

Abstract
Over the past decades evidence has accumulated clearly demonstrating a pivotal role for the sympathetic nervous system (SNS) and its neurotransmitters in regulating inflammation. The first part of this review provides the reader with an overview showing that the interaction of the SNS with the immune system to control inflammation is strongly context-dependent (for example, depending on the activation state of the immune cell or neuro-transmitter concentration). In the second part we focus on autoimmune arthritis as a well investigated example for sympathetically controlled inflammation to show that the SNS and catecholamines play a differential role depending on the time point of ongoing disease. A model will be developed to explain the proinflammatory effects of the SNS in the early phase and the anti-inflammatory effects of catecholamines in the later phase of autoimmune arthritis. In the final part, a conceptual framework is discussed that shows that a major purpose of increased SNS activity is nourishment of a continuously activated immune system at a systemic level using energy-rich fuels (glucose, amino acids, lipids), while uncoupling from central nervous regulation occurs at sites of inflammation by repulsion of sympathetic fibers and local adrenoceptor regulation. This creates zones of ‘permitted local inflammation’. However, if this ‘inflammatory configuration’ persists and is strong, as in autoimmunity, the effects are detrimental because of the resultant chronic catabolic state, leading to cachexia, high blood pressure, insulin resistance, and increased cardiovascular mortality, and so on. Today, the challenge is to translate this conceptual knowledge into clinical benefit.
Introduction
The sympathetic nervous system (SNS) is an integrative system that reacts to dangerous situations, and activation of the SNS is part of the classical ‘fight and flight’ response. This is common knowledge. However, the SNS is not active just in these extreme situations, but is part of constant regulatory machinery that keeps body functions in a steady-state equilibrium. Of course, the SNS is not alone in performing these tasks but is interwoven into complex regulatory circuits. Therefore, it is not possible to analyze the action of the SNS in inflammation without considering the other important players, like the hypothalamic-pituitary-adrenal (HPA) axis, and the sensory nervous system and vagal nervous system (VNS). For a detailed description of the functional anatomy of the autonomous (SNS and VNS) and sensory nervous system, as well as the HPA axis, we refer the reader to respective standard textbooks of physiology since this is established and common knowledge and a detailed description would go beyond the scope of this review. In the first part of this review we focus on important highlights concerning the SNS and inflammation. In the second part, the standalone facts will be integrated to try to understand the deeper meaning of this regulatory machinery in inflammatory disease. As an example, we refer to findings concerning neuroendocrine immune regulation in arthritis.
Review criteria
This review is based on a systematic search of the PubMed database using the search terms ‘sympathetic nervous system’, ‘peripheral nervous system’, ‘nerve fiber’, ‘neuroimmun*’, ‘norepinephrine’, ‘arthritis’, ‘collagen induced arthritis’, ‘rheumatoid arthritis’, ‘autoimmune diseases’, ‘autoimmunity’. Articles (including abstracts) published in English or German up to March 2014 were considered. All retrieved articles were screened for eligibility based on title, abstract, and full content.
The sympathetic nervous system and inflammation
It was noted some time ago that the SNS and inflammation are close partners. One of the first mentions of the influence of the SNS on inflammation can be found in an article from 1903. The authors performed surgical local sympathectomy of the ear of rabbits after provoking inflammation by inoculation with staphylococci. They concluded that ‘….relations of the sympathetic nerve … to the course of inflammation, … are due to some nervous functions of the sympathetic nerve other than… vasoconstriction and vasodilatation’ [1]. Already in 1936, Reilly speculated that endotoxin concentrates in sympathetic tissue and irritates sympathetic nerve fibers, which results in a systemic reaction that resembles symptoms of typhoid fever [2]. This view, of course, was very rudimentary but this theory already implied that there is some crosstalk between the SNS and inflammation, and that both systems interact with each other.
Today our understanding of this relationship is more detailed. When an antigen enters the body, local activation of immune cells leads to release of proinflammatory mediators, which are able to excite or lower thresholds of afferent nociceptive and afferent vagal nerve fibers [3]. If the neuronal signal strength is strong enough or if spillover of local inflammatory mediators into the circulation is robust enough, it signals to the brain, resulting in activation of the two major stress axes, the HPA axis and the SNS [3,4]. Cytokines like interleukin (IL)-1β [3,5] or tumor necrosis factor (TNF) [6] produced by locally activated innate immune cells are pivotal in this communication from immune system to central nervous system.
Vice versa, central sympathetic activity has a direct impact on inflammatory cytokines. In a study with hypertensive patients, central inhibition of the SNS decreased peripheral TNF serum levels [7]. In another study, sympathetic tone was positively correlated with IL-6 plasma levels [8]. Similarly, stress responses that modulate SNS activity have great impact on inflammation [9]. However, there might be a disruption of this communication between the brain and the immune system in the course of protracted inflammation, as shown in an arthritis model in rats [10]. This disruption is beneficial on a systemic level, which is discussed below.
In the mid-1980s, it was recognized that secondary lymphoid tissue is highly innervated by sympathetic nerve fibers and sympathetic nerve terminals are found in close proximity to immune cells, especially in primary and secondary lymphoid tissue [11]. Immune cells express receptors for neurotransmitters, for example, adrenoceptors (ARs), which are functional and translate neuronal signals into immune cell signals [12]. The communication between brain and inflamed area can be disturbed, for example, by a stroke, which results in asymmetric inflammation. This can lead to reduced inflammation on the paralyzed side in rheumatoid arthritis, which was already recognized in 1962 [13].
In this respect, it has been shown that patients with minor stroke [14] or poliomyelitis [15] show weaker delayed type hypersensitivity (DTH) responses on the paretic side. After excluding changes in blood flow, the authors of the latter study concluded that ‘… another mechanism, such as a direct effect of sympathetic transmitters on inflammatory cells, may mediate the putative effects of the SNS on DTH responses.’
Another clinically well recognized phenomenon after stroke is immunosuppression. In a rat model of stroke, the authors observed reduced infection rates following sympathectomy, indicating SNS-mediated immunosuppression [16], which might depend on the type of infectious agent [17].
The activation of the SNS in the context of an active immune system results in release of sympathetic neurotransmitters. Notably, sympathetic nerves release not only norepinephrine (NE) as the major neurotransmitter, but also ATP, neuropeptide Y (NPY), and nitric oxide [18]. All neurotransmitters have direct influence on immune cells, although NE is the best characterized in this respect. NPY, for example, has been shown to increase adhesion of human leukocytes to endothelial cells [19], and the NPY antagonist PP56 showed anti-inflammatory effects in acute carrageenan-induced arthritis and chronic adjuvant arthritis [20].
Sympathetic influence on immune cells can be direct, via ARs on immune cells [4], or indirect via regulating blood or lymph flow [21], regulating distribution [22] and production [23] of lymphocytes, or modulating the release of proinflammatory peptides [24], like substance P from sensory nerve endings, which among others express α-ARs [25] (Figure 1). Inflammatory cell recruitment and redistribution is also controlled by the SNS (Figure 1). One study showed that regulation of circadian changes in leukocyte distribution involves, among others, the activity of the SNS via β-ARs expressed on non-hematopoietic cells, leading to tissue-specific, differential circadian oscillations in the expression of endothelial cell adhesion molecules and chemokines [22]. Another study pointed out the role of SNS-dependent monocyte recruitment from the spleen in experimental peritoneal infection [17,26]. In addition, the generation of some leukocytes in the bone marrow is influenced by the SNS via β-ARs, resulting in preferential production of proinflammatory leukocyte populations [23].
Figure 1.
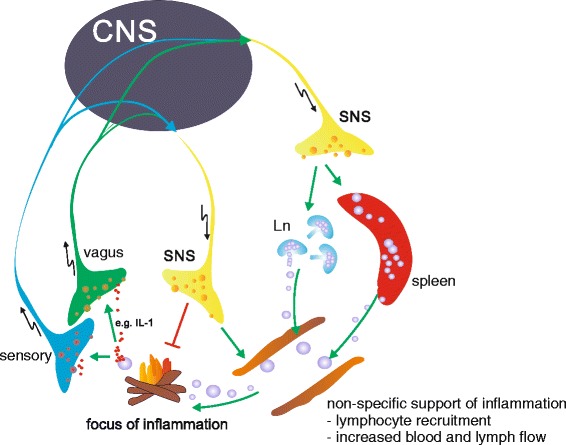
Basic neuronal anti-inflammatory reflex. Local inflammation (the fire) is detected by vagal and sensory nerve fibers, which express receptors for inflammatory mediators, like interleukin (IL)-1β (red dots). An afferent signal is generated and transmitted to the brain (central nervous sytem (CNS)), which in turn leads to activation of the sympathetic nervous system (SNS), which has a complex impact on inflammation. Local release of SNS neurotransmitters, like norepinephrine, at the site of inflammation or in secondary lymphoid organs has a net anti-inflammatory outcome. On the other hand, non-specific immune stimulatory processes on a systemic level are supported, like recruitment of leukocytes, increased blood and lymph flow, but also increasing antigen processing and presentation and provision of energy-rich fuels. Ln, lymph node.
