

Abstract
Advances in understanding the control mechanisms governing the behavior of cells in adherent mammalian tissue culture models are becoming increasingly dependent on modes of single-cell analysis. Methods which deliver composite data reflecting the mean values of biomarkers from cell populations risk losing subpopulation dynamics that reflect the heterogeneity of the studied biological system. In keeping with this, traditional approaches are being replaced by, or supported with, more sophisticated forms of cellular assay developed to allow assessment by high-content microscopy. These assays potentially generate large numbers of images of fluorescent biomarkers, which enabled by accompanying proprietary software packages, allows for multi-parametric measurements per cell. However, the relatively high capital costs and overspecialization of many of these devices have prevented their accessibility to many investigators.
Described here is a universally applicable workflow for the quantification of multiple fluorescent marker intensities from specific subcellular regions of individual cells suitable for use with images from most fluorescent microscopes. Key to this workflow is the implementation of the freely available Cell Profiler software1 to distinguish individual cells in these images, segment them into defined subcellular regions and deliver fluorescence marker intensity values specific to these regions. The extraction of individual cell intensity values from image data is the central purpose of this workflow and will be illustrated with the analysis of control data from a siRNA screen for G1 checkpoint regulators in adherent human cells. However, the workflow presented here can be applied to analysis of data from other means of cell perturbation (e.g., compound screens) and other forms of fluorescence based cellular markers and thus should be useful for a wide range of laboratories.
Keywords: Cellular Biology, Issue 94, Image analysis, High-content analysis, Screening, Microscopy, Individual cell analysis, Multiplexed assays
Introduction
The work presented here describes the use of the freely available software Cell Profiler to perform algorithm-guided breakdown of fluorescent microscopy images of adherent cells to identify individual cells and defined subcellular regions. This approach, referred to as image segmentation, allows the subsequent multi-parametric analysis of the imaged cells by quantifying fluorescently labeled markers localized to each cell or subcellular region (referred to as segmented objects). This workflow constitutes a basis for enabling high-content analysis and is intended to serve as a tool that can be further developed and modified to suit multi-parametric, individual cell analyses in laboratories without access to specialized high-content instruments or proprietary software. The files supplied with this manuscript include a test set of relevant raw image data, algorithm settings and supporting scripts to generate the analysis described. The provided algorithm settings for Cell Profiler are optimized for the example data set and the Discussion section details what adjustments may be necessary to enable use of image data from other studies.
Once quantitative data has been extracted using Cell Profiler, different laboratories may have different requirements for how to use the information presented by the individual cell values in the raw data; shown here is one approach by which gates are applied to the raw data for each assay. Using these gates, the data are transformed into binary terms of response, allowing visualization of trends linking different treatments with the subpopulations of cells undergoing response defined by the gates. The gates are set based on observations of the data distributions obtained for appropriate negative and positive controls for each relevant measurement. The use of gates is just one example of how to manage the raw, cell-based measurements. Also shown here is the use of nuclear DNA intensity measurements in their raw form as a continuous range of values in combination with the gated data. Other approaches to managing image analysis data should be considered, depending on the nature of the study; statistical alternatives to using gates for assigning cells to subpopulations have been reported2 and systematic comparisons of strategies to summarize high-content data across large numbers of parameters have been reported3.
High-content analyses of image data have found use in cellular studies of drug-response, reverse genetics and environmental stress signalling4–6. The merit of high-content analysis stems from the fact that algorithmic analysis of fluorescence microscopy data allows quantitative and spatial parameters to be considered simultaneously across individual cells7. In this way, cellular outcomes for multiple assays can be cross-referenced, differential behavior of assay-defined cell subpopulations can be tracked within experimental conditions and assays can include consideration of morphological variables. The strategies and analysis workflow discussed here, as for other high-content approaches, are capable of delivering multiplexed data that are cross-referenced to individual cells. High-content methods suit studies that generate fluorescent microscope images and are applicable to analysis of data ranging from tens of images produced in low throughput conventional fluorescence-based microscopy through to the thousands of images produced using automated high-content screening platforms.
The workflow is illustrated here with example data from which separate assays are measured in terms of either nuclear fluorescent marker intensities or nuclear/cytoplasmic translocation of a fluorescent reporter protein, respectively. The workflow is flexible in that these assays can be considered separately or in combination depending on each given research question by different investigators. The example data are produced as part of a RNA interference (RNAi) experiment (Figure 1). Small interfering RNA oligonucleotides (siRNA) are used to knockdown specific proteins in HCT116 human colorectal carcinoma cells which result in changes for two fluorescent reporters of cyclin-dependent kinase (CDK) activity. The CDK6-dependent phosphorylation of the nuclear retinoblastoma protein at serine 780 (P-S780 RB1) is assessed by antibody staining. In the same cells, a green fluorescent protein-tagged reporter of CDK2 activity (GFP-CDK2 reporter) is assessed by its nuclear to cytoplasmic ratio where in the absence of CDK2 activity the reporter resides in the nucleus and upon CDK2 activation shuttles into the cytoplasm8. Additionally, the nuclear DNA of each cell is stained using a DNA-intercalating dye, Bisbenzimide, which serves as a means to identify cells and define nuclei borders in the images as well as a measure of DNA abundance providing information on cell cycle position of the cell (Figure 2).
