
Based on the finding that TRPM2 induces a rise in intracellular Ca2+ that can activate the apoptosis program51, an important question in the present study is the extent to which TRPM2 activation-induced by ROS generation mediates apopotsis. Any apoptosis would counteract revascularization induced by TRPM2. However, Ca2+-mediated apopotosis occurred at greater than four fold higher threshold of Ca2+ signaling than induced by VEGF and required greater ROS generation than seen in the present studies51. In summary, we showed that TRPM2 activated by VEGF-induced ROS generation and the resulting activation of c-Src was required for angiogenesis and post-ischemic neovascularization in mice as described in Fig. 7.
Fig 7. Schematic representation describing the role of TRPM2 in mediating VEGF-induced angiogenesis.
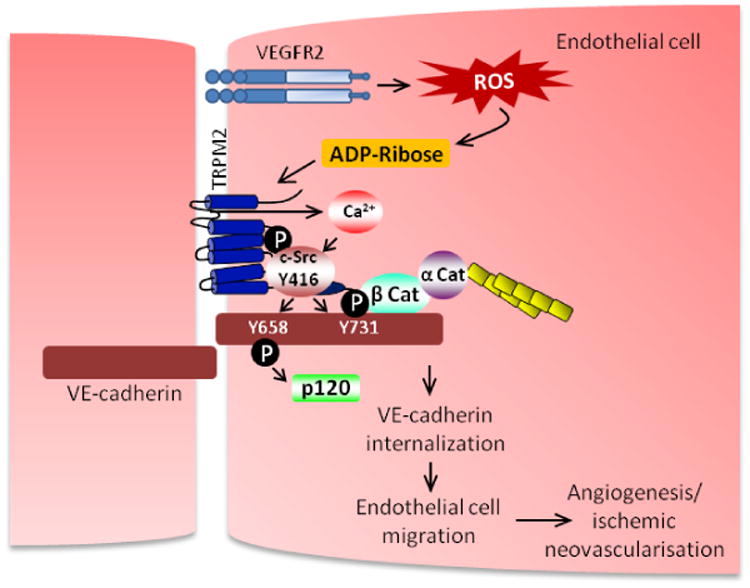
VEGF induced the association of TRPM2 with c-Src on VE-cadherin forming a signaling complex leading to TRPM2 mediated Ca2+ influx and c-Src phosphorylation on active Y416 site. Activation of c-Src phosphorylated VE-cadherin on Y658 and Y731 induced AJ disassembly and EC migration resulting in activation of angiogenesis program.
Supplementary Material
Significance.
TRPM2 is a ROS-sensitive calcium channel which is highly expressed in endothelial cells and has been implicated in increasing endothelial permeability in response to oxidants. However, the role of TRPM2 in angiogenesis has not been explored. The studies presented here provide compelling evidence that TRPM2 plays an important role in ischemic neovascularisation by promoting endothelial cell migration following c-Src activation. To define the TRPM2's pro-angiogenic function in ECs, studies were made in cultured ECs as well as TRPM2-/- mice. In cultured ECs, VEGF induced the formation of a signalplex consisting of TRPM2 and c-Src localized on VE-cadherin junctions. These studies provide novel insights of the heretofore unknown role of TRPM2 in mediating angiogenesis and arteriogenesis with the potential of developing novel therapeutics for reperfusion of ischemic disease.
Acknowledgments
We thank GlaxoSmithKline for providing the Trpm2−/− C57BL/6 mice used in these experiments.
Sources of Funding: This work was supported in part by NIH training grants HL045638 and HL077806, HL116976, HL077524, R21HL112293 and AHA Post Doctoral Fellowship 13POST16640000.