As a side note, there is a direct interrelation between the SNS and the sensory nervous system, since the sensory response is significantly modulated by sympathetic signaling (for example, [27]). Such findings have also been discussed in the context of understanding clinical entities like the complex regional pain syndrome (for example, [28]).
TNF was the first cytokine whose production was shown to be regulated by occupation of α-ARs or β-ARs by catecholamines [29,30]. Subsequently, a whole array of other cytokines and immune cells has been demonstrated to be influenced by AR stimulation, both in vitro and in vivo (for example, [31]). Selected examples of the direct modulation of immune cell function by sympathetic neurotransmitters are presented in Table 1.
Table 1.
Examples of direct sympathetic neurotransmitter immune cell interactions
| Cell type | Source | Receptor/neurotransmitter concentration | Costimulus | Main effect | Reference |
|---|---|---|---|---|---|
| Macrophage | Bone marrow, mouse | NE 10−6 M | LPS | Decrease CCR2 | [32] |
| NE 10−8 M, 10−6 M | None | Decrease BMM proliferation | |||
| NE 10−8 M | LPS | Increase maturation | |||
| NE 10−8 M, 10−6 M | None | Increase phagocytosis | |||
| NE 10−8 M, 10−6 M | LPS | Increase TNF | |||
| Spleen, mouse | NE 10−8 M | LPS | Increase TNF | [29,33] | |
| Spleen, mouse | NE 10−6 M | LPS | Decrease TNF | [29,33] | |
| Peritoneum, mouse | Neuropeptide Y | Increase HMGB1 | [34] | ||
| Dendritic cell | Bone marrow derived, mouse | β2-AR | NOD2 agonist | Increase IL6 | [35] |
| TLR2 agonist | Decrease IL-12 | ||||
| β2-AR | Increase IL-33 | [36] | |||
| α2-AR | Increase in antigen uptake | [37] | |||
| Human cord blood | NE 10−6 M | LPS | Decrease IL12p40, TNF, IL-6, IL-23 | [38] | |
| T cell | Spleen, mouse | β2-AR | None | Increase Treg apoptosis | [39] |
| β2-AR | Anti-CD3 | Decrease IL-2 in CD4 + CD62L+ cells | [40] | ||
| β2-AR | None | Increase in Treg mediated cell suppression | |||
| CIA, mouse | NE | ConA | Increase IFN-γ | [41] | |
| Splenic naïve T cells | β2-AR | Anti-CD3 | Decrease IL-2 | [42] | |
| Anti-CD28 | |||||
| β2-AR | Anti-CD3 | Increase IFN-γ per Th1 cell | [43] | ||
| Anti-CD28 | |||||
| IL-12 | |||||
| T cell clone | Neuropeptide Y | None | Decrease IFN-γ | [44] | |
| Increase IL-4 | |||||
| B cell | CIA, mouse | β2-AR | Anti-CD40/IL-4 | Increase IL-10 | [45] |
| β2-AR | Anti-CD40/IL-4 | Inhibits IL-7 receptor signaling | [46] | ||
| Naïve, splenic B cells | β2-AR | CD40L/IL-4 | Increase IgG1, IgE | [47,48] |
AR, adrenoceptor; BMM, bone marrow-derived macrophages; CCR2, C-C chemokine receptor type 2; CIA, collagen-induced arthritis; ConA, Concanavalin A; HMGB1, high-mobility-group-protein B1; IFN, interferon; Ig, immunoglobulin; IL, interleukin; LPS, lipopolysaccharide; NE, norepinephrine; NO2, nucleotide-binding oligomerization domain-containing protein 2; Th1, T helper 1; TLR2, toll-like receptor 2; TNF, tumor necrosis factor; Treg, regulatory T cell.
Also, pathogens use the sympathetic machinery to their advantage. For example, the cytomegalovirus immediate/early promotor can be stimulated directly via β2-ARs of monocytes, leading to reactivation of the virus [49]. NE release from sympathetic nerves in the gut is inhibited by infection with Trichinella spiralis to dampen the immune response against the pathogen [50].
The net effect of stimulating ARs on immune cells is not straightforward because it strongly depends on the context of exposure of receptive cells to sympathetic neurotransmitters; for example, the activation state of the cell [45,51], the proximity of the cell to the source of neurotransmitters (since this determines neurotransmitter concentration at the receptor; Figure 2), the presence of factors that modulate the adrenergic response [52], the pattern of AR expression on immune cells [53], or simply age [54].
Figure 2.
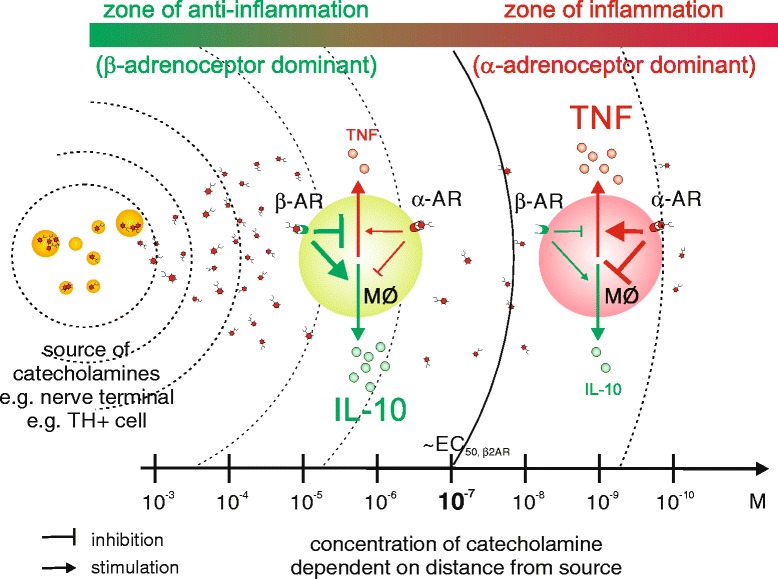
Catecholamine effects depend on the distance from catecholamine source. α- and β-adrenoceptors (ARs) show different binding affinities for catecholamines. Norepinephrine, the main neurotransmitter in the sympathetic nervous system (SNS), binds with higher affinity to α-ARs than β-ARs. Simultaneous expression of these receptors on immune cells (for example, macrophages (MΦ)) provides these cells with a passive means to determine the distance to the next catecholamine source. In close proximity to the catecholamine source (for example, sympathetic nerve terminal or catecholamine-producing tyrosine hydroxylase (TH)-positive cell) the concentration is high enough to activate β-ARs, whereas at a greater distance only α-ARs are activated. In the case of innate immune cells, like macrophages, this directly translates into anti-inflammatory (for example, increases in interleukin (IL)-10 via β-AR) or proinflammatory activity (for example, increases in tumor necrosis factor (TNF) via α-AR). Therefore, the simultaneous expression of α-ARs and β-ARs on immune cells provides a mean to regulate inflammatory processes dependent on the distance to the catecholamine source. We hypothesize that the body uses this system to promote local inflammation by repulsion of sympathetic nerve fibers from inflamed areas (zone of inflammation) and, at the same time, locally confines the inflammatory process by suppression of bystander activation in the zone of anti-inflammation.
Increasing the complexity of this matter, the VNS also has profound effects on inflammatory responses. The activity of the VNS is increased following endotoxemia. In this respect, an ‘anti-inflammatory reflex’ has been postulated, with the efferent vagus nerve acting in an anti-inflammatory manner via release of acetylcholine and activation of α7-nicotinic acetylcholine (nACh) receptors expressed on immune cells [55]. Since the spleen has no parasympathetic innervation, it has been hypothesized that the efferent part of the vagus activates splenic SNS fibers that release NE from SNS nerve endings in close proximity to immune cells. Upon stimulation of ARs on a subset of CD4 T cells, these cells release acetylcholine, which in turn has an immunosuppressive effect via α7-nACh receptors on macrophages [55]. However, this view has been challenged recently, since it has been shown by retrograde and anterograde staining and electrophysiological experiments that there is no neural connection from VNS to SNS projecting to the spleen [56]. This challenges the view that the vagus is indeed the effector arm of the ‘anti-inflammatory reflex’ [57]. Furthermore, it has been shown that the efferent arm of the ‘anti-inflammatory reflex’ to lipopolysaccharide challenge is primarily the splanchnic sympathetic nerve acting on immune cells in the spleen [58] (Figure 1).
Thus, there is no simple statement like ‘norepinephrine is anti- or pro-inflammatory’. It is better to say ‘norepinephrine modulates immune function in a context-dependent manner’. It gets even more complex when the release of co-transmitters, which is dependent on the firing rate of sympathetic nerve fibers [59], and neuroanatomical facts are taken into account, because all known co-transmitters like NPY, ATP, and nitric oxide are potent immune modulators and, thus, effects superimpose on each other. To answer the question about the role of the SNS in inflammation, research at the single cell level is important to understand basic regulatory mechanisms. However, the complexity of the interrelation between different factors is challenging. In addition, it has to be respected that the SNS also interacts with non-immune cells to modulate release of inflammatory mediators. For example, endothelial cells can be stimulated to increase release of IL-6 via NE and ATP from SNS nerve terminals [60].