The activities of CDK6 and CDK2 are detectable as cells transit from G1 to S phase of the cell cycle5 and succeed each other9,10 and, as such, close concordance between the two reporters in individual cells is expected. The demonstration dataset used here analyzes as an example the effect of siRNA targets CDK6, retinoblastoma protein (RB1) and a non-targeting negative control (Table 1). Knockdown of CDK6 should elicit both a decrease of the P-S780 RB1 epitope and an accumulation of cells in G1 phase of the cell cycle. The RB1 knockdown serves as a reagent control for the specificity of the phospho-S780 antibody. Fluorescence microscope images from formalin fixed11, fluorescently stained HCT116 tissue culture cells are used for algorithmic image analysis. The resulting numeric data is then used to cross-reference the reporters and gauge the impact of the different knockdown states.
The potential size of the data produced by this type of analysis can present a challenge to normal analysis tools. For example, the individual cell data can be larger than some spreadsheet software will accommodate. Included are Perl scripts which perform simple, highly-repetitive, supervised processing of the data to aid analysis of large datasets. The Perl scripts are written specifically for the output files produced by Cell Profiler, when processing image files with a specific file naming convention (Figure 3), and allow for variable numbers of fields per well to be used in the analysis. It is frequently important to gate individual cell assay data to track trends in cell subpopulations5 and shown here is the use of a Perl script to flag each cell based on a set gate predetermined for each assay type. Also included are optional Perl scripts which summarize the data outcome for individual wells (or conditions), delivering: percentage of cells within the set gate and the mean values of the raw assay scores. The latter, more homogeneous way of viewing the data, is valid where responses affect all or the majority of cells within a well. As discussed above, such assessment is less useful than that afforded by the individual cell data gating where response is confined to a subset of cells within a population.
The utility of the described workflow is not limited to perturbation by siRNA or the marker assays described. Studies have used this approach to assay responses in tissue culture experiments using combinations of siRNA, chemical inhibitors and radiation treatment and for assessment of markers other than CDK6 and CDK2 activity5.
Conceptually, the experimental strategy allows a variety of biologically useful subcellular regions to be automatically registered in individual cells present in fluorescent microscope images. As such, this approach can yield quantitative, multiplexed data revealing biological information that may be missed through techniques that focus on populations rather than individual cells. With minor modifications, the approach and analysis workflow described can yield quantitative, individual cell data for any fluorescence-based assay outputs and cell-biological responses, where quantitative assessment of DNA content, quantification of nuclear or cytoplasmic fluorescence or the shuttling of markers between these two compartments either individually or in a multiplexed manner is of interest. As publishing requirements increasingly tend towards submission of openly accessible raw data, access to and familiarity with free tools for microscopy image analysis such as those described here will also be of direct interest to labs looking to reanalyze published data.
Protocol
1. Experimental Perturbation and Cell Labeling for Response Markers (Reverse Transfection siRNA Screen)
In a sterile tissue culture hood pipette 70 µl of 200 nM siRNA in 1x siRNA buffer in wells of a sterile, plain 96 well plate. Dilute transfection lipid into 40 volumes of serum-free DMEM media and dispense 105 µl into each well containing siRNA. NOTE: Diluting 262.5 µl lipid into 10.5 ml of serum-free DMEM yields a master mix suitable for a whole 96 well plate of siRNA, delivering 2.6 µl of lipid per well. Use of 200 nM siRNA starting concentration at this step will deliver a working concentration of 20 nM in step 1.3, but procedures will work for working concentrations down to 5 nM, with the starting concentration adjusted accordingly (i.e. 50 nM). Lower working concentration may reduce off-target false positive scores, although they can reduce the magnitude of on-target response, leading to increase of on-target false negative rates.
Mix the plate by gentle vibration for ten minutes at room temperature. Sub-divide the resulting 175 µl into three 50 µl replicates per target onto an opaque, tissue culture treated, 96-well plate with a transparent base.
Reverse transfect by dispensing 8,000 cells per well in 150 µl DMEM containing 10% serum directly onto the 50 µl lipid-siRNA complexes. Use HCT116 human colorectal cells stably expressing a GFP-tagged marker reporting CDK2 activity5,8. No further mixing is necessary. Seal the plate with a sterile, adhesive breathable membrane to control humidity and prevent plate ‘edge-effects’ and place the plate into a humidified incubator at 37 °C, 5% CO2 for 48 hr.
Aspirate the media such that a small residual amount of media remains in the wells. Fix the cells by adding 100 µl of 4% buffered formaldehyde to each well and incubate in a fume hood for 10 min at room temperature.
