

Abstract
A fusion protein based on the S-layer protein SbpA from Bacillus sphaericus CCM 2177 and the enzyme laminarinase (LamA) from Pyrococcus furiosus was designed and overexpressed in Escherichia coli. Due to the construction principle, the S-layer fusion protein fully retained the self-assembly capability of the S-layer moiety, while the catalytic domain of LamA remained exposed at the outer surface of the formed protein lattice. The enzyme activity of the S-layer fusion protein monolayer obtained upon recrystallization on silicon wafers, glass slides and different types of polymer membranes was determined colorimetrically and related to the activity of sole LamA that has been immobilized with conventional techniques. LamA aligned within the S-layer fusion protein lattice in a periodic and orientated fashion catalyzed twice the glucose release from the laminarin polysaccharide substrate in comparison to the randomly immobilized enzyme. In combination with the good shelf-life and the high resistance towards temperature and diverse chemicals, these novel composites are regarded a promising approach for site-directed enzyme immobilization.
Keywords: S-layer, Enzyme immobilization, Self-assembly, Extremophilic enzyme, Monolayer, Nanobiotechnology
1. Introduction
Immobilization plays a critical role for the performance, activity and costs of industrially used enzymatic reaction systems. Immobilization prevents loss of enzyme in the process stream and allows re-use and separation of enzyme from the products of the catalytic reaction. On the other hand, transfer of an enzyme from the soluble into the insoluble state through immobilization affects its stability and activity (Chaplin and Bucke, 1990; Aslam and Dent, 1998). Thus, the choice of the right immobilization technique is essential for the application potential of an enzyme.
Currently used immobilization methods are based on the adsorption and covalent binding of enzymes to water-insoluble carriers, the incorporation of enzymes into semi-permeable gels, and the enclosing of enzymes in polymer membranes (Fig. 1a-c) (Angenendt, 2005; Chaplin and Bucke, 1990; Zhu and Snyder, 2003). Self-assembly systems open new possibilities for enzyme immobilization. As a novel approach, we have designed a chimeric protein by fusing an enzyme to a crystalline bacterial cell surface (S-layer) protein (Fig. 1d). The suitability of S-layer protein lattices as matrix for the covalent immobilization of enzymes has already been demonstrated in our laboratory (Neubauer et al., 1993, 1994, 1996). S-layers, in general, are monomolecular proteinaceous arrays that represent the outermost cell envelope component of many prokaryotic organisms (Sára and Sleytr, 2000; Sleytr et al., 1999, 2002, 2005, 2007a; Sleytr and Beveridge, 1999). They are composed of identical (glycol) protein species that are aligned into 2D crystalline arrays. Isolated S-layer subunits usually have the intrinsic capability to self-assemble in suspension, on the air–liquid interface, on lipid films or on solid supports (Sleytr et al., 1999, 2004, 2005, 2007b). By genetic engineering, discrete functions could be introduced into S-layer arrays (Breitwieser et al., 2002; Huber et al., 2006; Ilk et al., 2002, 2004; Moll et al., 2002; Pleschberger et al., 2003, 2004; Völlenkle et al., 2004). Based on the high density and regular display of the introduced functions, a broad spectrum of applications of S-layers fusion proteins is envisaged, particularly in the fields of biotechnology, molecular nanotechnology and biomimetics (Sára et al., 2006; Sleytr et al., 2002, 2004, 2007a). Regarding enzyme immobilization, the advantages offered by the S-layer self-assembly system are (i) the requirement of only a simple, one-step incubation process for site-directed immobilization without preceding surface activation of the support, (ii) the provision of a cushion to the enzyme through the S-layer moiety of the fusion protein preventing denaturation, and, consequently, loss of enzyme activity upon immobilization, (iii) the principal applicability of the “S-layer tag” to any enzyme, and (iv) the high flexibility for variation of enzymatic groups within a single S-layer array by co-crystallization of different enzyme/S-layer fusion proteins.
Fig. 1.

Comparison of different enzyme immobilization methods. (a) Random immobilization via covalent binding; (b) random adsorptive binding; (c) random physical adsorption within a 3D gel structure; (d) novel approach for site-directed immobilization of an enzyme via the S-layer self-assembly technique, allowing orientated and dense surface display of the enzyme in its native conformation and ensuring accessibility for the substrate; (e) legend.
In this study, the feasibility of fusing a hyperthermophilic enzyme to an S-layer protein was investigated. Extremophilic enzymes from microbial resources, in general, attract special interest because of their frequently unusual catalytic capabilities and process-related properties such as stability. For the set-up of a hyperthermophilic biocatalytic system, a 200-amino acid deletion mutant of the 1268 amino acid S-layer protein SbpA of Bacillus sphaericus CCM 2177 was used. This approach is based on previous structure–function studies revealing that this C-terminal deletion of SbpA results in significant increase of the spatial accessibility of the C-terminus, while fully retaining the protein’s self-assembly capability (Ilk et al., 2002) into a square S-layer lattice with a center-to-center spacing of the tetrameric morphological units of 13.1 nm (Fig. 4a, inset). An advantage of the SbpA system for nanobiotechnological applications is the dependence of its in vitro recrystallization on the presence of calcium ions (Györvary et al., 2003), allowing control over lattice formation by varying the calcium ion concentration. As extremophilic enzyme fusion partner for SbpA the laminarinase LamA (LamA, PF0076) derived from Pyrococcus furiosus was chosen. LamA is an endoglucanase displaying its main hydrolytic activity on the β-1,3-glucose polymer laminarin. It is extremely thermostable (half life time at 100 °C, 16 h) and thermoactive (temperature optimum, 100–105 °C), and, as a particular property, it is remarkably resistant to denaturation retaining a significant extent of its secondary structure in 8 M guanidinium hydrochloride (GHCl). These properties are paralleled by a notable stability at extremely low pH (~3) (Chiaraluce et al., 2002; Gueguen et al., 1997; Van Lieshout et al., 2004).