Another approach to understand the role of the SNS in inflammation is to investigate the overall effect of SNS activity on clinical outcomes. Well-known clinical phenomena, like the reactivation or first occurrence of chronic inflammatory disorders like colitis or asthma during or after episodes of psychological stress, have been directly linked to the activation of the autonomic nervous system [61,62]. Influence of the SNS on inflammation at a systemic level has been demonstrated for several disease models and entities like sepsis [17], colitis [63], allergic asthma [47,61], chronic eye inflammation [64], arthritis [51,65], endometriosis [66], T helper type 1-mediated skin diseases [67], influenza A [68], Chagas disease [69], and chronic regional pain syndrome [70].
Evidence has also accumulated to show that chronic activation of the SNS by changing function of immune cells contributes to hypertrophy and fibrosis of the heart [71]. Similarly, in a mouse model of primary biliary cirrhosis, blockade of sympathetic activity improved fibrosis [72]. It has been shown in a restraint stress paradigm influenza model that the sympathetic component of the stress response, possibly due to limiting otherwise detrimental specific effector cell activation, together with glucocorticoids are responsible for better survival after experimental infection [73].
There is also evidence that different forms of cancer might be influenced by the SNS, including from experimental animal data, epidemiological studies that show the use of beta-blockers is beneficial for breast cancer and melanoma, and studies showing that psychological stress might play a role in the pathogenesis of some cancers [74]. Taken together, these studies show that the SNS plays an important role in several immune-mediated or immune-related diseases.
Clinical models demonstrate that influencing the sympathetic response impacts on the outcome. In a model of acute septic inflammation, the adrenergic system has a profound influence on cell proliferation, apoptosis, and circulating immune cell subpopulations [75]. In a model of polymicrobial sepsis by cecal ligation and puncture, mechanisms through α-ARs increase mortality. In the same system, it has been described that tyrosine hydroxylase (TH) is markedly increased in sympathetic fibers of the small intestine-associated SNS, resulting in enhanced NE release [76]. Therefore, not only is response of immune cells to SNS stimuli highly context-dependent, but the nervous system itself also underlies plasticity depending on the inflammatory context.
From our point of view, arthritis is the best investigated disease entity concerning the influence of the SNS on the inflammatory process. Therefore, the next section focuses on this chronic disease to introduce current concepts of SNS influence on inflammation.
The sympathetic nervous system and arthritis
Sympathectomy in patients with rheumatoid arthritis was reported as early as 1927 (mentioned in [77]), followed by several reports showing that pain as well as joint swelling improved upon sympathectomy (for example, [77]). In a double blind study in 1986, however, overall pain decreased but no changes were recorded with respect to morning stiffness or joint tenderness [78]. This is in contrast to reports in animal models that sympathectomy leads to less severe disease - for example, in carrageenan-induced arthritis [79] or adjuvant arthritis in rats [80]. In the latter model, spontaneous hypertensive rats, which show higher activity of the SNS, developed more severe arthritis [81]. It seems that this proinflammatory effect of the SNS on early adjuvant arthritis is caused by an increase in T helper type 1 lymphocyte (Th1) and Th17 responses [82].
A proinflammatory activity of the SNS was also shown in the collagen type II model of arthritis [51]. In this model, proinflammatory CD4 + CD25 + FOXP3- cells induced this effect [83] (Figure 3). These results in human and animal studies seem to be contradictory. However, these divergent results can be explained by the importance of the time point of sympathetic intervention. This was clearly shown in the collagen type II model of arthritis in DBA/1 mice, where early sympathectomy leads to less severe disease, but late sympathectomy in the chronic phase of the disease clearly has the opposite effect, resulting in increased disease activity [51]. How can one explain this dichotomy?
Figure 3.
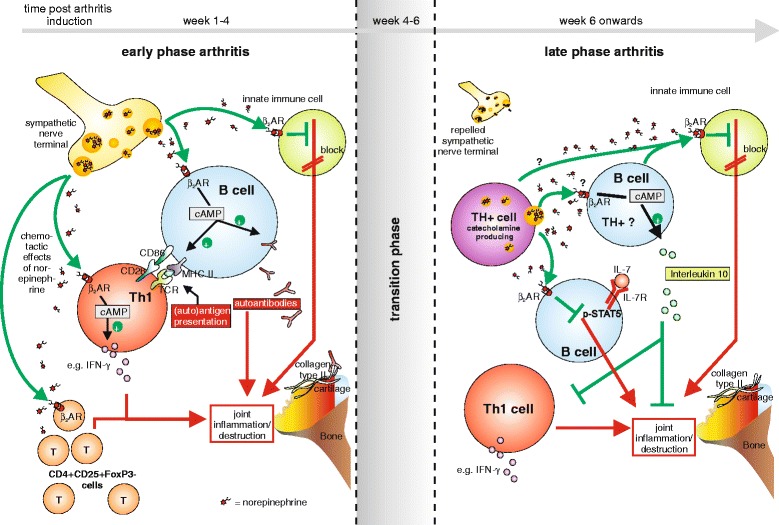
Current model of sympathetic nervous system influence in arthritis. In early arthritis (left panel), the sympathetic nervous system (SNS) supports inflammation in the joint through a proinflammatory influence on adaptive immune cells; for example, increased specific antibody production by B cells and increased proinflammatory activity of T cells. The SNS also inhibits innate immune cells via stimulation of β2 adrenoceptors (β2ARs), although the net outcome of SNS influence in the early phase is proinflammatory. Then, during the transition phase, we hypothesize that the influence of the SNS changes from pro- to anti-inflammatory. In the later stages, central regulation of the inflammatory process is less important, since sympathetic nerve fibers are repelled from the inflamed area and secondary lymphoid organs. However, local sympathetic influence becomes increasingly important, indicated by the appearance of catecholamine-producing, tyrosine hydroxylase-positive (TH+) cells, which have a dominant anti-inflammatory effect. Possible mechanisms of action are paracrine and autocrine in manner; for example, inhibiting proinflammatory interleukin (IL)-7 receptor-positive B cells, increasing the activity of IL-10-producing anti-inflammatory B cells, or inhibiting innate immune cells via β2AR-mediated effects. AR, adrenoceptor; cAMP, cyclic adenosine monophosphate; CD, cluster of differentiation; FoxP3, forkhead box P3; IFN, interferon; MHC, major histocompatibility complex; pSTAT5, phosphorylated-signal transducer and activator of transcription 5; TCR, T-cell receptor; Th1, T helper 1 cell.
It has long been known that innervation, which is usually dense in synovial tissue, is lost during experimental inflammation and in chronic inflammatory conditions [84]. However, more recent studies showed that the loss of innervation is a specific process and affects mainly sympathetic nerves fibers, whereas sensory nerves remain in the inflamed region [85], an observation reproducible in many inflammatory conditions of humans and rodents. Recent research demonstrates an active process possibly involving specific nerve repellent factors [86].
As a compensatory mechanism for this deprivation of sympathetic neurotransmitters in the joint, cells that are capable of producing neurotransmitters accumulate [87]. These TH-positive catecholamine-producing cells modulate inflammation dependent on the model used. In a model of lung injury, α2-dependent proinflammatory effects of catecholamine-producing phagocytes were postulated [88]. On the other hand, in multiple sclerosis [89] and human and experimental arthritis [87,90,91], catecholamine-producing cells have anti-inflammatory potential. These TH-positive cells are sensitive to sympathectomy with 6-hydroxydopamine (a neurotoxin) or anti-dopamine beta hydroxylase antibodies [90]. Since TH-positive cells dominate the later phase of collagen type II-induced arthritis in the joint (they are also present in synovial inflammation in chronic rheumatoid arthritis), it is not surprising that depletion of these cells by sympathectomy leads to aggravation of arthritis in the late phase [51]. At the moment, however, the mechanism of anti-inflammatory action has not been fully established in arthritis. Possibly, cAMP content in TH-positive cells is increased by autocrine mechanisms. In this respect, it has been shown for regulatory T cells (Tregs) that cAMP can be used as a direct immunosuppressive agent by transferring cAMP molecules from Tregs via gap junctions into target cells [92]. Due to high concentrations of neurotransmitters in the vicinity of TH-positive cells, however, stimulation of β2-ARs on innate immune cells might be the dominant immunosuppressive mechanism (Table 1, Figures 3 and 4).
Figure 4.