Remove the fixing solution by aspirating the plate. At this point either stop the experiment by washing the plate three times with 100 µl phosphate-buffered saline (PBS) and then store sealed, under 100 µl of PBS in the dark at 4 °C for up to a week, or proceed with the permeabilization of the cells. NOTE: We recommend processing plates as soon as possible after fixation, and generally prefer storage of fully processed plates. Biocidal preservatives such as thimerosal, sodium azide, or commercial alternatives may be added to prevent micoroganismal growth. Addition of phosphatase inhibitors helps to preserve phospho-epitopes, and other means to preserve protein modification states may be useful in relevant assay contexts
Remove PBS from the plate and permeabilize the cells by adding 100 µl of permeabilization solution. Incubate for 10 min at room temperature without shaking. Aspirate the permeabilization solution using a multichannel pipette. Repeat this step three times.
Block the cells by adding 100 µl block solution per well for 30 min at room temperature. Remove the block solution by aspirating the plate, then probe with 50 µl of anti P-S780 RB1 antibody diluted 500-fold in the block solution for 2 hr in the dark at room temperature.
Wash the plate three times with 100 µl plate wash solution, leaving the solution on the plate for 5 min each time. Probe the plate overnight in the dark at 4 °C with 50 µl fluorescently-tagged secondary antibody diluted 1,000-fold in block solution supplemented with 2 µM of the chromatin-specific DNA dye Bisbenzimide. Wash the plate three times as before and store sealed, under 100 µl PBS in the dark at 4 °C. Image the plate within two weeks.
2. Imaging and Image Segmentation
Use a confocal or spinning-disk fluorescence microscope with a 20X objective to take separate 16-bit, greyscale TIFF images in three channels corresponding to the DNA dye, GFP and immuno-staining fluorophores. Capture many non-overlapping image sets, referred to here as frames, to image approximately 1,000-2,000 cells per well.
Name the image files systematically so that each file name is a unique combination of ‘experiment name’, ‘well address’, ‘frame number’ and ‘channel identifier’, in this order (Figure 3). The example data set uses “Blue” (chromatin DNA staining) or “Green” (GFP) or “Red” (the immuno-stained fluorophore) as channel identifiers. The well address, frame number and channel identifier are further on referred to as the image metadata. Use the underscore symbol to avoid confusing well and frame metadata.
Name the files with these metadata elements in the specified order. This is necessary to ensure that the subsequent software steps correctly group sets of images for analysis.
Download and install the freeware Cell Profiler, Active Perl Community Edition, R statistical programming environment and RStudio. Accept all default options during installation; PC users installing Active Perl should enable all options relating to PATH, file extension association and script mapping where prompted. Active Perl is optional for Mac users, but they will otherwise need to run the Perl script in step 3.2 from the Terminal command line rather than using icon clicking.
Open the Cell Profiler software, click ‘File’, ‘Import Pipeline’ then ‘From file’ and select the file 3_channels_pipeline.cppipe (Figures S1A & S1B). The file contains instructions necessary for the software to interpret image file metadata from the file name convention described. Cell Profiler now relates the images, extracts nuclear DNA and antibody intensities from these and uses the GFP channel to calculate the ratio of nuclear versus cytoplasm intensity for each cell detected (Figures 4 & 5).
Click the button ‘View output settings’ in the lower left corner of the Cell Profiler window. At the top of the new screen are text boxes labeled ‘Default Input Folder’ and ‘Default Output Folder’. One at a time, click the folder-icons to the right of these boxes and select the location of the image files for analysis and the destination for the extracted data, respectively (Figure S1C).
Begin the image analysis by pressing the ‘Analyze Images’ button in the lower left corner of Cell Profiler. At the bottom of the screen observe the remaining time for the data extraction, the ‘Stop Analysis’ and ‘Pause’ buttons. If required pause the analysis by selecting the ‘Pause’ button at any time, which is useful when watching the images being analyzed (described in step 2.8).
Optionally, open the windows for any of the image analysis steps by clicking the eye-icons in the panel on the extreme left of the program window (Figure S1D). Observe the ‘IdentifyPrimaryObjects’ window and those for ‘Secondary’ and ‘Tertiary Objects’ to check that the current settings in Cell Profiler to perform image segmentation are suitable (see Figure 1 and Discussion for advice on modifying these settings).
Click ‘Ok’ in the message box that appears when the analysis is complete. Go to the location ‘Default Output Folder’ where all the data files with the results are saved as comma-separated-value (.csv) files (Figure S2A).
3. Data Extraction
Find the new file ‘Nuclei.csv ’, which is included among the output from Cell Profiler. This file contains individual cell data for fluorescent nuclear antibody intensity, nuclear DNA intensity and GFP-CDK2 reporter ratio values (Figures 6A & S2A). NOTE: Different laboratories will want to process this type of data to suit the nature of their own assays. Suggested for the current data is the gating of the cells from each treatment condition according to the antibody data and the GFP-CDK2 reporter values using the provided Perl script ‘2_gate_classifier.pl’.