Fig. 4.

(a) Transmission electron micrograph of a negatively stained preparation of LamA/SbpA self-assembled in solution into a monomolecular array (bar, 100 nm). Inset showing the square lattice symmetry of the S-layer enzyme fusion protein with a lattice constant of 13.1 nm (enzyme moiety in blue); (b) fluorescent microscopic image of microspheres coated with LamA/SbpA after binding of an anti-vsv-g FITC conjugate (bar, 3 μm), indicating surface exposure of the enzymatic group; (c) AFM deflection image of the S-layer enzyme fusion protein after recrystallization on a silicon wafer, measured in contact mode in aqueous solution (bar, 100 nm); (d) electron micrograph of a porous membrane (bar, 1 μm) with a schematic representation of immobilized LamA/SbpA.
Due to the robustness of the chosen enzyme, the LamA/SbpA construct is well suited for the evaluation of the S-layer self-assembly system for enzyme immobilization. The studies performed with this S-layer fusion protein include (i) determination of enzyme activity when immobilized on different supports, with a focus on membraneous supports, (ii) analysis of the effects of inter- and intramolecular chemical cross-linking of LamA/S-layer subunits, and (iii) overall comparison of the S-layer based immobilization technique with currently used enzyme immobilization techniques.
2. Material and methods
Unless otherwise listed, all solvents and reagents were purchased from Sigma–Aldrich, St. Louis, MO.
2.1. Overexpression, isolation and purification of LamA/SbpA
To construct the S-layer fusion protein (Fig. 2), the PCR product encoding the 263-amino acid enzyme LamA (molecular mass, 33,123 Da) from P. furiosus possessing an 11-amino acid C-terminal vsv-g tag, spaced by a flexible linker (Ser-Ala-Ser-Ser-Gly-Gly-Gly-Gly-Ser-Ala) was cloned via a Gly-Gly linker into plasmid pET28a(+) (Novagen, Madison, WI), harboring the sequence encoding the S-layer protein SbpA31-1068 (molecular mass, 109,728 Da). The expression construct was provided by Guy de Roo (CatchMabs, Wageningen, NL). Protein overexpression following a standard protocol described in the pET System Manual (Novagen) was done by steady-state cultivation in a 4-l fermenter using E. coli BL21(DE3)star (Invitrogen, Vienna, Austria) as host. Protein expression was monitored by SDS-PAGE with Coomassie staining (Laemmli, 1970). Western blotting was performed to confirm the presence of the LamA portion of the fusion protein using monoclonal mouse anti-vsv-g-peroxidase conjugate (Völlenkle et al., 2004).
Fig. 2.
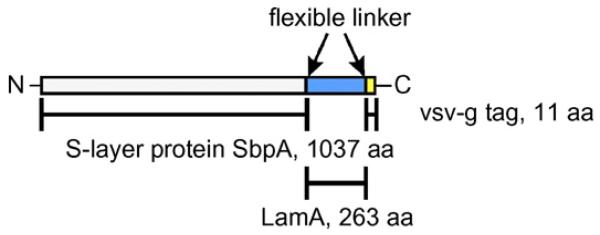
Schematic representation of the LamA/SbpA fusion protein.
For isolation of the LamA/SbpA protein, the B-PER® reagent (Pierce, Rockford, IL) was used following the protocol provided by the manufacturer. Extraction of S-layer fusion protein with 50 mM Tris–HCl/150 mM NaCl/5 M GHCl, pH 7.2 and purification by gel permeation chromatography (GPC) were performed as described previously (Pleschberger et al., 2003).
2.2. Investigation of the self-assembly and recrystallization property of LamA/SbpA
To investigate the capability of the LamA/SbpA protein to self-assemble in solution, 3 mg of purified protein were dissolved in 1 ml of 5 M GHCl in 0.5 mM Tris–HCl, pH 7.2, and the solution was dialyzed against 10 mM CaCl2 in MilliQ® water for 18 h at 4 °C. Negative staining of the suspension was performed as described previously (Pum et al., 1989). Recrystallization of LamA/SbpA monomers, obtained after dialyzation of the GHCl extract against distilled water for 3 h at 4 °C and centrifugation of the dialysate at 16,000 × g for 5 min at 4 °C (Avanti® J-25, Beckman Coulter, Fullerton, CA), on 300-mesh copper grids, coated with pioloform and carbon, was investigated by transmission electron microscopy (Pum et al., 1989). For recrystallization on silicon wafers with a native silicon oxide layer (p-type, 100 orientation; 7 mm × 7 mm; IMEC, Leuven, Belgium), wafers were washed with 70% ethanol and MilliQ® water, immersed with 100 μl of a 0.1 mg ml−1-LamA/SbpA protein solution in 0.5 mM Tris–HCl/10 mM CaCl2, pH 9.0, and incubated at 25 °C for 1–4 h. Atomic force microscopy (AFM) analysis was performed as described previously (Györvary et al., 2003).