Non Standard Abbreviations and Acronyms
- 3ABA
3 Amino benzamide
- ADP-ribose
Adenosine diphosphate ribose
- AJs
adherens junctions
- BM
Bone Marrow
- c-Src
Cellular Src Kinase
- Ca2+
Calcium
- DCF-DA
2′,7′-dichlorfluorescein-diacetate (DCFH-DA)
- DPQ
3,4-Dihydro-5[4-(1-piperindinyl)butoxy]-1(2H)-isoquinoline
- ECs
Endothelial Cells
- ERK
Extracellular signal-regulated kinase
- GC
Gastrocnemius muscle
- H2O2
Hydrogenperoxide
- NOX2
NADPH oxidase homolog 2
- PARP
poly ADP ribose polymerase
- PEG-Catalase
polyethylene glycol catalase
- PLA
Proximal Ligation Assay
- pVEC
Phosphorylated Vascular Endothelial Cadherin
- ROS
Reactive Oxygen Species
- SEM
Standard Error of Mean
- TER
Trans Endothelial Resistance
- TRPM2
Transient Receptor Potential Melastatin-2
- Tyr
Tyrosine residue
- VEGF
Vascular Endothelial Growth Factor
- VEGFR2
Vascular Endothelial Growth Factor Receptor-2
- VE-cadherin
Vascular Endothelial Cadherin
- WT
Wild Type mice
Footnotes
Disclosures: None
References
- 1.Potente M, Gerhardt H, Carmeliet P. Basic and therapeutic aspects of angiogenesis. Cell. 2011;146:873–887. doi: 10.1016/j.cell.2011.08.039. [DOI] [PubMed] [Google Scholar]
- 2.Carmeliet P. Mechanisms of angiogenesis and arteriogenesis. Nat Med. 2000;6:389–395. doi: 10.1038/74651. [DOI] [PubMed] [Google Scholar]
- 3.Limbourg A, Korff T, Napp LC, Schaper W, Drexler H, Limbourg FP. Evaluation of postnatal arteriogenesis and angiogenesis in a mouse model of hind-limb ischemia. Nat Protoc. 2009;4:1737–1746. doi: 10.1038/nprot.2009.185. [DOI] [PubMed] [Google Scholar]
- 4.Urao N, Sudhahar V, Kim SJ, Chen GF, McKinney RD, Kojda G, Fukai T, Ushio-Fukai M. Critical role of endothelial hydrogen peroxide in post-ischemic neovascularization. PLoS One. 2013;8:e57618. doi: 10.1371/journal.pone.0057618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Urao N, Razvi M, Oshikawa J, McKinney RD, Chavda R, Bahou WF, Fukai T, Ushio-Fukai M. IQGAP1 is involved in post-ischemic neovascularization by regulating angiogenesis and macrophage infiltration. PLoS One. 2010;5:e13440. doi: 10.1371/journal.pone.0013440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ferrara N. Vascular endothelial growth factor. Arterioscler Thromb Vasc Biol. 2009;29:789–791. doi: 10.1161/ATVBAHA.108.179663. [DOI] [PubMed] [Google Scholar]
- 7.Bravi L, Dejana E, Lampugnani MG. VE-cadherin at a glance. Cell Tissue Res. 2014;355:515–522. doi: 10.1007/s00441-014-1843-7. [DOI] [PubMed] [Google Scholar]
- 8.Bentley K, Franco CA, Philippides A, Blanco R, Dierkes M, Gebala V, Stanchi F, Jones M, Aspalter IM, Cagna G, Westrom S, Claesson-Welsh L, Vestweber D, Gerhardt H. The role of differential VE-cadherin dynamics in cell rearrangement during angiogenesis. Nat Cell Biol. 2014;16:309–321. doi: 10.1038/ncb2926. [DOI] [PubMed] [Google Scholar]
- 9.Lambeng N, Wallez Y, Rampon C, Cand F, Christe G, Gulino-Debrac D, Vilgrain I, Huber P. Vascular endothelial-cadherin tyrosine phosphorylation in angiogenic and quiescent adult tissues. Circ Res. 2005;96:384–391. doi: 10.1161/01.RES.0000156652.99586.9f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Esser S, Lampugnani MG, Corada M, Dejana E, Risau W. Vascular endothelial growth factor induces VE-cadherin tyrosine phosphorylation in endothelial cells. J Cell Sci. 1998;111:1853–1865. doi: 10.1242/jcs.111.13.1853. [DOI] [PubMed] [Google Scholar]
- 11.Potter MD, Barbero S, Cheresh DA. Tyrosine phosphorylation of VE-cadherin prevents binding of p120- and beta-catenin and maintains the cellular mesenchymal state. J Biol Chem. 2005;280:31906–31912. doi: 10.1074/jbc.M505568200. [DOI] [PubMed] [Google Scholar]