Copy the provided Perl script file ‘2_gate_classifier.pl’ into the same folder as the ‘Nuclei.csv’ data file (Figure S2A). Double-click the icon for the Perl script and, when prompted, type the full name of the data file followed by a ‘.csv’ filename name for the file in which the cells are to be gated and finally the gate values for the antibody fluorescence and GFP-CDK2 reporter data. NOTE: How to principally determine gate settings and apply these for analysis of data are discussed below in the Representative Data section and Figure 6 (to analyze the data provided use ‘0.004’ and ‘1.5’, respectively). Mac users should run the Perl script from the Terminal command line by typing: ‘perl 2_gate_classifier.pl’.
Observe the newly created file which combines the raw individual cell assay values from the original Cell Profiler data with sub-population labels that show how each cell from each well performs against both gates (Figure 6C).
Plot the data for each experimental condition using the individual cell subpopulation labels by opening the RStudio software. Click ‘File’ and ‘Open File’, then select the provided ‘analysis.r’ file. Observe the commands to plot Figures 6B, 7 and 8 in the upper left window of RStudio (Figure S2B). In the upper left window, between the double quote symbols on lines 5 and 6, type the computer address of the folder containing the gated data. Include the drive letter and the name of the file itself, respectively (e.g., “C:/analysis folder/analysis output” and “nuclei_gated.csv”). NOTE: If RStudio is used for the first time on a given computer, the R graphics package ‘ggplot2’ will need to be installed first. This is a once only step for a new installation of RStudio, after which this step becomes redundant. To install ‘ggplot2’, click the tab called ‘Packages’ above the window in the lower right corner of RStudio, click the ‘Install Packages’ button that appears beneath this. A new window will appear. Type ‘ggplot2’ (omitting quotations) into the ‘Packages’ space in this new window and finally click the ‘Install’ button to close the window, install the necessary ggplot2 functions and return to the main RStudio window to continue from step 3.6.
Highlight lines 1 through 17 in the upper left window of RStudio, then click the ‘Run’ button. This will enter the experimental data, threshold values and well location details into R (Figure S2C). R will now temporarily hold the relevant data for plotting.
Highlight individual blocks of the remaining code beneath line 17 and create the corresponding plots by clicking the ‘Run’ button as before. Observe the plots in the window in the lower right corner of RStudio and save number of formats by clicking the ‘Export’ button (Figure S2D).
While closing RStudio, click ‘Don’t Save’ when prompted. This prevents confusion on the next use of RStudio, which will otherwise hold data from the previous session.
Representative Results
The example set of images generated using the reverse-transfection siRNA screening protocol have been prepared for and analyzed using Cell Profiler software. The resulting numerical raw data is such that every cell is individually represented, traceable back to its image and well of origin and measured for several fluorescence intensity parameters (Figure 6A). For each cell identified the mean nuclear fluorescence intensity for the P-S780 RB1 antibody and the integrated DNA intensity for the DNA dye-defined nuclear masks are determined. Mean GFP intensity values for nucleus and cytoplasm regions of each cell are also recorded allowing the calculation of nuclear versus cytoplasmic fluorescence of the GFP-CDK2 reporter. Downstream of these algorithmic fluorescence intensity measurements use is made of these individual cell data to define gates for two assays, nuclear antibody staining and GFP-CDK2 reporter. Subsequent annotation of the cells on the basis of assay outcome and use of these labels to enable specific subpopulations to be further characterized by a third measurement (nuclear DNA content) is described.
Histogram plots of the raw fluorescence intensity data gathered for each assay are an effective way of assessing how cell subpopulations behave under different conditions. The histograms in Figure 6B show the population distributions of individual cell data from triplicate wells for each RNAi knockdown condition. To the left are the data for nuclear antibody intensity and on the right are the corresponding data for the GFP-CDK2 reporter. The P-S780 RB1 antibody data reveals that the cells broadly exist in two populations with regard to this post-translational modification and that cell populations with loss of RB1 phosphorylated on S780 can be distinguished as a left-hand peak of nuclear intensity which is enriched when CDK6 is knocked down by siRNA. This same left-hand peak is seen when RB1 itself is the RNAi target, reflecting the outright removal of the protein and thereby P-S780 RB1 staining. In contrast, the same experimental conditions for the same cells, when observed via the GFP-CDK2 reporter assay, show a different dynamic in the individual cell data. A continuous distribution is observed, with only a single peak, but siRNA which disturbs the cell cycle (siCDK6) and causes accumulation in G1 phase results in an extension of the right-hand shoulder of that distribution (i.e. indicating enhanced presence of cells showing an increase in the nuclear/cytoplasm GFP ratio, plotted on the X-axis).