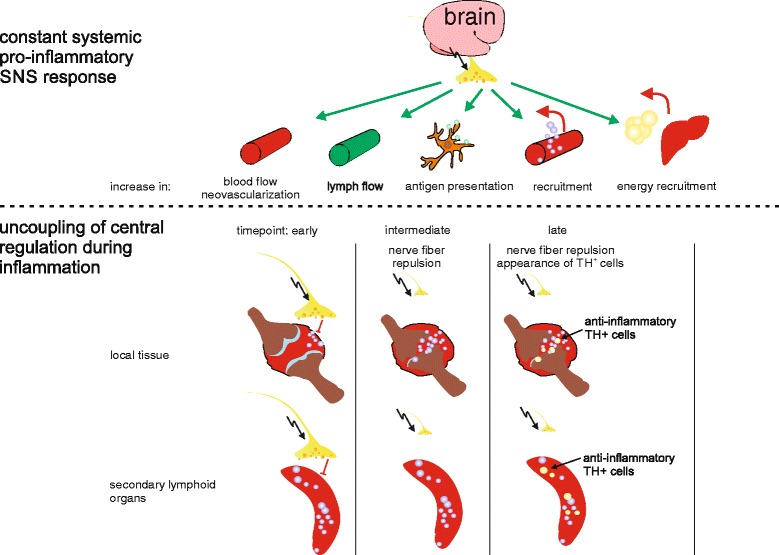
Morphologic adaptation to persistent inflammation. Centrally controlled increase of sympathetic nervous system (SNS) activity is a basic response to inflammation. The constant increase in SNS activity supports inflammation in several ways; for example, increasing blood flow, lymph flow, antigen presentation, and liberation of energy-rich fuels like lipids and glucose from adipose tissue and liver. However, the specific interaction with immune cells in secondary lymphoid organs and at local sites of inflammation (for example, joints) shows a net anti-inflammatory effect. Therefore, to mount an effective immune response, non-specific support of inflammation on a systemic level is maintained, while the anti-inflammatory influence on a local level is decreased and uncoupled from central regulation through repulsion of sympathetic nerve fibers and the appearance of tyrosine hydroxylase (TH) + catecholamine-producing cells during the inflammatory process. In the end, a systemic proinflammatory configuration is established, which helps to optimally clear the antigen. However, if inflammation persists, like during chronic inflammation, this constant increase in SNS activity and resultant catabolic state is detrimental to the body and results in known disease sequelae of chronic inflammatory conditions, like cachexia, diabetes, hyperlipidemia, high blood pressure, increased cardiovascular risk, and so on.
An influence on adaptive immune cells like B cells has also been shown. In the collagen-induced arthritis model, B cells expressing IL-7 receptor are proinflammatory [46]. However, stimulation of β2-AR on B cells results in loss of proinflammatory activity by inhibiting IL-7 receptor downstream signaling (Figure 3). Another possible explanation for the anti-inflammatory effects of TH-positive cells is increased anti-inflammatory function, which is augmented by catecholamines in an autocrine or paracrine manner via ARs. In collagen type II-induced arthritis, it has been shown that a subpopulation of B cells might play a role in this respect [45]. NE via β2-AR increased IL-10 production from B cells from arthritic animals (Figure 3), and these cells were anti-inflammatory when re-injected into arthritic animals [45]. One might speculate that these B cells, which can be TH-positive, are stimulated by catecholamines produced by TH-positive cells in the joint in an autocrine/paracrine manner (Figure 3).
The purpose of activating the sympathetic nervous system in inflammation - exemplified by synovial inflammation
So far, we introduced a new model of neuroimmune regulation specified in arthritis. All these elaborate mechanistic and structural adaptations during inflammation need to serve some purpose, however, otherwise they would not have been positively selected during evolution. In recent hypothetical modeling, a framework was developed that tries to explain the underlying meaning.
An activated immune system needs a significant amount of energy above that required for the normal non-inflamed state [93]. The activation of the SNS and the HPA axis at the beginning of inflammation helps to provide enough energy, because activation of these axes mobilizes energy-rich fuels mainly by increasing lipolysis, glycogenolysis, muscle protein breakdown, and gluconeogenesis (Figure 4). At the beginning of an inflammatory innate immune response, the SNS but also HPA axis support inflammation by non-specific means; for example, mobilization of leukocytes [22,26], increasing blood pressure and heart rate, increasing lymph flow [21], plasma extravasation [94], antigen uptake and presentation [37] (Figure 4). In this initial phase of inflammation, SNS activity also ‘programs’ adaptive immune cells via β2-AR - for example, B cells to produce increased amounts of antibodies and T cells to produce more or less cytokines dependent on the context of activation [95]. This mainly proinflammatory action takes place on a systemic level in secondary lymphoid organs like the spleen and lymph nodes, where immune cells are programmed and then released to attack the intruder.
At the local site of inflammation, however, SNS activity contributes primarily to anti-inflammatory mechanisms, mainly by direct influence of neurotransmitters on immune cells [4]. Besides the local promotion of regulatory B cells (see above), also macrophages stimulated via the β2-ARs acquire an anti-inflammatory M2 phenotype [96] and β2-AR stimulation also inhibits TNF production [97] (Figure 2). On the other hand, stimuli via α-ARs are proinflammatory: for example, α2-AR stimulation increases reactive oxygen species in macrophages [98]. Therefore, the net outcome of stimulating ARs on immune cells strongly depends on the receptor engaged and, therefore, on the receptor expression pattern (which might change during the course of inflammation [4,45]) and neurotransmitter concentration, because NE binds preferentially to α-ARs, only binding to β-ARs at high concentrations (for example, [99]). However, why do some immune cells, like macrophages, express both α-ARs and β-ARs, which will counteract each other in terms of immunoregulation? One possible explanation is that, due to the different binding affinities of NE to these AR subtypes, this system can be used as a distance detector to the source of catecholamines.
In this respect, repulsion of sympathetic nerve fibers from inflamed tissue makes sense, since it is not favorable to inhibit the immune response (high concentrations of catecholamines preferentially stimulate anti-inflammatory β-ARs) before the antigen is cleared (Figure 4). Therefore, this distance detector system (simultaneous expression of α-AR and β-AR on immune cells) provides a means for the body to define sites of permitted inflammation (low SNS fiber density, low catecholamine concentration) and, on the other hand, prevent uncontrolled spreading of inflammation by preventing bystander activation (high SNS fiber density, high catecholamine concentration) (Figure 2).
To get an impression of the contribution of SNS to local anti-inflammatory mechanisms, the eye is a good example. The eye is known as an exceptional immune-privileged site, dominated by anti-inflammatory mechanisms. It has been shown that sympathetic denervation of the eye leads to a decrease in anti-inflammatory molecules, like tumor growth factor-β, which results in a complete loss of the immune-privileged status [100]. Therefore, repulsion of SNS fibers from inflamed tissue is an effective means to increase local inflammation (Figures 2 and 4). This has been positively selected during evolution to clear invading microbes but not to serve chronic autoimmune inflammation.
We hypothesize that catecholamine-producing cells start to play a role in the later inflammatory phase, possibly as a compensatory mechanism for the local loss of SNS fibers. These TH-positive cells can be anti-inflammatory as described above. One might argue that it is easier to just shut down SNS activity at the systemic level than to repel nerve fibers from local inflamed tissue, but SNS activity stays high during many chronic inflammatory conditions (for example, [101]). Concerning the energetic aspect discussed above, this is beneficial in terms of providing enough energy to feed the activated immune system on a systemic level. In contrast to the SNS activity, which is still high in chronic inflammation, HPA axis activity is relatively reduced, not down to normal, but to a level without immunosuppression, to not disturb the local immune response (Figures 3 and 4).
Overall, the system takes on an ‘inflammation configuration’, including repulsion of sympathetic nerve fibers from local inflamed tissue to create an area of permitted inflammation, high SNS activity on a systemic level, and reduced HPA activity without local immunosuppression, but provision of energy-rich fuels is still maintained and important (Figure 4).
These processes are positively selected during evolution to serve short-term acute inflammation [93,102]. If these processes persist for too long, they cause harm because the body is in a constant state of catabolism and volume overload. Known disease sequelae in chronic inflammatory conditions can be explained by this constant activation of the SNS and HPA axis and the resultant catabolic state, like cachexia, high blood pressure, insulin resistance, and so on [93,102].
Potential clinical and therapeutic implications for chronic inflammatory processes
From the current conceptual and experimental knowledge, certain hypotheses can be derived about potential clinical and therapeutic approaches that might improve clinical practice. Clinical data applying the current knowledge specifically on sympathetic regulation of inflammation is scarce. However, one promising approach that underscores the importance of sympathetic downstream signaling in anti-inflammation is the inhibition of phosphodiesterase (PDE)4, an enzyme that degrades cAMP. Increasing cAMP by inhibiting this enzyme shows promising results in psoriatic arthritis, which led to the approval of the PDE inhibitor apremilast for this disease entity [103]. PDE inhibitors are also currently being tested for several other clinical entities; for example, psoriasis, rheumatoid arthritis, and Behcet’s syndrome [103]. Taking into consideration that a general increase in cAMP might also support detrimental effects as discussed above, it is noteworthy that PDE4 is the predominant PDE isoform expressed in immune cells [104]. However, whether increasing cAMP by pharmacologic PDE inhibition will support disease sequelae is not clear at the moment and further research is needed. Right now, neuroimmunology in the sense presented in this review is on the verge of clinical translation. In terms of sympathetic control of inflammatory arthritis a possible approach is to follow the success seen in animal models and put effort into developing novel cellular therapies; for example, after induction of TH in certain immune cells or treatment of B cells with sympathetic stimuli to increase their regulatory potential. On the other hand, the systemic permanent overactivation of the SNS as discussed above could also be a potential target for intervention; for example, by psychological or pharmacological means. However, clinical data are missing at the moment and further research is warranted. For this research an approach to support local activation of sympathetic mechanisms, like increasing cAMP in immune cells (for example, PDE4 inhibition) but on the other hand decreasing systemic SNS activation to prevent disease sequelae, needs to be the focus.