To investigate the spatial accessibility of the LamA moiety within the fusion protein, amino modified cellulose microspheres (diameter, 3 μm; Frauenhofer Institute of Applied Polymer Research, Golm, D) and glass slides (Elka, micro slides cleaned, Glaswarenfabrik Karl Hecht KG, Sondheim, D) were coated with LamA/SbpA protein. Microspheres were activated with glycidylpropyltrimethoxysilane and pre-treated with ammonia, resulting in a positive surface charge with free amino groups. The particles were extensively washed with MilliQ® water prior to use. For coating, 300 μl of microspheres (corresponding to 170 μg) and a cleaned glass slides were incubated under rotation with 1 ml of a 0.1 mg ml−1-LamA/SbpA protein solution in 0.5 mM Tris–HCl/10 mM CaCl2, pH 9, at 25 °C for 4 h. Subsequently, coated microspheres and the glass slide were washed twice with MilliQ® water and immobilized LamA/SbpA was detected via fluorescence originating from anti-vsv-g FITC conjugate (1:250 in PBS/Triton; QED Bioscience, San Diego, CA) bound to the C-terminal vsv-g tag of the fusion protein. To avoid unspecific binding, coated microspheres and coated slides were blocked with 2% TopBlock (VWR International, Luzern, CH) in PBS containing 0.1% Triton X-100 at 4 °C over night. Twenty-five microliters of the suspension and 50 μl of anti-vsv-g FITC conjugate were mixed and incubated at 25 °C for 1 h followed by fluorescence measurement using a Nikon eclipse E400 fluorescence microscope (Nikon, Tokyo, JP).
2.3. Enzyme assay
LamA activity was quantified by the 3,5-dinitrosalicylic acid method via the amount of glucose released from the polymeric substrate laminarin (Summer and Somers, 1949). Briefly, the reaction was performed at 90 °C in 1 ml of 100 mM sodium phosphate buffer, pH 6.0, containing 1% (w v−1) of laminarin and 14 pM of Lam A, supplied as LamA/SbpA protein, either in sole or immobilized state, or as sole LamA (positive control). The specific enzyme activity was defined as the amount of Lam A required to release 1 μmol of glucose per min (U mg−1), taking into account the difference in molecular masses of LamA/SbpA and sole LamA. For comparison of the immobilization efficiency of LamA/SbpA on solid supports (silicon, glass, membranes) the enzyme activity was expressed in mM per cm2 of support, calculated from a total reaction time of 1 h. Protein concentrations in solutions and on solid supports were determined by the BCA (Bradford, 1976) and Lowry assay (Lowry et al., 1951), respectively.
2.4. Immobilization of LamA/SbpA and sole LamA on planar surfaces and membranes
Prior to immobilization, glass slides (Elka, micro slides cleaned, Glaswarenfabrik Karl Hecht KG, Sondheim, D) were treated with an alkaline detergent (2% solution, Hellmanex, Hellman, Müllheim/Baden, D) for 20 min at 25 °C followed by washing with MilliQ® water to remove dirt and fat traces. Silicon wafers were washed in 70% ethanol for 20 min at 25 °C followed by washing with MilliQ® water. For adsorptive immobilization on glass slides and silicon wafers, the supports were incubated with 500 μl of a 1 nmol ml−1-solution of Lam A in 1 M potassium phosphate buffer, pH 8.0, at 25 °C for 4 h. One molar potassium phosphate buffer, pH 8.0 is common for promoting noncovalent interactions between enzyme and carrier. By using this buffer system, it was simultaneously accounted for an adequate (optimization not shown) and for a standard immobilization protocol for enzymes. Recrystallization of LamA/SbpA on glass slides and silicon wafers was done according to a standard procedure (Györvary et al., 2003). For covalent binding of protein onto epoxy-activated glass surfaces (microarray slides, epoxysilane, Nunc, Rochester, NY), slides were washed with 1 M potassium phosphate buffer, pH 8.0, followed by incubation with 500 μl of a 1 nmol ml−1-protein solution in the same buffer.
For immobilization of LamA/SbpA on membranes, non-activated membranes for adhesive binding (polyethylenesulfonate, Whatman Dassel, D; cellulose actetate, polypropylene, stabilized reinforced cellulose, Sartorius, Goettingen, D) and an epoxy-activated membrane for covalent binding (Sartobind®Epoxy 75, with stabilized reinforced cellulose matrix; Sartorius) with a pore size of 0.45 μm were used. For reproducible coating, membranes (d = 2.5 cm) were mounted in a stirred ultrafiltration cell (Amicon, model 8010, Millipore, Billerica, MA) and washed with immobilization buffer (0.5 mM Tris–HCl/10 mM CaCl2, pH 9.0, for LamA/SbpA, and 1 M potassium phosphate buffer, pH 8.0, for sole LamA) at 25 °C. Subsequently, 2 ml of a 1.4 nmol ml−1-protein solution in immobilization buffer were applied onto the pre-wetted membranes followed by incubation for 4 h at 25 °C. Non-immobilized protein was removed by washing with MilliQ® water at 25 °C.
For stabilization of LamA/SbpA immobilized on glass slides and Sartobind®Epoxy 75 membrane, inter- and intra molecular cross-linking with the homobifunctional cross-linker dimethylpimelimidate (DMP; Pierce, Rockford, IL) was performed as described previously (Völlenkle et al., 2004).