Also shown on the histograms of Figure 6B are the gate values (vertical bars) that are chosen on the basis of the distributions of both sets of assay data. The rule used for the P-S780 RB1 antibody data is to define the gate position as the half-height:maximum-width position on the left shoulder of the main (right) peak when considering the negative control cell data (non-targeting siRNA). Data highlighted red are cells with reduced and absent P-S780 RB1, which are identified with this gate. A similar gate positioned on the opposite shoulder of the ratio value distribution is used for the GFP-CDK2 reporter. The resulting high-ratio subpopulation cells, which lack or feature reduced CDK2 activity, are shown in green. To illustrate multiplexed analysis of the two assays Figure 6C shows the implementation of both gate values using the 2_gate_classifier.pl perl script to convert the raw data (Figure 6A) into the annotated file below. This new file includes the original data alongside a new column of class labels for each cell and the two gate values used to distinguish them (in this case gates of 0.004 for the antibody data and 1.5 for the GFP-CDK2 reporter were used, respectively).
Having classified the individual cells from each knockdown condition on the basis of the two assays it is now possible to use these class labels to assist the annotation of plots of the assay data. Figure 7 shows scatter plots of the individual cell data for the P-S780 RB1 and GFP-CDK2 assays from the example data set for all three RNAi conditions. Numbers annotating the quadrants on the scatterplots show the relative percentages of each gated subpopulation to the whole for that knockdown context and are generated in R using the class labels described above. These plots reveal that, compared to cells transfected with non-targeting siRNA (Figure 7B), cells transfected with siCDK6 reveal a net data distribution shifted both downward on the Y-axis (indicating absence of RB1 phosphorylation at serine 780) and to the right on the X-axis (indicating low CDK2 activity, Figure 7C). Both of these shifts are expected for knockdown of this target. In contrast to this, the data from siRB1 transfected cells (Figure 7A) show a loss of the antibody staining in keeping with loss of the epitope, but little effect in the data distribution for the CDK2 reporter compared to controls transfected with non-targeting siRNA, suggesting no great effect on the GFP-CDK2 reporter arises from RB1 knockdown.
To further explore the use of individual cell data, subpopulation classification and assay multiplexing Figure 8 shows the scatter plot for the siCDK6 data from Figure 7C alongside paired histogram profiles for integrated DNA intensity. The pairs of histograms relate to opposing halves of the entire population, divided on the basis of either antibody intensity (right of the scatterplot) or GFP-CDK2 reporter ratio values (above the scatterplot). Quantification of nuclear DNA intensity for these populations shows two peaks characteristic of 2N and 4N DNA content as left and right peaks, respectively. The intentions of the gates shown in Figures 6, 7 and 8 are such that cells identified as low for P-S780 RB1 (labeled: P-S780-) or with a high ratio value from the GFP-CDK2 reporter (labeled: G1) will be in G1 phase of the cell cycle. Indeed, the DNA profile histograms for subpopulations identified with either of these assays predominantly contain cells with 2N DNA content. DNA profiles of the oppositely gated population (labeled: P-S780+ or Non-G1) contains cells with distributions ranging from 2N to 4N, in keeping with such cells adopting a range of cell cycle positions post-G1 phase.
Although the focus here has been the generation and analysis of individual cell data from fluorescently stained images, it is also useful to be able to take these data and summarize each assay on a well-by-well basis to monitor variability between replicates and the performance of all the wells for a given assay across a whole plate of data. Figure 9 shows the data from each siRNA treatment summarized as the mean values from triplicate wells for the percent cells within the gates applied to A) the P-S780 RB1 data and B) the GFP-CDK2 reporter data. The values plotted in A and B are produced by two additional Perl scripts provided with this manuscript; ‘antibody_fluorescence_summary.pl’ and ‘G1assay_summary.pl’, respectively. These scripts use the raw data created by Cell Profiler (Nuclei.csv) and report data per well as i) total cells measured per well, ii) number of cells within the gate, iii) percent cells within the gate and iv) the arithmetic mean of the measured, raw data for that well. This is included as an option suitable for looking across large sets of assay data, prior to focusing on individual treatment data using multiplexed assessment of individual cell data as illustrated in Figures 7 and 8. The charts displayed here plot ‘iii) percent cells within the gate’ for both assays, which suit the non-normal data distributions seen for the P-S780 RB1 and GFP-CDK2 data in the histograms in Figure 6B. These scripts also calculate ‘iv) the arithmetic mean of the measured, raw data for that well’, which would suit analysis of data for homogeneous population responses and normal data distribution before and after experimental perturbation.