Conclusion
Inflammation causes increased activity of the SNS with release of NE and co-transmitters in lymphoid organs and inflamed local sites. Immune cells carry receptors (for example, ARs) to detect and process signals from the SNS. The reaction of the immune cell to neurotransmitters is variable depending on the context of receptor engagement (activation state of the cell, expression pattern of neurotransmitter receptors, microenvironment, cytokine milieu, and distance from the catecholamine source (concentration)).
On a systemic level, the signals from the SNS are proinflammatory in the initial phase of inflammation, whereas anti-inflammatory effects are dominant in the late or chronic phases of an inflammatory response, at least in collagen-induced arthritis. Upon initiating an inflammatory process, the body adopts an ‘inflammatory configuration’ with increased systemic SNS and HPA axis activity. This reaction can be interpreted as an ‘energy appeal reaction’ resulting in the provision of enough energy-rich fuels, like glucose and free fatty acids, to fulfill the needs of an activated immune system.
If inflammation becomes chronic, as in chronic inflammatory illness, the system changes into a ‘chronic inflammatory condition’ that is characterized by 1) still increased systemic activity of the SNS, 2) still increased activity of the HPA axis but without immunosuppression (glucocorticoid receptor desensitization and inadequacy), and 3) local repulsion of SNS fibers from inflamed tissue, including lymphoid organs, to create zones of permitted inflammation. The immune response is more or less uncoupled from central regulation to avoid the anti-inflammatory influence of the brain. All mechanisms ensure an optimal fight against an antigen.
These adaptations are evolutionarily positively selected to clear the antigen, usually an intruding microbe. However, if a ‘chronic inflammatory configuration’ persists, as in autoimmunity, the effects are detrimental because of the persistently increased SNS activity, HPA activity, and the resultant chronic catabolic state. This leads to known comorbidities in chronic inflammatory disease, like cachexia, high blood pressure, insulin resistance, and increased cardiovascular mortality. The challenge is now to translate this conceptual knowledge into clinical benefit.
Abbreviations
- AR
Adrenoceptor
- DTH
Delayed type hypersensitivity
- HPA
Hypothalamic-pituitary-adrenal
- IL
Interleukin
- nACh
Nicotinic acetylcholine
- NE
Norepinephrine
- NPY
Neuropeptide Y
- PDE
Phosphodiesterase
- SNS
Sympathetic nervous system
- TH
Tyrosine hydroxylase
- TNF
Tumor necrosis factor
- Treg
Regulatory T cell
- VNS
Vagal nervous system
Footnotes
Competing interests
The authors declare that they have no competing interests.
Contributor Information
Georg Pongratz, Email: georg.pongratz@klinik.uni-regensburg.de.
Rainer H Straub, Email: rainer.straub@ukr.de.
References
- 1.Meltzer SJ, Meltzer C. On a difference in the influence upon inflammation between the section of the sympathetic nerve and the removal of the sympathetic ganglion. J Med Res. 1903;10:135–141. [PMC free article] [PubMed] [Google Scholar]
- 2.Hopkin DA, Laplane R. James Reilly and the autonomic nervous system. A prophet unheeded? Ann R Coll Surg Engl. 1978;60:108–116. [PMC free article] [PubMed] [Google Scholar]
- 3.Besedovsky H, del Rey A, Sorkin E, Dinarello CA. Immunoregulatory feedback between interleukin-1 and glucocorticoid hormones. Science. 1986;233:652–654. doi: 10.1126/science.3014662. [DOI] [PubMed] [Google Scholar]
- 4.Nance DM, Sanders VM. Autonomic innervation and regulation of the immune system (1987–2007) Brain Behav Immun. 2007;21:736–745. doi: 10.1016/j.bbi.2007.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Goehler LE, Relton JK, Dripps D, Kiechle R, Tartaglia N, Maier SF, Watkins LR. Vagal paraganglia bind biotinylated interleukin-1 receptor antagonist: a possible mechanism for immune-to-brain communication. Brain Res Bull. 1997;43:357–364. doi: 10.1016/S0361-9230(97)00020-8. [DOI] [PubMed] [Google Scholar]
- 6.Zielinski MR, Dunbrasky DL, Taishi P, Souza G, Krueger JM. Vagotomy attenuates brain cytokines and sleep induced by peripherally administered tumor necrosis factor-alpha and lipopolysaccharide in mice. Sleep. 2013;36:227–1238. doi: 10.5665/sleep.2892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pöyhönen-Alho MK, Manhem K, Katzman P, Kibarskis A, Antikainen RL, Erkkola RU, Tuomilehto JO, Ebeling PE, Kaaja RJ. Central sympatholytic therapy has anti-inflammatory properties in hypertensive postmenopausal women. J Hypertens. 2008;26:2445–2449. doi: 10.1097/HJH.0b013e328311cf37. [DOI] [PubMed] [Google Scholar]
- 8.Bernstein IM, Damron D, Schonberg AL, Shapiro R. The relationship of plasma volume, sympathetic tone, and proinflammatory cytokines in young healthy nonpregnant women. Reprod Sci. 2009;16:980–985. doi: 10.1177/1933719109338876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Straub RH, Kalden JR. Stress of different types increases the proinflammatory load in rheumatoid arthritis. Arthritis Res Ther. 2009;11:114. doi: 10.1186/ar2712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.del Rey A, Wolff C, Wildmann J, Randolf A, Hahnel A, Besedovsky HO, Straub RH. Disrupted brain-immune system-joint communication during experimental arthritis. Arthritis Rheum. 2008;58:3090–3099. doi: 10.1002/art.23869. [DOI] [PubMed] [Google Scholar]
- 11.Felten DL, Felten SY, Bellinger DL, Carlson SL, Ackerman KD, Madden KS, Olschowki JA, Livnat S. Noradrenergic sympathetic neural interactions with the immune system: structure and function. Immunol Rev. 1987;100:225–260. doi: 10.1111/j.1600-065X.1987.tb00534.x. [DOI] [PubMed] [Google Scholar]
- 12.Carr DJJ, Blalock JE. Neuropeptide hormones and receptors common to the immune and neuroendocrine systems: bidirectional pathway of intersystem communication. In: Ader R, Felten DL, Cohen N, editors. Psychoneuroimmunology. 2. San Diego: Academic Press, Inc; 1991. pp. 573–588. [Google Scholar]
- 13.Thompson M, Bywaters EG. Unilateral rheumatoid arthritis following hemiplegia. Ann Rheum Dis. 1962;21:370–377. doi: 10.1136/ard.21.4.370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tarkowski E, Naver H, Wallin BG, Blomstrand C, Tarkowski A. Lateralization of T-lymphocyte responses in patients with stroke. Effect of sympathetic dysfunction? Stroke. 1995;26:57–62. doi: 10.1161/01.STR.26.1.57. [DOI] [PubMed] [Google Scholar]
- 15.Tarkowski E, Naver H, Wallin BG, Blomstrand C, Grimby G, Tarkowski A. Lateralization of cutaneous inflammatory responses in patients with unilateral paresis after poliomyelitis. J Neuroimmunol. 1996;67:1–6. doi: 10.1016/0165-5728(96)00011-2. [DOI] [PubMed] [Google Scholar]
- 16.Prass K, Meisel C, Höflich C, Braun J, Halle E, Wolf T, Ruscher K, Victorov IV, Priller J, Dirnagl U, Volk HD, Meisel A. Stroke-induced immunodeficiency promotes spontaneous bacterial infections and is mediated by sympathetic activation reversal by poststroke T helper cell type 1-like immunostimulation. J Exp Med. 2003;198:725–736. doi: 10.1084/jem.20021098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Straub RH, Pongratz G, Weidler C, Linde HJ, Kirschning CJ, Glück T, Schölmerich J, Falk W. Ablation of the sympathetic nervous system decreases gram-negative and increases gram-positive bacterial dissemination: key roles for tumor necrosis factor/phagocytes and interleukin-4/lymphocytes. J Infect Dis. 2005;192:560–572. doi: 10.1086/432134. [DOI] [PubMed] [Google Scholar]
- 18.Burnstock G. Cotransmission in the autonomic nervous system. Handb Clin Neurol. 2013;117:23–35. doi: 10.1016/B978-0-444-53491-0.00003-1. [DOI] [PubMed] [Google Scholar]