2.5. Exposure of LamA/SbpA to different temperatures and chemicals
Glass slides and Sartobind®Epoxy 75 membrane with immobilized LamA/SbpA (cross-linked) and LamA, respectively, were incubated in MilliQ® water at 90 °C, and in 0.1 M NaOH, 6 M urea, and 20% ethanol, respectively, at 25 °C. In addition, LamA/SbpA covered glass slides (with and without epoxy activation) as well as slides with covalently bound LamA were frozen in an ethanol bath and lyophilized using a vacuum pump over night (0.1 mbar; Lyovac GT3, Leybold-Heraeus, Hürth, D). The composites were also autoclaved in MilliQ® water at 121 °C (1 bar) for 25 min. After exposure, samples were regenerated in MilliQ® water at 4 °C for at least 1 h prior to determination of LamA activity.
2.6. Investigation of the operational stability and protein leakage of LamA/SbpA
LamA activity of LamA/SbpA immobilized on glass slides was measured eight times within 20 days. After each cycle, the slides were washed with MilliQ® water and protein leakage was determined by measuring the protein content of the supernatant. For evaluation of the storage stability, glass slides with immobilized LamA/SbpA were stored at 25 °C in MilliQ® water containing 0.02% of sodium azide and the enzyme activity was monitored over a time span of 3 weeks.
3. Results and discussion
3.1. Overexpression, isolation and purification of LamA/SbpA
The designed S-layer fusion protein comprises truncated SbpA31-1068 S-layer protein of B. sphaericus CCM 2177 to which the enzyme LamA, carrying a C-terminal vsv-g tag via interspacing Ser-Ala-Ser-Ser-Gly-Gly-Gly-Gly-Ser-Ala, was fused via a Gly-Gly linker. The chimeric sequence was cloned into pET28a(+) and overexpressed in E. coli BL21(DE3)star. The additional band of a 2-h expression culture appearing on an SDS-PA gel upon Coomassie staining corresponded to the calculated molecular mass for LamA/SbpA of 141.9 kDa (Fig. 3a).
Fig. 3.

Overexpression and purification of the LamA/SbpA fusion protein. (a) SDS-PAGE analysis after Coomassie staining. Lane 1, Bench mark ladder (All blue, Biorad laboratories, Hercules, CA); lane 2, E. coli BL21star(DE3) expression culture harboring plasmid pET28a.sbpA.lamA before induction; lane 3, E. coli BL21star(DE3) expression culture harboring plasmid pET28a.sbpA.lamA after 2 h of induction; lane 4, purified LamA/SbpA fusion protein. (b) Western blot analysis of LamA/SbpA using anti-vsv-g-peroxidase conjugate.
For isolation of LamA/SbpA, the biomass harvested from a 4-l fermentation was treated with B-PER® reagent. As previously observed for the sole rSbpA protein, the fusion protein accumulated as inclusion bodies (Ilk et al., 2002). LamA/SbpA was extracted by treatment with 5 M GHCl (3 ml g−1 inclusion bodies) and finally enriched by GPC to high purity with an overall yield of 600 mg l−1. The integrity of the S-layer fusion protein was confirmed by binding of anti-vsv-g peroxidase conjugate to the C-terminal vsv-g tag in a Western blot experiment (Fig. 3b).
3.2. Investigation of the self-assembly and recrystallization properties of LamA/SbpA
As shown by transmission electron microscopy of negatively stained preparations, LamA/SbpA had the capability to self-assemble in suspension into monomolecular, regular arrays (Fig. 4a) and to recrystallize on EM grids as closed monolayers (not shown). Self-assembled and recrystallized LamA/SbpA exhibited the square lattice structure characteristic of native SbpA with a center-to-center spacing of tetrameric morphological units of 13.1 nm (Sára and Sleytr, 2000; Sleytr, 1970).
Due to the specific physicochemical properties of the N-terminal part of the SbpA moiety (Ilk et al., 2002), the S-layer fusion protein attached with its inner surface to the different supports that were included in the present study (compare with Table 1), thereby leaving the C-terminally fused LamA moiety exposed to the ambient environment. The defined orientation of LamA/SbpA was demonstrated via labeling with anti-vsv-g-FITC conjugate after recrystallization on glass slides and cellulose microspheres. Green luminescence of the planar slide surface (not shown) as well as of microspheres indicated exposure of the enzymatic moiety at the surface (Fig. 4b), while uncoated particles and glass slides showed no fluorescence (not shown).
Table 1.
Enzyme activity (mM/cm2) of S-layer fusion protein LamA/SbpA and sole enzyme LamA when immobilized on different solid supports
| LamAa 33,123c |
SbpA31-1068/LamAb 141,919c |
|
|---|---|---|
| Silicon wafer | 0.1 ± 0.0d | 6.1 ± 0.4 |
| Epoxy-activated glass slide | 3.4 ± 0.1 | 6.2 ± 0.4 |
| Glass slides | 0.4 ± 0.1 | 6.8 ± 0.1 |
| PES membrane | 0.1 ± 0.0 | 5.1 ± 0.2 |
| Cellulose acetate membrane | 2.7 ± 0.4 | 4.8 ± 0.2 |
| Sartobind®Epoxy 75 membrane | 13.1 ± 0.3 | 16.5 ± 0.5 |
| Cellulose membrane | 1.1 ± 0.6 | 11.6 ± 1.3 |
| Polypropylene membrane | 0.0 ± 0.0 | 3.2 ± 0.5 |
Sole enzyme LamA was covalently bound to epoxy-activated glass slides and to the Sartobind®Epoxy 75 membrane. On all other solid supports, it was attached adhesively.
In the absence of Ca2+ ions, monomeric LamA/SbpA was covalently bound in random orientation to epoxy-activated glass slides. On all other solid supports, crystallized arrays were formed due to the presence of calcium ions.
Molecular weight.
Measurements were performed in quadruplicate and standard deviations were calculated.