- 12.Monaghan-Benson E, Burridge K. The regulation of vascular endothelial growth factor-induced microvascular permeability requires Rac and reactive oxygen species. J Biol Chem. 2009;284:25602–25611. doi: 10.1074/jbc.M109.009894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tojo T, Ushio-Fukai M, Yamaoka-Tojo M, Ikeda S, Patrushev N, Alexander RW. Role of gp91phox (Nox2)-containing NAD(P)H oxidase in angiogenesis in response to hindlimb ischemia. Circulation. 2005;111:2347–2355. doi: 10.1161/01.CIR.0000164261.62586.14. [DOI] [PubMed] [Google Scholar]
- 14.Urao N, Inomata H, Razvi M, Kim HW, Wary K, McKinney R, Fukai T, Ushio-Fukai M. Role of nox2-based NADPH oxidase in bone marrow and progenitor cell function involved in neovascularization induced by hindlimb ischemia. Circ Res. 2008;103:212–220. doi: 10.1161/CIRCRESAHA.108.176230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li J, Cubbon RM, Wilson LA, Amer MS, McKeown L, Hou B, Majeed Y, Tumova S, Seymour VA, Taylor H, Stacey M, O'Regan D, Foster R, Porter KE, Kearney MT, Beech DJ. Orai1 and CRAC channel dependence of VEGF-activated Ca2+ entry and endothelial tube formation. Circ Res. 2011;108:1190–1198. doi: 10.1161/CIRCRESAHA.111.243352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yu PC, Gu SY, Bu JW, Du JL. TRPC1 is essential for in vivo angiogenesis in zebrafish. Circ Res. 2010 Apr 16;106:1221–1232. doi: 10.1161/CIRCRESAHA.109.207670. [DOI] [PubMed] [Google Scholar]
- 17.Hecquet CM, Ahmmed GU, Vogel SM, Malik AB. Role of TRPM2 channel in mediating H2O2-induced Ca2+ entry and endothelial hyperpermeability. Circ Res. 2008;102:347–355. doi: 10.1161/CIRCRESAHA.107.160176. [DOI] [PubMed] [Google Scholar]
- 18.Takahashi N, Kozai D, Kobayashi R, Ebert M, Mori Y. Roles of TRPM2 in oxidative stress. Cell Calcium. 2011;50:279–287. doi: 10.1016/j.ceca.2011.04.006. [DOI] [PubMed] [Google Scholar]
- 19.Perraud AL, Fleig A, Dunn CA, Bagley LA, Launay P, Schmitz C, Stokes AJ, Zhu Q, Bessman MJ, Penner R, Kinet JP, Scharenberg AM. ADP-ribose gating of the calcium-permeable LTRPC2 channel revealed by Nudix motif homology. Nature. 2001;411:595–599. doi: 10.1038/35079100. [DOI] [PubMed] [Google Scholar]
- 20.Shah A, Xia L, Goldberg H, Lee KW, Quaggin SE, Fantus IG. Thioredoxin-interacting Protein Mediates High Glucose-induced Reactive Oxygen Species Generation by Mitochondria and the NADPH Oxidase, Nox4, in Mesangial Cells. Journal of Biological Chemistry. 2013;288:6835–6848. doi: 10.1074/jbc.M112.419101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sano Y, Inamura K, Miyake A, Mochizuki S, Yokoi H, Matsushime H, Furuichi K. Immunocyte Ca2+ influx system mediated by LTRPC2. Science. 2001;293:1327–1330. doi: 10.1126/science.1062473. [DOI] [PubMed] [Google Scholar]
- 22.Mei ZZ, Mao HJ, Jiang LH. Conserved cysteine residues in the pore region are obligatory for human TRPM2 channel function. Am J Physiol Cell Physiol. 2006;291:C1022–C1028. doi: 10.1152/ajpcell.00606.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Eliceiri BP, Paul R, Schwartzberg PL, Hood JD, Leng J, Cheresh DA. Selective requirement for Src kinases during VEGF-induced angiogenesis and vascular permeability. Mol Cell. 1999;4:915–924. doi: 10.1016/s1097-2765(00)80221-x. [DOI] [PubMed] [Google Scholar]
- 24.Soderberg O, Gullberg M, Jarvius M, Ridderstrale K, Leuchowius KJ, Jarvius J, Wester K, Hydbring P, Bahram F, Larsson LG, Landegren U. Direct observation of individual endogenous protein complexes in situ by proximity ligation. Nat Methods. 2006;3:995–1000. doi: 10.1038/nmeth947. [DOI] [PubMed] [Google Scholar]