- 19.Sung CP, Arleth AJ, Feuerstein GZ. Neuropeptide Y upregulates the adhesiveness of human endothelial cells for leukocytes. Circ Res. 1991;68:314–318. doi: 10.1161/01.RES.68.1.314. [DOI] [PubMed] [Google Scholar]
- 20.Claxson A, Morris C, Blake D, Sirén M, Halliwell B, Gustafsson T, Löfkvist B, Bergelin I. The anti-inflammatory effects of D-myo-inositol-1.2.6-trisphosphate (PP56) on animal models of inflammation. Agents Actions. 1990;29:68–70. doi: 10.1007/BF01964724. [DOI] [PubMed] [Google Scholar]
- 21.Howarth D, Burstal R, Hayes C, Lan L, Lantry G. Autonomic regulation of lymphatic flow in the lower extremity demonstrated on lymphoscintigraphy in patients with reflex sympathetic dystrophy. Clin Nucl Med. 1999;24:383–387. doi: 10.1097/00003072-199906000-00001. [DOI] [PubMed] [Google Scholar]
- 22.Scheiermann C, Kunisaki Y, Lucas D, Chow A, Jang JE, Zhang D, Hashimoto D, Merad M, Frenette PS. Adrenergic nerves govern circadian leukocyte recruitment to tissues. Immunity. 2012;37:290–301. doi: 10.1016/j.immuni.2012.05.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Powell ND, Sloan EK, Bailey MT, Arevalo JM, Miller GE, Chen E, Kobor MS, Reader BF, Sheridan JF, Cole SW. Social stress up-regulates inflammatory gene expression in the leukocyte transcriptome via beta-adrenergic induction of myelopoiesis. Proc Natl Acad Sci U S A. 2013;110:16574–16579. doi: 10.1073/pnas.1310655110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Merhi M, Helme RD, Khalil Z. Age-related changes in sympathetic modulation of sensory nerve activity in rat skin. Inflamm Res. 1998;47:239–244. doi: 10.1007/s000110050324. [DOI] [PubMed] [Google Scholar]
- 25.Dawson LF, Phillips JK, Finch PM, Inglis JJ, Drummond PD. Expression of alpha1-adrenoceptors on peripheral nociceptive neurons. Neuroscience. 2011;175:300–314. doi: 10.1016/j.neuroscience.2010.11.064. [DOI] [PubMed] [Google Scholar]
- 26.Seeley EJ, Barry SS, Narala S, Matthay MA, Wolters PJ. Noradrenergic neurons regulate monocyte trafficking and mortality during gram-negative peritonitis in mice. J Immunol. 2013;190:4717–4724. doi: 10.4049/jimmunol.1300027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Drummond PD. The effect of sympathetic activity on thermal hyperalgesia in capsaicin-treated skin during body cooling and warming. Eur J Pain. 2001;5:59–67. doi: 10.1053/eujp.2001.0224. [DOI] [PubMed] [Google Scholar]
- 28.Birklein F, Kunzel W, Sieweke N. Despite clinical similarities there are significant differences between acute limb trauma and complex regional pain syndrome I (CRPS I) Pain. 2001;93:165–171. doi: 10.1016/S0304-3959(01)00309-8. [DOI] [PubMed] [Google Scholar]
- 29.Spengler RN, Allen RM, Remick DG, Strieter RM, Kunkel SL. Stimulation of alpha-adrenergic receptor augments the production of macrophage-derived tumor necrosis factor. J Immunol. 1990;145:1430–1434. [PubMed] [Google Scholar]
- 30.Severn A, Rapson NT, Hunter CA, Liew FY. Regulation of tumor necrosis factor production by adrenaline and beta-adrenergic agonists. J Immunol. 1992;148:3441–3445. [PubMed] [Google Scholar]
- 31.Szabó C, Haskó G, Zingarelli B, Németh ZH, Salzman AL, Kvetan V, Pastores SM, Vizi ES. Isoproterenol regulates tumour necrosis factor, interleukin-10, interleukin-6 and nitric oxide production and protects against the development of vascular hyporeactivity in endotoxaemia. Immunology. 1997;90:95–100. doi: 10.1046/j.1365-2567.1997.00137.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Xiu F, Stanojcic M, Jeschke MG. Norepinephrine inhibits macrophage migration by decreasing CCR2 expression. PLoS One. 2013;8:e69167. doi: 10.1371/journal.pone.0069167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Szelenyi J, Kiss JP, Vizi ES. Differential involvement of sympathetic nervous system and immune system in the modulation of TNF-alpha production by alpha2- and beta-adrenoceptors in mice. J Neuroimmunol. 2000;103:34–40. doi: 10.1016/S0165-5728(99)00234-9. [DOI] [PubMed] [Google Scholar]
- 34.Zhou JR, Zhang LD, Wei HF, Wang X, Ni HL, Yang F, Zhang T, Jiang CL. Neuropeptide Y induces secretion of high-mobility group box 1 protein in mouse macrophage via PKC/ERK dependent pathway. J Neuroimmunol. 2013;260:55–59. doi: 10.1016/j.jneuroim.2013.04.005. [DOI] [PubMed] [Google Scholar]
- 35.Manni M, Granstein RD, Maestroni G. beta2-Adrenergic agonists bias TLR-2 and NOD2 activated dendritic cells towards inducing an IL-17 immune response. Cytokine. 2011;55:380–386. doi: 10.1016/j.cyto.2011.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yanagawa Y, Matsumoto M, Togashi H. Adrenoceptor-mediated enhancement of interleukin-33 production by dendritic cells. Brain Behav Immun. 2011;25:1427–1433. doi: 10.1016/j.bbi.2011.04.012. [DOI] [PubMed] [Google Scholar]
- 37.Yanagawa Y, Matsumoto M, Togashi H. Enhanced dendritic cell antigen uptake via alpha2 adrenoceptor-mediated PI3K activation following brief exposure to noradrenaline. J Immunol. 2010;185:5762–5768. doi: 10.4049/jimmunol.1001899. [DOI] [PubMed] [Google Scholar]
- 38.Goyarts E, Matsui M, Mammone T, Bender AM, Wagner JA, Maes D, Granstein RD. Norepinephrine modulates human dendritic cell activation by altering cytokine release. Exp Dermatol. 2008;17:188–196. doi: 10.1111/j.1600-0625.2007.00677.x. [DOI] [PubMed] [Google Scholar]
- 39.Wirth T, Westendorf AM, Bloemker D, Wildmann J, Engler H, Mollerus S, Wadwa M, Schäfer MK, Schedlowski M, del Rey A. The sympathetic nervous system modulates CD4Foxp3 regulatory T cells via noradrenaline-dependent apoptosis in a murine model of lymphoproliferative disease. Brain Behav Immun. 2014;38:100–110. doi: 10.1016/j.bbi.2014.01.007. [DOI] [PubMed] [Google Scholar]
- 40.Guereschi MG, Araujo LP, Maricato JT, Takenaka MC, Nascimento VM, Vivanco BC, Reis VO, Keller AC, Brum PC, Basso AS. Beta2-adrenergic receptor signaling in CD4+ Foxp3+ regulatory T cells enhances their suppressive function in a PKA-dependent manner. Eur J Immunol. 2013;43:1001–1012. doi: 10.1002/eji.201243005. [DOI] [PubMed] [Google Scholar]
- 41.Straub RH, Rauch L, Fassold A, Lowin T, Pongratz G. Neuronally released sympathetic neurotransmitters stimulate splenic interferon-gamma secretion from T cells in early type II collagen-induced arthritis. Arthritis Rheum. 2008;58:3450–3460. doi: 10.1002/art.24030. [DOI] [PubMed] [Google Scholar]
- 42.Ramer-Quinn DS, Swanson MA, Lee WT, Sanders VM. Cytokine production by naive and primary effector CD4+ T cells exposed to norepinephrine. Brain Behav Immun. 2000;14:239–255. doi: 10.1006/brbi.2000.0603. [DOI] [PubMed] [Google Scholar]
- 43.Swanson MA, Lee WT, Sanders VM. IFN-gamma production by Th1 cells generated from naive CD4+ T cells exposed to norepinephrine. J Immunol. 2001;166:232–240. doi: 10.4049/jimmunol.166.1.232. [DOI] [PubMed] [Google Scholar]
- 44.Kawamura N, Tamura H, Obana S, Wenner M, Ishikawa T, Nakata A, Yamamoto H. Differential effects of neuropeptides on cytokine production by mouse helper T cell subsets. Neuroimmunomodulation. 1998;5:9–15. doi: 10.1159/000026321. [DOI] [PubMed] [Google Scholar]
- 45.Pongratz G, Melzer M, Straub RH. The sympathetic nervous system stimulates anti-inflammatory B cells in collagen-type II-induced arthritis. Ann Rheum Dis. 2012;71:432–439. doi: 10.1136/ard.2011.153056. [DOI] [PubMed] [Google Scholar]
- 46.Pongratz G, Anthofer JM, Melzer M, Anders S, Grassel S, Straub RH. IL-7 receptor alpha expressing B cells act proinflammatory in collagen-induced arthritis and are inhibited by sympathetic neurotransmitters. Ann Rheum Dis. 2014;73:306–312. doi: 10.1136/annrheumdis-2012-202944. [DOI] [PubMed] [Google Scholar]
- 47.Pongratz G, McAlees JW, Conrad DH, Erbe RS, Haas KM, Sanders VM. The level of IgE produced by a B cell is regulated by norepinephrine in a p38. J Immunol. 2006;177:2926–2938. doi: 10.4049/jimmunol.177.5.2926. [DOI] [PubMed] [Google Scholar]