3.3. Determination of the enzyme activity of LamA/SbpA
The enzyme activity of the S-layer fusion protein was determined by its ability to release glucose from the polysaccharide laminarin and compared to the sole enzyme.
Water soluble LamA/SbpA protein as present in the absence of calcium ions revealed a specific activity of 344 U mg−1, while the value for sole LamA was 727 U mg−1. Thus, the S-layer fusion protein had approximately 50% of the specific enzyme activity of the sole enzyme. The difference in activity might be due to different diffusion rates between the substrate and sole LamA and the fused enzyme, respectively, or to partial denaturation of the SbpA moiety at the high temperature (i.e., 90 °C) applied during the enzyme assay.
3.4. Exploitation of the SbpA lattice for enzyme immobilization on planar supports
After the enzyme activity of the LamA/SbpA construct had been demonstrated in principle, the performance of the S-layer fusion protein upon immobilization on diverse supports, including silicon wafers and glass slides was investigated (Table 1).
Investigation of silicon wafers coated with LamA/SbpA was based on the assumption that LamA that is periodically aligned and concomitantly surface exposed within the S-layer matrix is capable of hydrolyzing the laminarin substrate. Lowry assays of LamA/SbpA protein lattices that have been detached from the silicon wafers revealed that 0.004 nmol of LamA/SbpA occupied a surface area of 1 cm2, which corresponded to the calculated amount of a tetrameric protein monolayer (four molecules LamA/SbpA per 171.61 nm2). Monolayer formation was also confirmed by AFM evidence (Fig. 4c). The specific enzyme activity determined for the LamA/SbpA lattices recrystallized as a monolayer on a silicon wafer was 774 U mg−1, which matched the specific enzyme activity of sole LamA. Obviously, alignment of the catalytic epitopes within the fusion protein lattice resulted in better accessibility for the substrate than when provided as soluble LamA/SbpA fusion protein. Generally, immobilization transfers enzymes in a randomly orientated, insoluble state with a limited reaction space and may lead to partial denaturation at the sites of adhesion to the support. It is conceivable that with S-layer fusion proteins, the S-layer moiety acts as a cushion preventing denaturation of the enzyme moiety upon immobilization.
To support the above assumption, LamA and LamA/SbpA were immobilized in a random fashion on epoxy-activated glass slides and the enzyme activity of the composites was determined. This experimental approach was possible, because SbpA lattice formation can be controlled by the calcium ion concentration of the medium. For direct comparison with the results obtained for sole LamA, which per se is randomly immobilized, calcium was omitted for random immobilization of LamA/SbpA (Györvary et al., 2003; Ilk et al., 2002). As expected, LamA/SbpA revealed higher enzyme activity than sole LamA, with values being approximately twice as high (for accurate data including standard deviations see Table 1). At the current state of knowledge, the similarity of LamA activities obtained for the S-layer fusion protein upon recrystallization and upon immobilization via epoxy groups may be explained by steric factors of substrate diffusion to the surface exposed catalytic epitopes, which, in either case, are obviously very similar.
The additional advantage of SbpA as a fusion partner for LamA through provision of a regular display matrix for the enzymatic function was demonstrated after formation of the LamA/SbpA lattice on non-activated glass slides. These composites revealed approximately twice the glucose release of LamA immobilized on an epoxy-activated glass surface. When using non-activated glass slides for adhesive immobilization of LamA, the S-layer fusion protein was even 17-fold more active than sole LamA (Table 1). This finding clearly demonstrates that supports with low binding capacity for an enzyme can be utilized when applying the S-layer fusion protein approach.
3.5. Immobilization of LamA/SbpA on membranes
In addition to planar supports, porous polymer membranes were investigated as immobilization matrix for the LamA/SbpA fusion protein (Fig. 4d). Enzyme immobilization on membranes constitutes an interesting area of applied research, because such microporous composites favor easy flow of substrates and products (Bora et al., 2006), and may be integrated in more complex processes, where combination of a catalytic function with a conventional filtration function is required (Hilal et al., 2006). Among the attractive features of enzyme-immobilized membrane reactors are easy control, straightforward scaling up, prolonged enzyme activity, high flow rates and reduced costs (Lopez and Matson, 1997). For comparative immobilization studies, LamA/SbpA and sole LamA were adhesively bound to several commercially available membranes (for specifications of the membranes included in this study see Section 2) and the glucose release per membrane unit area was determined. For all membranes tested, there was a significantly higher enzyme activity when the LamA/SbpA protein was immobilized as opposed to sole LamA immobilization. Best results were obtained for the polyethylensulfonate membrane with an approximately 50-fold higher enzyme activity. All data are summarized in Table 1. Overall differences of fusion protein series and LamA activity within the sole LamA series reflect different surface and structure properties of the matrix materials.
3.6. Effects of temperature and chemicals on the catalytic activity of immobilized LamA/SbpA in comparison to sole LamA
Previous studies have demonstrated that S-layer lattices can be stabilized with the homobifunctional cross-linker DMP (Völlenkle et al., 2004). Comparative studies on the chemical resistance and temperature resistance of cross-linked LamA/SbpA and sole LamA was investigated upon immobilization on glass slides. For these experiments, it was crucial to note that cross-linking of the S-layer fusion protein led only to a 10% reduction of enzyme activity compared to the native (non-cross-linked) LamA/SbpA monolayer.