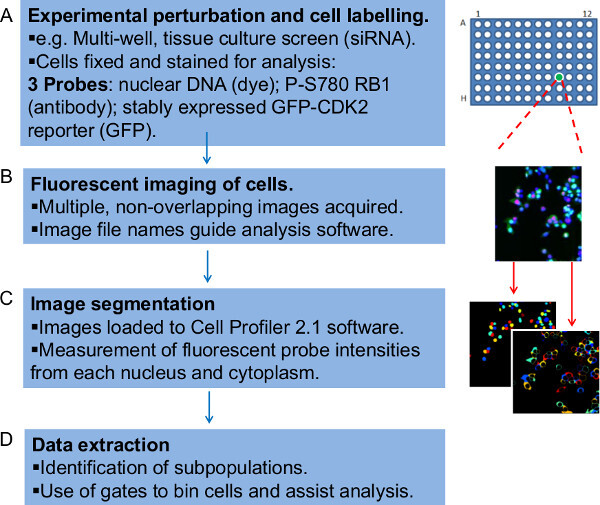 Figure 1: Overview of the steps in the workflow to quantitatively analyze fluorescently labeled microscope image data. The workflow is represented here as four steps. (A) First it is necessary to experimentally prepare cells for fluorescent imaging. The example described here is that of a screen in which siRNA-treated adherent human tumor cells are grown for 48 hours, fixed and stained on a 96-well tissue culture plate. Different RNAi conditions are present in triplicate in separate wells within the plate. Cells are stained with a DNA dye, an antibody specific for RB1 phosphorylated on the CDK4 and 6 selective target site Serine-780 (P-S780 RB1) and they also stably express a GFP-CDK2-reporter, reporting G1 cell cycle exit. Collectively these fluorescent probes constitute two assays separately assessed within the workflow. (B) Parallel microscope images for each fluorescent probe (channel) are generated and named such as to include details by which the image analysis software can organize the data. (C) The image files are loaded into the Cell Profiler software, which algorithmically identifies individual cells and the associated pairs of nuclei and cytoplasm before yielding intensity measurements for the three fluorescent probes detected in each. (D) Finally, a Perl script is used to organize the raw quantitative data produced. This step applies gates to the fluorescence intensity data for each cell, effectively binning the cells into subpopulations, which can be plotted, tracked and cross-examined. Please click here to view a larger version of this figure.
Figure 1: Overview of the steps in the workflow to quantitatively analyze fluorescently labeled microscope image data. The workflow is represented here as four steps. (A) First it is necessary to experimentally prepare cells for fluorescent imaging. The example described here is that of a screen in which siRNA-treated adherent human tumor cells are grown for 48 hours, fixed and stained on a 96-well tissue culture plate. Different RNAi conditions are present in triplicate in separate wells within the plate. Cells are stained with a DNA dye, an antibody specific for RB1 phosphorylated on the CDK4 and 6 selective target site Serine-780 (P-S780 RB1) and they also stably express a GFP-CDK2-reporter, reporting G1 cell cycle exit. Collectively these fluorescent probes constitute two assays separately assessed within the workflow. (B) Parallel microscope images for each fluorescent probe (channel) are generated and named such as to include details by which the image analysis software can organize the data. (C) The image files are loaded into the Cell Profiler software, which algorithmically identifies individual cells and the associated pairs of nuclei and cytoplasm before yielding intensity measurements for the three fluorescent probes detected in each. (D) Finally, a Perl script is used to organize the raw quantitative data produced. This step applies gates to the fluorescence intensity data for each cell, effectively binning the cells into subpopulations, which can be plotted, tracked and cross-examined. Please click here to view a larger version of this figure.
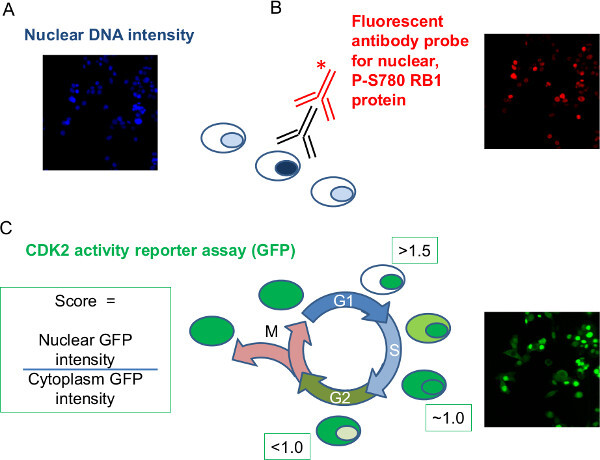 Figure 2: Experimental data to be obtained by image analysis. The fixed, siRNA-treated, fluorescently labeled cells from the example data set were imaged and corresponding intensity measurements taken per cell. Representative image data are shown for each parameter recorded during image analysis. (A) Nuclear DNA intensity: The intensity of staining of nuclear DNA dye is used to yield a measure of DNA per nucleus. (B) Nuclear intensity of phospho-RB1: Immuno-staining specific for P-S780 RB1 using a primary (black) antibody and fluorescently tagged secondary antibody (red) enable an intensity measurement of RB1 phosphorylation at S780 per nucleus. (C) GFP-CDK2 reporter: The cells used stably express a GFP-tagged reporter protein that translocates between the nucleus and cytoplasm in a set pattern with the cell cycle. Dual measurement of the paired nuclear and cytoplasmic GFP intensity for each cell allows calculation of a ratio per cell that can be used to distinguish G1 phase from the rest of the cell cycle. Three siRNA targets will be used to illustrate the analysis; a non-targeting negative control siRNA; CDK6 siRNA as a positive control in perturbing RB1 phosphorylation and cell cycle progress; RB1 siRNA to establish antibody specificity. Please click here to view a larger version of this figure.