- 48.Podojil JR, Sanders VM. Selective regulation of mature IgG1 transcription by CD86 and beta 2-adrenergic receptor stimulation. J Immunol. 2003;170:5143–5151. doi: 10.4049/jimmunol.170.10.5143. [DOI] [PubMed] [Google Scholar]
- 49.Prösch S, Wendt CE, Reinke P, Priemer C, Oppert M, Krüger DH, Volk HD, Döcke WD. A novel link between stress and human cytomegalovirus (HCMV) infection: sympathetic hyperactivity stimulates HCMV activation. Virology. 2000;272:357–365. doi: 10.1006/viro.2000.0367. [DOI] [PubMed] [Google Scholar]
- 50.Swain MG, Blennerhassett PA, Collins SM. Impaired sympathetic nerve function in the inflamed rat intestine. Gastroenterology. 1991;100:675–682. doi: 10.1016/0016-5085(91)80011-w. [DOI] [PubMed] [Google Scholar]
- 51.Harle P, Mobius D, Carr DJ, Scholmerich J, Straub RH. An opposing time-dependent immune-modulating effect of the sympathetic nervous system conferred by altering the cytokine profile in the local lymph nodes and spleen of mice with type II collagen-induced arthritis. Arthritis Rheum. 2005;52:1305–1313. doi: 10.1002/art.20987. [DOI] [PubMed] [Google Scholar]
- 52.Li W, Knowlton D, Woodward WR, Habecker BA. Regulation of noradrenergic function by inflammatory cytokines and depolarization. J Neurochem. 2003;86:774–783. doi: 10.1046/j.1471-4159.2003.01890.x. [DOI] [PubMed] [Google Scholar]
- 53.Lorton D, Lubahn C, Bellinger DL. Potential use of drugs that target neural-immune pathways in the treatment of rheumatoid arthritis and other autoimmune diseases. Curr Drug Targets Inflamm Allergy. 2003;2:1–30. doi: 10.2174/1568010033344499. [DOI] [PubMed] [Google Scholar]
- 54.Donoso V, Gomez CR, Orriantia MA, Pérez V, Torres C, Coddou C, Nelson P, Maisey K, Morales B, Fernandez R, Imarai M, Huidobro-Toro JP, Sierra F, Acuña-Castillo C. The release of sympathetic neurotransmitters is impaired in aged rats after an inflammatory stimulus: a possible link between cytokine production and sympathetic transmission. Mech Ageing Dev. 2008;129:728–734. doi: 10.1016/j.mad.2008.09.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Olofsson PS, Rosas-Ballina M, Levine YA, Tracey KJ. Rethinking inflammation: neural circuits in the regulation of immunity. Immunol Rev. 2012;248:188–204. doi: 10.1111/j.1600-065X.2012.01138.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bratton BO, Martelli D, McKinley MJ, Trevaks D, Anderson CR, McAllen RM. Neural regulation of inflammation: no neural connection from the vagus to splenic sympathetic neurons. Exp Physiol. 2012;97:1180–1185. doi: 10.1113/expphysiol.2011.061531. [DOI] [PubMed] [Google Scholar]
- 57.Martelli D, McKinley MJ, McAllen RM. The cholinergic anti-inflammatory pathway: A critical review. Auton Neurosci. 2013;182:65–69. doi: 10.1016/j.autneu.2013.12.007. [DOI] [PubMed] [Google Scholar]
- 58.Martelli D, Yao ST, McKinley MJ, McAllen RM. Reflex control of inflammation by sympathetic nerves, not the vagus. J Physiol. 2014;592:1677–1686. doi: 10.1113/jphysiol.2013.268573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Todorov LD, Mihaylova-Todorova ST, Bjur RA, Westfall DP. Differential cotransmission in sympathetic nerves: role of frequency of stimulation and prejunctional autoreceptors. J Pharmacol Exp Ther. 1999;290:241–246. [PubMed] [Google Scholar]
- 60.Stohl LL, Zang JB, Ding W, Manni M, Zhou XK, Granstein RD. Norepinephrine and adenosine-5’-triphosphate synergize in inducing IL-6 production by human dermal microvascular endothelial cells. Cytokine. 2013;64:605–612. doi: 10.1016/j.cyto.2013.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Chen E, Miller GE. Stress and inflammation in exacerbations of asthma. Brain Behav Immun. 2007;21:993–999. doi: 10.1016/j.bbi.2007.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Saunders PR, Miceli P, Vallance BA, Wang L, Pinto S, Tougas G, Kamath M, Jacobson K. Noradrenergic and cholinergic neural pathways mediate stress-induced reactivation of colitis in the rat. Auton Neurosci. 2006;124:56–68. doi: 10.1016/j.autneu.2005.12.002. [DOI] [PubMed] [Google Scholar]
- 63.Straub RH, Grum F, Strauch U, Capellino S, Bataille F, Bleich A, Falk W, Schölmerich J, Obermeier F. Anti-inflammatory role of sympathetic nerves in chronic intestinal inflammation. Gut. 2008;57:911–921. doi: 10.1136/gut.2007.125401. [DOI] [PubMed] [Google Scholar]
- 64.Steinle JJ. Sympathetic neurotransmission modulates expression of inflammatory markers in the rat retina. Exp Eye Res. 2007;84:118–125. doi: 10.1016/j.exer.2006.09.006. [DOI] [PubMed] [Google Scholar]
- 65.Lorton D, Lubahn C, Klein N, Schaller J, Bellinger DL. Dual role for noradrenergic innervation of lymphoid tissue and arthritic joints in adjuvant-induced arthritis. Brain Behav Immun. 1999;13:315–334. doi: 10.1006/brbi.1999.0564. [DOI] [PubMed] [Google Scholar]
- 66.Arnold J, de Arellano ML B, Rüster C, Vercellino GF, Chiantera V, Schneider A, Mechsner S. Imbalance between sympathetic and sensory innervation in peritoneal endometriosis. Brain Behav Immun. 2012;26:132–141. doi: 10.1016/j.bbi.2011.08.004. [DOI] [PubMed] [Google Scholar]
- 67.Manni M, Maestroni GJ. Sympathetic nervous modulation of the skin innate and adaptive immune response to peptidoglycan but not lipopolysaccharide: involvement of beta-adrenoceptors and relevance in inflammatory diseases. Brain Behav Immun. 2008;22:80–88. doi: 10.1016/j.bbi.2007.06.016. [DOI] [PubMed] [Google Scholar]
- 68.Grebe KM, Takeda K, Hickman HD, Bailey AL, Embry AC, Bennink JR, Yewdell JW. Cutting edge: Sympathetic nervous system increases proinflammatory cytokines and exacerbates influenza A virus pathogenesis. J Immunol. 2010;184:540–544. doi: 10.4049/jimmunol.0903395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Machado MP, Rocha AM, de Oliveira LF, de Cuba MB, de Oliveira LI, Castellano LR, Silva MV, Machado JR, Nascentes GA, Paiva LH, Savino W, Junior VR, Brum PC, Prado VF, Prado MA, Silva EL, Montano N, Ramirez LE, VJ D d S. Autonomic nervous system modulation affects the inflammatory immune response in mice with acute Chagas disease. Exp Physiol. 2012;97:1186–1202. doi: 10.1113/expphysiol.2012.066431. [DOI] [PubMed] [Google Scholar]
- 70.Schlereth T, Drummond PD, Birklein F. Inflammation in CRPS: role of the sympathetic supply. Auton Neurosci. 2013;182:102–107. doi: 10.1016/j.autneu.2013.12.011. [DOI] [PubMed] [Google Scholar]
- 71.Levick SP, Murray DB, Janicki JS, Brower GL. Sympathetic nervous system modulation of inflammation and remodeling in the hypertensive heart. Hypertension. 2010;55:270–276. doi: 10.1161/HYPERTENSIONAHA.109.142042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Strack I, Schulte S, Varnholt H, Schievenbusch S, Töx U, Wendland K, Steffen HM, Drebber U, Dienes HP, Odenthal M. beta-Adrenoceptor blockade in sclerosing cholangitis of Mdr2 knockout mice: antifibrotic effects in a model of nonsinusoidal fibrosis. Lab Invest. 2011;91:252–261. doi: 10.1038/labinvest.2010.162. [DOI] [PubMed] [Google Scholar]
- 73.Hermann G, Beck FM, Tovar CA, Malarkey WB, Allen C, Sheridan JF. Stress-induced changes attributable to the sympathetic nervous system during experimental influenza viral infection in DBA/2 inbred mouse strain. J Neuroimmunol. 1994;53:173–180. doi: 10.1016/0165-5728(94)90027-2. [DOI] [PubMed] [Google Scholar]
- 74.Fitzgerald PJ. Beta blockers, norepinephrine, and cancer: an epidemiological viewpoint. Clin Epidemiol. 2012;4:151–156. doi: 10.2147/CLEP.S33695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Oberbeck R, Schmitz D, Wilsenack K, Schüler M, Pehle B, Schedlowski M, Exton MS. Adrenergic modulation of survival and cellular immune functions during polymicrobial sepsis. Neuroimmunomodulation. 2004;11:214–223. doi: 10.1159/000078439. [DOI] [PubMed] [Google Scholar]