When immobilized on glass slides in a cross-linked state, LamA/SbpA maintained a much higher enzyme activity upon exposure to 6 M urea and 0.1 M NaOH at 25 °C and to a temperature of 90 °C. Cross-linked LamA/SbpA withstood even treatment with 6 M urea for 8 h at 25 °C without loss of enzyme activity (Fig. 5a) and protein content per square unit. In contrast, LamA covalently bound to epoxy-activated glass slides showed a rapid reduction of glucose release with retaining only 15% of its original value under the same conditions. Investigation of the effect of 0.1 M NaOH, which is commonly used as sanitizing agent (Bengio et al., 1996), on the enzyme activity revealed that on glass slides neither native LamA/SbpA nor covalently bound LamA was stable at 25 °C. The loss of enzymatic activity of LamA/SbpA corresponded to the desorption of the S-layer moiety from the surface. However, cross-linking with DMP could retain half of the LamA activity of the S-layer fusion protein after approximately 30 min of exposure (Fig. 5b). The LamA/SbpA protein was also superior to sole LamA when the enzyme activity was determined upon exposure to 90 °C. The sole enzyme LamA exhibited a half life time of 3.8 h, whereas the S-layer fusion protein retained around half of the enzymatic activity after 40 h (Fig. 5c). The protective effect of the S-layer moiety prevented the loss of enzymatic activity, however it could not withstand the temperature exposure itself resulting in gradual denaturation and desorption from the surface. A protein leakage of 60% was determined after exposure to 90 °C for 40 h.
Fig. 5.

Comparative stability studies on LamA/SbpA recrystallized and cross-linked on glass slides and LamA covalently bound onto epoxy-activated glass slides. Enzyme activity upon exposure to (a) 6 M urea (■] LamA/SbpA, □ LamA), (b) 0.1 M NaOH (● LamA/SbpA, ○ LamA), and (c) 90 °C (▲ LamA/SbpA, △LamA). Enzyme activity is given as percentage of original activity of the respective composite.
In 20% ethanol, the “stability” of either enzyme system (sole LamA and S-layer fusion protein) was constant for 14 days, without decrease of enzyme activity. Freeze dried samples of LamA/SbpA immobilized on glass slides fully maintained the enzyme activity upon rewetting, whereas sole LamA covalently bound glass slides revealed a 26% loss of activity upon regeneration. Autoclaving of both LamA/SbpA and LamA coated glass slides completely abolished the enzyme activity.
3.7. Investigation of the operational stability and protein leakage of LamA/SbpA
The ability of the immobilized enzyme to retain activity after repeated exposure to the analyte is an important technological aspect of enzymes. The time course of enzyme activity of recrystallized and cross-linked LamA/SbpA protein compared to adhesively bound LamA after repeated use is given in Fig. 6. The S-layer fusion protein retained 31% of its original enzyme activity after eight cycles within 20 days, whereas the activity of sole LamA dropped to 10% of its original value, when assayed under identical conditions. With the leakage of the S-layer fusion protein during recycling studies being only 4%, the S-layer system fulfils the requirement for separation of the enzyme system from reaction mixtures.
Fig. 6.

Recycling of LamA immobilized as S-layer enzyme fusion protein (■) on silicon wafers in comparison to adhesively bound sole enzyme LamA (□). Enzyme activity is given as percentage of original activity of the respective composite.
To account for the storage stability, which is another important issue for the application potential of an enzyme, LamA/SbpA recrystallized on glass slides was stored in MilliQ® water at 25 °C with addition of a preservative and the enzyme activity was monitored over a period of 3 weeks. During that time, no decrease of enzyme activity of either LamA/SbpA or sole LamA was detectable.
4. Conclusions
Methods for organizing functional materials at the nanometer level are essential for the development of novel fabrication techniques (Sleytr et al., 2007a, 2007b). In particular, molecular self-assembly systems that exploit the molecular scale manufactory precision of biological systems are prime candidates in nanobiotechnology. In the present study, the enzyme/S-layer fusion protein LamA/SbpA31-1068 was constructed comprising the S-layer protein SbpA from B. sphaericus CCM 2177 and the enzyme laminarinase LamA from P. furiosus. By exploiting the intrinsic self-assembly property of the S-layer protein moiety, the chimeric protein was used for spatial control over display of enzyme activity on planar and porous supports. The results obtained in this study clearly demonstrate that S-layer based bottom up self-assembly systems for functionalizing solid supports with a catalytic function could have significant advantages over processes based on random immobilization of sole enzymes. A balanced comparison of advantages and disadvantages of the S-layer self-assembly system for enzyme immobilization is given in Table 2.
Table 2.
Advantages and disadvantages of the S-layer fusion protein based enzyme immobilization system
| Advantages |
| No preceding surface activation of the support needed |
| One-step incubation process |
| Stabilization of the enzymatic group within the S-layer lattice |
| S-layer cushion prevents denaturation of enzyme upon immobilization on solid supports |
| Higher enzymatic activity per square unit |
| Applicability to any functional group |
| No complex optimization of immobilization conditions |
| Disadvantages |
| Genetic engineering of the enzyme |
| Refolding step needed |
For future studies on process development using S-layer based enzyme immobilization with regard to specific production requirements, the use of S-layer enzyme fusion proteins in continuous enzymatic reactions will constitute an important aspect that has to be evaluated.
5. Note added in proof
During the reviewing process of this manuscript a paper dealing with the conceptuation of novel types of biocatalysts based on the S-layer self-assembly system was published by our Center (Scḧaffer et al., 2007).
Acknowledgements
This work was supported by Nano-S Biotechnology GmbH, CatchMabs BV, European Union Project NMP4-CT-2004-013523 (NAS-SAP), and the US Air Force Office of Scientific Research (project BIOCAT FA9550-07-1-0313).