Figure 2: Experimental data to be obtained by image analysis. The fixed, siRNA-treated, fluorescently labeled cells from the example data set were imaged and corresponding intensity measurements taken per cell. Representative image data are shown for each parameter recorded during image analysis. (A) Nuclear DNA intensity: The intensity of staining of nuclear DNA dye is used to yield a measure of DNA per nucleus. (B) Nuclear intensity of phospho-RB1: Immuno-staining specific for P-S780 RB1 using a primary (black) antibody and fluorescently tagged secondary antibody (red) enable an intensity measurement of RB1 phosphorylation at S780 per nucleus. (C) GFP-CDK2 reporter: The cells used stably express a GFP-tagged reporter protein that translocates between the nucleus and cytoplasm in a set pattern with the cell cycle. Dual measurement of the paired nuclear and cytoplasmic GFP intensity for each cell allows calculation of a ratio per cell that can be used to distinguish G1 phase from the rest of the cell cycle. Three siRNA targets will be used to illustrate the analysis; a non-targeting negative control siRNA; CDK6 siRNA as a positive control in perturbing RB1 phosphorylation and cell cycle progress; RB1 siRNA to establish antibody specificity. Please click here to view a larger version of this figure.
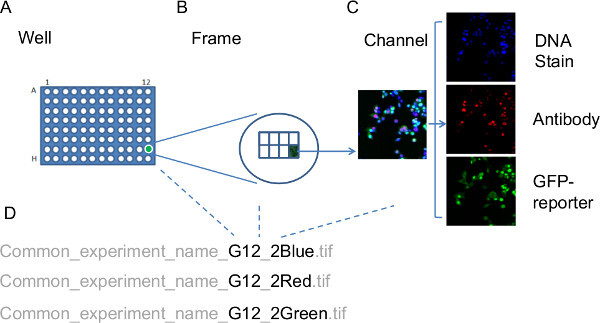 Figure 3: Organization of image files prior to image analysis. The images taken from the tissue culture plate are named systematically in order to allow the image analysis software to relate the image data back to the original experimental context. This information is placed within the filename for each image. (A) As each well on the experiment plate may correspond to different RNAi targets or treatments, the well address forms part of the filename. (B) The frame number is part of the filename as each well is imaged to collect multiple, non-overlapping frames. (C) Fluorescent probes from each frame are imaged separately; consequently the filenames also need to reflect which channel each image relates to. (D) Example filenames, relating to well (G12), Frame (2) with each image representing one of the channels (Blue, Red, Green). Dotted lines link the filename elements to the relevant schematic representations for Well, Frame and Channel, respectively. Please click here to view a larger version of this figure.
Figure 3: Organization of image files prior to image analysis. The images taken from the tissue culture plate are named systematically in order to allow the image analysis software to relate the image data back to the original experimental context. This information is placed within the filename for each image. (A) As each well on the experiment plate may correspond to different RNAi targets or treatments, the well address forms part of the filename. (B) The frame number is part of the filename as each well is imaged to collect multiple, non-overlapping frames. (C) Fluorescent probes from each frame are imaged separately; consequently the filenames also need to reflect which channel each image relates to. (D) Example filenames, relating to well (G12), Frame (2) with each image representing one of the channels (Blue, Red, Green). Dotted lines link the filename elements to the relevant schematic representations for Well, Frame and Channel, respectively. Please click here to view a larger version of this figure.