- 76.Zhou M, Hank SH, Wang P. Increased gut-derived norepinephrine release in sepsis: up-regulation of intestinal tyrosine hydroxylase. Biochim Biophys Acta. 2004;1689:212–218. doi: 10.1016/j.bbadis.2004.03.008. [DOI] [PubMed] [Google Scholar]
- 77.Kidd BL, Cruwys S, Mapp PI, Blake DR. Role of the sympathetic nervous system in chronic joint pain and inflammation. Ann Rheum Dis. 1992;51:1188–1191. doi: 10.1136/ard.51.11.1188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Levine JD, Fye K, Heller P, Basbaum AI, Whiting-O’Keefe Q. Clinical response to regional intravenous guanethidine in patients with rheumatoid arthritis. J Rheumatol. 1986;13:1040–1043. [PubMed] [Google Scholar]
- 79.Aloe L, Tuveri MA, Levi-Montalcini R. Studies on carrageenan-induced arthritis in adult rats: presence of nerve growth factor and role of sympathetic innervation. Rheumatol Int. 1992;12:213–216. doi: 10.1007/BF00302155. [DOI] [PubMed] [Google Scholar]
- 80.Levine JD, Moskowitz MA, Basbaum AI. The contribution of neurogenic inflammation in experimental arthritis. J Immunol. 1985;135:843s–847s. [PubMed] [Google Scholar]
- 81.Levine JD, Dardick SJ, Roizen MF, Helms C, Basbaum AI. Contribution of sensory afferents and sympathetic efferents to joint injury in experimental arthritis. J Neurosci. 1986;6:3423–3429. doi: 10.1523/JNEUROSCI.06-12-03423.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Ebbinghaus M, Gajda M, Boettger MK, Schaible HG, Brauer R. The anti-inflammatory effects of sympathectomy in murine antigen-induced arthritis are associated with a reduction of Th1 and Th17 responses. Ann Rheum Dis. 2012;71:253–261. doi: 10.1136/ard.2011.150318. [DOI] [PubMed] [Google Scholar]
- 83.Harle P, Pongratz G, Albrecht J, Tarner IH, Straub RH. An early sympathetic nervous system influence exacerbates collagen-induced arthritis via CD4 + CD25+ cells. Arthritis Rheum. 2008;58:2347–2355. doi: 10.1002/art.23628. [DOI] [PubMed] [Google Scholar]
- 84.Mapp PI, Walsh DA, Garrett NE, Kidd BL, Cruwys SC, Polak JM, Blake DR. Effect of three animal models of inflammation on nerve fibres in the synovium. Ann Rheum Dis. 1994;53:240–246. doi: 10.1136/ard.53.4.240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Miller LE, Justen HP, Scholmerich J, Straub RH. The loss of sympathetic nerve fibers in the synovial tissue of patients with rheumatoid arthritis is accompanied by increased norepinephrine release from synovial macrophages. FASEB J. 2000;14:2097–2107. doi: 10.1096/fj.99-1082com. [DOI] [PubMed] [Google Scholar]
- 86.Fassold A, Falk W, Anders S, Hirsch T, Mirsky VM, Straub RH. Soluble neuropilin-2, a nerve repellent receptor, is increased in rheumatoid arthritis synovium and aggravates sympathetic fiber repulsion and arthritis. Arthritis Rheum. 2009;60:2892–2901. doi: 10.1002/art.24860. [DOI] [PubMed] [Google Scholar]
- 87.Capellino S, Weber K, Gelder M, Harle P, Straub RH. First appearance and location of catecholaminergic cells during experimental arthritis and elimination by chemical sympathectomy. Arthritis Rheum. 2012;64:1110–1118. doi: 10.1002/art.33431. [DOI] [PubMed] [Google Scholar]
- 88.Flierl MA, Rittirsch D, Nadeau BA, Chen AJ, Sarma JV, Zetoune FS, McGuire SR, List RP, Day DE, Hoesel LM, Gao H, Van Rooijen N, Huber-Lang MS, Neubig RR, Ward PA. Phagocyte-derived catecholamines enhance acute inflammatory injury. Nature. 2007;449:721–725. doi: 10.1038/nature06185. [DOI] [PubMed] [Google Scholar]
- 89.Cosentino M, Fietta AM, Ferrari M, Rasini E, Bombelli R, Carcano E, Saporiti F, Meloni F, Marino F, Lecchini S. Human CD4 + CD25+ regulatory T cells selectively express tyrosine hydroxylase and contain endogenous catecholamines subserving an autocrine/paracrine inhibitory functional loop. Blood. 2007;109:632–642. doi: 10.1182/blood-2006-01-028423. [DOI] [PubMed] [Google Scholar]
- 90.Capellino S, Cosentino M, Wolff C, Schmidt M, Grifka J, Straub RH. Catecholamine-producing cells in the synovial tissue during arthritis: modulation of sympathetic neurotransmitters as new therapeutic target. Ann Rheum Dis. 2010;69:1853–1860. doi: 10.1136/ard.2009.119701. [DOI] [PubMed] [Google Scholar]
- 91.Jenei-Lanzl Z, Capellino S, Kees F, Fleck M, Lowin T, Straub RH: Anti-inflammatory effects of cell-based therapy with tyrosine hydroxylase-positive catecholaminergic cells in experimental arthritis.Ann Rheum Dis 2013, doi:10.1136/annrheumdis-2013-203925. [DOI] [PubMed]
- 92.Bopp T, Becker C, Klein M, Klein-Hessling S, Palmetshofer A, Serfling E, Heib V, Becker M, Kubach J, Schmitt S, Stoll S, Schild H, Staege MS, Stassen M, Jonuleit H, Schmitt E. Cyclic adenosine monophosphate is a key component of regulatory T cell-mediated suppression. J Exp Med. 2007;204:1303–1310. doi: 10.1084/jem.20062129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Straub RH, Cutolo M, Buttgereit F, Pongratz G. Energy regulation and neuroendocrine-immune control in chronic inflammatory diseases. J Intern Med. 2010;267:543–560. doi: 10.1111/j.1365-2796.2010.02218.x. [DOI] [PubMed] [Google Scholar]
- 94.Lo EJ, Green PG, Miao FJ, Relchling DB, Levine JD. Bradykinin-induced neurogenic migration of neutrophils into the rat knee joint. Neuroreport. 1999;10:3821–3824. doi: 10.1097/00001756-199912160-00018. [DOI] [PubMed] [Google Scholar]
- 95.Sanders VM. The beta2-adrenergic receptor on T and B lymphocytes: do we understand it yet? Brain Behav Immun. 2012;26:195–200. doi: 10.1016/j.bbi.2011.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Grailer JJ, Haggadone MD, Sarma JV, Zetoune FS, Ward PA. Induction of M2 regulatory macrophages through the beta-adrenergic receptor with protection during endotoxemia and acute lung injury. J Innate Immun. 2014;6:607–618. doi: 10.1159/000358524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Stanojevic S, Dimitrijevic M, Kustrimovic N, Mitic K, Vujic V, Leposavic G. Adrenal hormone deprivation affects macrophage catecholamine metabolism and beta2-adrenoceptor density, but not propranolol stimulation of tumour necrosis factor-alpha production. Exp Physiol. 2013;98:665–678. doi: 10.1113/expphysiol.2012.070524. [DOI] [PubMed] [Google Scholar]
- 98.Deo SH, Jenkins NT, Padilla J, Parrish AR, Fadel PJ. Norepinephrine increases NADPH oxidase-derived superoxide in human peripheral blood mononuclear cells via alpha-adrenergic receptors. Am J Physiol Regul Integr Comp Physiol. 2013;305:R1124–R1132. doi: 10.1152/ajpregu.00347.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Buxton IL, Brunton LL. Alpha-adrenergic receptors on rat ventricular myocytes: characteristics and linkage to cAMP metabolism. Am J Physiol. 1986;251:H307–H313. doi: 10.1152/ajpheart.1986.251.2.H307. [DOI] [PubMed] [Google Scholar]
- 100.Vega JL, Keino H, Masli S. Surgical denervation of ocular sympathetic afferents decreases local transforming growth factor-beta and abolishes immune privilege. Am J Pathol. 2009;175:1218–1225. doi: 10.2353/ajpath.2009.090264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Dekkers JC, Geenen R, Godaert GL, Bijlsma JW, van Doornen LJ. Elevated sympathetic nervous system activity in patients with recently diagnosed rheumatoid arthritis with active disease. Clin Exp Rheumatol. 2004;22:63–70. [PubMed] [Google Scholar]
- 102.Straub RH. Evolutionary medicine and chronic inflammatory state - known and new concepts in pathophysiology. J Mol Med (Berl) 2012;90:523–534. doi: 10.1007/s00109-012-0861-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Poole RM, Ballantyne AD. Apremilast: first global approval. Drugs. 2014;74:825–837. doi: 10.1007/s40265-014-0218-4. [DOI] [PubMed] [Google Scholar]
- 104.Schafer P. Apremilast mechanism of action and application to psoriasis and psoriatic arthritis. Biochem Pharmacol. 2012;83:1583–1590. doi: 10.1016/j.bcp.2012.01.001. [DOI] [PubMed] [Google Scholar]