- 25.Harvey R, Hehir KM, Smith AE, Cheng SH. pp60c-src variants containing lesions that affect phosphorylation at tyrosines 416 and 527. Mol Cell Biol. 1989;9:3647–3656. doi: 10.1128/mcb.9.9.3647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nanes BA, Chiasson-MacKenzie C, Lowery AM, Ishiyama N, Faundez V, Ikura M, Vincent PA, Kowalczyk AP. p120-catenin binding masks an endocytic signal conserved in classical cadherins. J Cell Biol. 2012;199:365–380. doi: 10.1083/jcb.201205029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Keese CR, Wegener J, Walker SR, Giaever I. Electrical wound-healing assay for cells in vitro. Proc Natl Acad Sci U S A. 2004;101:1554–1559. doi: 10.1073/pnas.0307588100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Baker M, Robinson SD, Lechertier T, Barber PR, Tavora B, D'Amico G, Jones DT, Vojnovic B, Hodivala-Dilke K. Use of the mouse aortic ring assay to study angiogenesis. Nat Protoc. 2012;7:89–104. doi: 10.1038/nprot.2011.435. [DOI] [PubMed] [Google Scholar]
- 29.Deng Y, Larrivee B, Zhuang ZW, Atri D, Moraes F, Prahst C, Eichmann A, Simons M. Endothelial RAF1/ERK activation regulates arterial morphogenesis. Blood. 2013;121:3988–3996. doi: 10.1182/blood-2012-12-474601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Semenza GL. Angiogenesis in ischemic and neoplastic disorders. Annu Rev Med. 2003;54:17–28. doi: 10.1146/annurev.med.54.101601.152418. [DOI] [PubMed] [Google Scholar]
- 31.Faber JE, Chilian WM, Deindl E, van RN, Simons M. A Brief Etymology of the Collateral Circulation. Arterioscler Thromb Vasc Biol. 2014;34:1854–1859. doi: 10.1161/ATVBAHA.114.303929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bentley K, Franco CA, Philippides A, Blanco R, Dierkes M, Gebala V, Stanchi F, Jones M, Aspalter IM, Cagna G, Westrom S, Claesson-Welsh L, Vestweber D, Gerhardt H. The role of differential VE-cadherin dynamics in cell rearrangement during angiogenesis. Nat Cell Biol. 2014;16:309–321. doi: 10.1038/ncb2926. [DOI] [PubMed] [Google Scholar]
- 33.Poteser M, Graziani A, Rosker C, Eder P, Derler I, Kahr H, Zhu MX, Romanin C, Groschner K. TRPC3 and TRPC4 associate to form a redox-sensitive cation channel. Evidence for expression of native TRPC3-TRPC4 heteromeric channels in endothelial cells. J Biol Chem. 2006;281:13588–13595. doi: 10.1074/jbc.M512205200. [DOI] [PubMed] [Google Scholar]
- 34.Balzer M, Lintschinger B, Groschner K. Evidence for a role of Trp proteins in the oxidative stress-induced membrane conductances of porcine aortic endothelial cells. Cardiovasc Res. 1999;42:543–549. doi: 10.1016/s0008-6363(99)00025-5. [DOI] [PubMed] [Google Scholar]
- 35.Gong H, Gao X, Feng S, Siddiqui MR, Garcia A, Bonini MG, Komarova Y, Vogel SM, Mehta D, Malik AB. Evidence of a common mechanism of disassembly of adherens junctions through Galpha13 targeting of VE-cadherin. J Exp Med. 2014;211:579–591. doi: 10.1084/jem.20131190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ha CH, Bennett AM, Jin ZG. A novel role of vascular endothelial cadherin in modulating c-Src activation and downstream signaling of vascular endothelial growth factor. J Biol Chem. 2008;283:7261–7270. doi: 10.1074/jbc.M702881200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Potter MD, Barbero S, Cheresh DA. Tyrosine phosphorylation of VE-cadherin prevents binding of p120- and beta-catenin and maintains the cellular mesenchymal state. J Biol Chem. 2005;280:31906–31912. doi: 10.1074/jbc.M505568200. [DOI] [PubMed] [Google Scholar]