References
- Angenendt P. Progress in protein and antibody microarray technology. Drug Discov. Today. 2005;10:503–511. doi: 10.1016/S1359-6446(05)03392-1. [DOI] [PubMed] [Google Scholar]
- Aslam M, Dent A. Bioconjugation: Protein Coupling Techniques for the Biomedical Sciences. Macmillan Reference Ltd; London, UK: 1998. [Google Scholar]
- Bengio S, Lettner H, Jungbauer A. Aseptic chromatography processing. Dream or reality? Ann. NY Acad. Sci. 1996;782:432–440. [Google Scholar]
- Bora U, Sharma P, Krishnamoorthy K, Nahar P. Photoreactive cellulose membrane—A novel matrix for covalent immobilization of biomolecules. J. Biotechnol. 2006;126:220–229. doi: 10.1016/j.jbiotec.2006.04.013. [DOI] [PubMed] [Google Scholar]
- Bradford MM. A rapid and sensitive method for quantitation of microgram quantities of protein utilizing the principle of protein-dye-binding. Anal. Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- Breitwieser A, Egelseer EM, Moll D, Ilk N, Hotzy C, Bohle B, Ebner C, Sleytr UB, Sára M. A recombinant bacterial cell surface (S-layer)-major birch pollen allergen-fusion protein (rSbsC/Bet v1) maintains the ability to self-assemble into regularly structured monomolecular lattices and the functionality of the allergen. Protein Eng. 2002;15:243–249. doi: 10.1093/protein/15.3.243. [DOI] [PubMed] [Google Scholar]
- Chaplin M, Bucke C. Enzyme Technology. Cambridge University Press; UK: 1990. [Google Scholar]
- Chiaraluce R, Van Der Oost J, Lebbink HJ, Kaper T, Consalvi V. Persistence of tertiary structure in 7.9 M guandidnium chloride: the case of endo-β-1,3-glucanase from Pyrococcus furiosus. Biochem. J. 2002;41:14624–14632. doi: 10.1021/bi026498u. [DOI] [PubMed] [Google Scholar]
- Gueguen Y, Voorhorst WGB, Van Der Oost J, De Vos WM. Molecular and biochemical characterization of an endo-β-1,3-glucanase of the hyperthermophilic archaeon Pyrococcus furiosus. J. Biol. Chem. 1997;272:31258–31264. doi: 10.1074/jbc.272.50.31258. [DOI] [PubMed] [Google Scholar]
- Györvary ES, Stein O, Pum D, Sleytr UB. Self-assembly and recrystallization of bacterial S layer proteins at silicon supports imaged in real time by atomic force microscopy. J. Microsc. 2003;212:300–306. doi: 10.1111/j.1365-2818.2003.01270.x. [DOI] [PubMed] [Google Scholar]
- Hilal N, Kochkodan V, Nigmatullin R, Goncharuk V, Al-Khatib L. Lipase-immobilized biocatalytic membranes for enzymatic esterification: comparison of various approaches to membrane preparation. J. Membr. Sci. 2006;268:198–207. [Google Scholar]
- Huber C, Liu J, Egelseer EM, Moll D, Knoll W, Sleytr UB, Sára M. Heterotetramers formed by an S-layer-streptavidin fusion protein and core-streptavidin as nanoarrayed template for biochip development. Small. 2006;2:142–150. doi: 10.1002/smll.200500147. [DOI] [PubMed] [Google Scholar]
- Ilk N, Völlenkle C, Egelseer EM, Breitwieser A, Sleytr UB, Sára M. Molecular characterization of the S-layer gene, sbpA, of B. sphaericus CCM 2177 and production of a functional S-layer fusion protein with the ability to recrystallize in a defined orientation while presenting the fused allergen. Appl. Environ. Microbiol. 2002;68:3251–3260. doi: 10.1128/AEM.68.7.3251-3260.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ilk N, Küpcü S, Moncayo G, Klimt S, Ecker RC, Hofer-Warbinek R, Egelseer EM, Sleytr UB, Sára M. A functional chimaeric S-layer-enhanced green fluorescent protein to follow the uptake of S-layer-coated liposomes into eukaryotic cells. Biochem. J. 2004;379:441–448. doi: 10.1042/BJ20031900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- Lopez JL, Matson SL. A multiphase/extractive enzyme membrane reactor for production of diltiazem chiral intermediate. J. Membr. Sci. 1997;125:189–211. [Google Scholar]
- Lowry O, Rosebrough N, Farr A, Randall R. Protein measurements with Folin-phenol reagent. J. Biol. Chem. 1951;193:265–275. [PubMed] [Google Scholar]
- Moll D, Huber C, Schlegel B, Pum D, Sleytr UB, Sára M. S-layer-streptavidin fusion proteins as template for nanopatterned molecular arrays. Proc. Natl. Acad. Sci. U.S.A. 2002;99:14646–14651. doi: 10.1073/pnas.232299399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neubauer A, Pum D, Sleytr UB. An amperometric glucose sensor based on isoporous crystalline protein membranes as immobilization matrix. Anal. Lett. 1993;26:1347–1360. [Google Scholar]
- Neubauer A, Hödl C, Pum D, Sleytr UB. A multistep enzyme sensor for sucrose based on S layer microparticles as immobilization matrix. Anal. Lett. 1994;27:849–865. [Google Scholar]