- 38.Hatanaka K, Simons M, Murakami M. Phosphorylation of VE-cadherin controls endothelial phenotypes via p120-catenin coupling and Rac1 activation. Am J Physiol Heart Circ Physiol. 2011;300:H162–H172. doi: 10.1152/ajpheart.00650.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Haidari M, Zhang W, Caivano A, Chen Z, Ganjehei L, Mortazavi A, Stroud C, Woodside DG, Willerson JT, Dixon RA. Integrin alpha2beta1 mediates tyrosine phosphorylation of vascular endothelial cadherin induced by invasive breast cancer cells. J Biol Chem. 2012;287:32981–32992. doi: 10.1074/jbc.M112.395905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nakamura Y, Patrushev N, Inomata H, Mehta D, Urao N, Kim HW, Razvi M, Kini V, Mahadev K, Goldstein BJ, McKinney R, Fukai T, Ushio-Fukai M. Role of protein tyrosine phosphatase 1B in vascular endothelial growth factor signaling and cell-cell adhesions in endothelial cells. Circ Res. 2008;102:1182–1191. doi: 10.1161/CIRCRESAHA.107.167080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lanahan AA, Lech D, Dubrac A, Zhang J, Zhuang ZW, Eichmann A, Simons M. PTP1b is a Physiologic Regulator of VEGF Signaling in Endothelial Cells. Circulation. 2014;130:902–909. doi: 10.1161/CIRCULATIONAHA.114.009683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Meng TC, Fukada T, Tonks NK. Reversible oxidation and inactivation of protein tyrosine phosphatases in vivo. Mol Cell. 2002;9:387–399. doi: 10.1016/s1097-2765(02)00445-8. [DOI] [PubMed] [Google Scholar]
- 43.Lee M, Choy WC, Abid MR. Direct sensing of endothelial oxidants by vascular endothelial growth factor receptor-2 and c-Src. PLoS One. 2011;6:e28454. doi: 10.1371/journal.pone.0028454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tiozzo R, Monti D, Straface E, Capri M, Croce MA, Rainaldi G, Franceschi C, Malorni W. Antiproliferative activity of 3-aminobenzamide in A431 carcinoma cells is associated with a target effect on cytoskeleton. Biochem Biophys Res Commun. 1996;225:826–832. doi: 10.1006/bbrc.1996.1258. [DOI] [PubMed] [Google Scholar]
- 45.Kim SY, Moon KA, Jo HY, Jeong S, Seon SH, Jung E, Cho YS, Chun E, Lee KY. Anti-inflammatory effects of apocynin, an inhibitor of NADPH oxidase, in airway inflammation. Immunol Cell Biol. 2012;90:441–448. doi: 10.1038/icb.2011.60. [DOI] [PubMed] [Google Scholar]
- 46.Bogeski I, Bozem M, Sternfeld L, Hofer HW, Schulz I. Inhibition of protein tyrosine phosphatase 1B by reactive oxygen species leads to maintenance of Ca2+ influx following store depletion in HEK 293 cells. Cell Calcium. 2006;40:1–10. doi: 10.1016/j.ceca.2006.03.003. [DOI] [PubMed] [Google Scholar]
- 47.Sternfeld L, Krause E, Schmid A, Anderie I, Latas A, Al-Shaldi H, Kohl A, Evers K, Hofer HW, Schulz I. Tyrosine phosphatase PTP1B interacts with TRPV6 in vivo and plays a role in TRPV6-mediated calcium influx in HEK293 cells. Cellular Signalling. 2005;17:951–960. doi: 10.1016/j.cellsig.2004.11.012. [DOI] [PubMed] [Google Scholar]
- 48.Craige SM, Chen K, Pei Y, Li C, Huang X, Chen C, Shibata R, Sato K, Walsh K, Keaney JF., Jr NADPH oxidase 4 promotes endothelial angiogenesis through endothelial nitric oxide synthase activation. Circulation. 2011;124:731–740. doi: 10.1161/CIRCULATIONAHA.111.030775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Scholz D, Ziegelhoeffer T, Helisch A, Wagner S, Friedrich C, Podzuweit T, Schaper W. Contribution of arteriogenesis and angiogenesis to postocclusive hindlimb perfusion in mice. J Mol Cell Cardiol. 2002;34:775–787. doi: 10.1006/jmcc.2002.2013. [DOI] [PubMed] [Google Scholar]
- 50.Di A, Gao XP, Qian F, Kawamura T, Han J, Hecquet C, Ye RD, Vogel SM, Malik AB. The redox-sensitive cation channel TRPM2 modulates phagocyte ROS production and inflammation. Nat Immunol. 2012;13:29–34. doi: 10.1038/ni.2171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hecquet CM, Zhang M, Mittal M, Vogel SM, Di A, Gao X, Bonini MG, Malik AB. Cooperative Interaction of trp Melastatin Channel TRPM2 with its Splice Variant TRPM2-S is Essential for Endothelial Cell Apoptosis. Circ Res. 2014;114:469–479. doi: 10.1161/CIRCRESAHA.114.302414. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.