- Neubauer A, Pum D, Sleytr UB, Klimant I, Wolfbeis OS. Fiberoptic glucose biosensor using enzyme membranes with 2-D crystalline structure. Biosens. Bioelectron. 1996;11:317–325. [Google Scholar]
- Pleschberger M, Neubauer A, Egelseer EM, Weigert S, Lindner B, Sleytr UB, Muyldermans S, Sára M. Generation of a functional monomolecular protein lattice consisting of an s-layer fusion protein comprising the variable domain of a camel heavy chain antibody. Bioconjug. Chem. 2003;14:440–448. doi: 10.1021/bc025603+. [DOI] [PubMed] [Google Scholar]
- Pleschberger M, Saerens D, Weigert S, Sleytr UB, Muyldermans S, Sára M, Egelseer EM. An S-layer heavy chain camel antibody fusion protein for generation of a nanopatterned sensing layer to detect the prostate-specific antigen by surface plasmon resonance technology. Bioconjug. Chem. 2004;15:664–671. doi: 10.1021/bc049964w. [DOI] [PubMed] [Google Scholar]
- Pum D, Sára M, Sleytr UB. Structure, surface charge, and self-assembly of the S-layer lattice from Bacillus coagulans E38-66. J. Bacteriol. 1989;171:5296–5303. doi: 10.1128/jb.171.10.5296-5303.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sára M, Sleytr UB. S-layer proteins. J. Bacteriol. 2000;182:859–868. doi: 10.1128/jb.182.4.859-868.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sára M, Egelseer EM, Huber C, Ilk N, Pleschberger M, Pum D, Sleytr UB. S-layer proteins: potential applications in nano (bio) technology. In: Rehm B, editor. Microbial Bionanotechnology: Biological Self-Assembly Systems and Biopolymer-Based Nanostructures. Horizon Scientific Press; Hethersett, Norwich: 2006. pp. 307–338. [Google Scholar]
- Scḧaffer C, Novotny R, Küpcü S, Zayni S, Scheberl A, Friedmann J, Sleytr UB, Messner P. Novel biocatalysts based on S-layer self-assembly of Geobacillus stearothermophilus NRS 2004/3a: a nanobiotechnological approach. Small. 2007;3:1549–1559. doi: 10.1002/smll.200700200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sleytr UB. Gefrier̈atzung verschiedener Sẗamme von Bacillus sphaericus. Arch. Mikrobiol. 1970;72:238–251. [PubMed] [Google Scholar]
- Sleytr UB, Messner P, Pum D, Sára M. Crystalline bacterial cell surface layers (S-layers): from supramolecular cell structure to biomimetics and nanotechnology. Angew. Chem. Int. Ed. 1999;38:1034–1054. doi: 10.1002/(SICI)1521-3773(19990419)38:8<1034::AID-ANIE1034>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- Sleytr UB, Beveridge TJ. Bacterial S-layers. Trends Microbiol. 1999;7:253–260. doi: 10.1016/s0966-842x(99)01513-9. [DOI] [PubMed] [Google Scholar]
- Sleytr UB, Sára M, Pum D, Schuster B, Messner P, Scḧaffer C. Self assembly protein systems: microbial S-layers. In: Steinbüchel A, Fahnestock S, editors. Biopolymers. Wiley–VCH; Weinheim: 2002. pp. 285–338. [Google Scholar]
- Sleytr UB, Egelseer EM, Pum D, Schuster B. S-layers. In: Niemeyer CM, Mirkin CA, editors. Nanobiotechnology: Concepts, Applications, and Perspectives. Wiley-VCH; Weinheim: 2004. pp. 77–92. [Google Scholar]
- Sleytr UB, Sára M, Pum D, Schuster B. Crystalline bacterial cell surface layers (S layers): a versatile self-assembly system. In: Ciferri A, editor. Supramolecular Polymers. CRC Press, Taylor & Francis Group; Boca Raton: 2005. pp. 583–616. [Google Scholar]
- Sleytr UB, Huber C, Ilk N, Pum D, Schuster B, Egelseer E. S-layers as tool kit for nanobiotechnological applications. FEMS Microbiol. Lett. 2007;267:131–144. doi: 10.1111/j.1574-6968.2006.00573.x. [DOI] [PubMed] [Google Scholar]
- Sleytr UB, Egelseer EM, Ilk N, Pum D, Schuster B. S-Layers as basic building block for a molecular construction kit. FEBS J. 2007;274:323–334. doi: 10.1111/j.1742-4658.2006.05606.x. [DOI] [PubMed] [Google Scholar]
- Summer JB, Somers GF. Dinitrosalicylic Method for Glucose: Laboratory Experiments in Biological Chemistry. Academic Press, Inc.; NY: 1949. pp. 38–39. [Google Scholar]
- Van Lieshout J, Faijes M, Nieto J, Van Der Oost J, Planas A. Hydrolase and glycosynthase activity of endo-1,3,-β-glucanase from thermophile Pyrococcus furiosus. Archea. 2004;1:285–292. doi: 10.1155/2004/731548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Völlenkle C, Weigert S, Ilk N, Egelseer E, Weber V, Loth F, Falkenhagen D, Sleytr UB, Sára M. Construction of a functional S-layer fusion protein comprising an immunoglobulin G-binding domain for development of specific adsorbents for extracorporeal blood purification. Appl. Environ. Microbiol. 2004;70:1514–1521. doi: 10.1128/AEM.70.3.1514-1521.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu H, Snyder M. Protein chip technology. Curr. Opin. Chem. Biol. 2003;7:55–63. doi: 10.1016/s1367-5931(02)00005-4. [DOI] [PubMed] [Google Scholar]