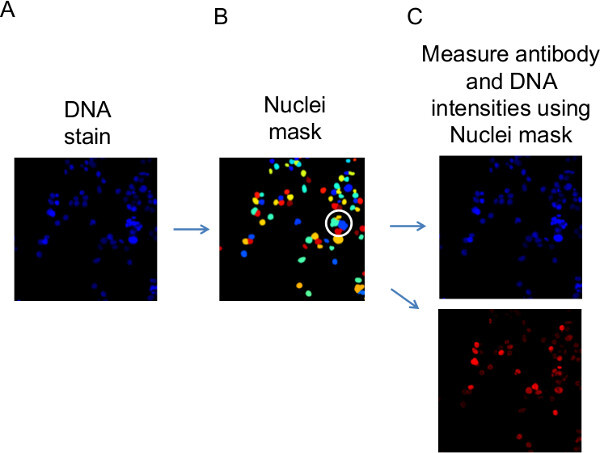 Figure 4: Use of Cell Profiler to measure nuclear DNA and antibody staining. With the settings in the provided pipeline file (3_channels_pipeline.cppipe), the Cell Profiler image analysis software measures fluorescence intensity values for nuclear DNA and antibody-binding relating to individual cells. (A) Nuclei are identified in the ‘blue’ channel image of stained DNA. (B) The positions of the DNA stained nuclei are temporarily held in a ‘Nuclei mask’. The Nuclei mask is then overlaid onto (C) the blue and red channel images (DNA and antibody fluorescence data, respectively) and the fluorescence values from image segments that overlap with the mask are recorded against each identified cell. Successful identification of separate, neighboring nuclei can be visually assessed in the appearance of the Nuclei mask. For illustration, shown circled in this mask image, are examples where the chosen settings for the algorithm have mis-identified neighboring nuclei as a single nucleus. Adjusting the algorithm settings to minimize these events is introduced in the Discussion section. Please click here to view a larger version of this figure.
Figure 4: Use of Cell Profiler to measure nuclear DNA and antibody staining. With the settings in the provided pipeline file (3_channels_pipeline.cppipe), the Cell Profiler image analysis software measures fluorescence intensity values for nuclear DNA and antibody-binding relating to individual cells. (A) Nuclei are identified in the ‘blue’ channel image of stained DNA. (B) The positions of the DNA stained nuclei are temporarily held in a ‘Nuclei mask’. The Nuclei mask is then overlaid onto (C) the blue and red channel images (DNA and antibody fluorescence data, respectively) and the fluorescence values from image segments that overlap with the mask are recorded against each identified cell. Successful identification of separate, neighboring nuclei can be visually assessed in the appearance of the Nuclei mask. For illustration, shown circled in this mask image, are examples where the chosen settings for the algorithm have mis-identified neighboring nuclei as a single nucleus. Adjusting the algorithm settings to minimize these events is introduced in the Discussion section. Please click here to view a larger version of this figure.
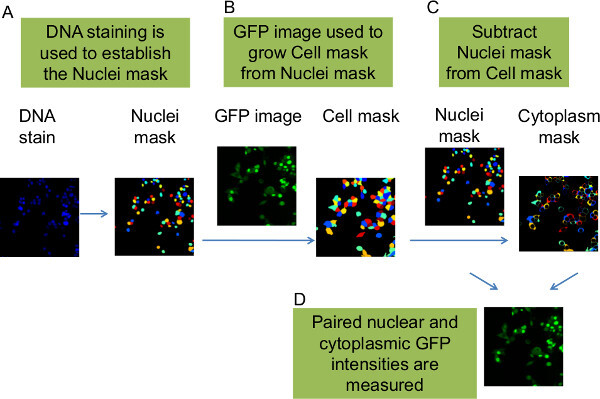 Figure 5: Use of Cell Profiler to measure nuclear and cytoplasmic GFP intensities. The GFP-tagged CDK2 reporter translocates between the nucleus and cytoplasm in relation to the cell cycle position of the cells. At the same time that Cell Profiler calculates the DNA and antibody nuclear intensities per cell (Figure 4), it also calculates the nuclear to cytoplasm ratio of GFP intensities for each cell. (A) The DNA dye data for each image is used to generate a Nuclei mask. (B) Cell Profiler uses the Nuclei mask in conjunction with the GFP image from the GFP-CDK2 reporter to seed the position of each cell and then expands to each cell’s perimeter to estimate the whole footprint of each cell. This becomes a new, ‘Cell mask’. (C) The nuclei mask is subtracted from the Cell mask to yield a donut-like series of cytoplasm outlines, which become the ‘Cytoplasm mask’. (D) The nuclei mask and cytoplasm mask are used by Cell Profiler to measure pairs of nuclear and cytoplasmic GFP values. These paired values are then used by Cell Profiler to calculate ratios, which inform as to each cell’s position in the cell cycle. Please click here to view a larger version of this figure.
Figure 5: Use of Cell Profiler to measure nuclear and cytoplasmic GFP intensities. The GFP-tagged CDK2 reporter translocates between the nucleus and cytoplasm in relation to the cell cycle position of the cells. At the same time that Cell Profiler calculates the DNA and antibody nuclear intensities per cell (Figure 4), it also calculates the nuclear to cytoplasm ratio of GFP intensities for each cell. (A) The DNA dye data for each image is used to generate a Nuclei mask. (B) Cell Profiler uses the Nuclei mask in conjunction with the GFP image from the GFP-CDK2 reporter to seed the position of each cell and then expands to each cell’s perimeter to estimate the whole footprint of each cell. This becomes a new, ‘Cell mask’. (C) The nuclei mask is subtracted from the Cell mask to yield a donut-like series of cytoplasm outlines, which become the ‘Cytoplasm mask’. (D) The nuclei mask and cytoplasm mask are used by Cell Profiler to measure pairs of nuclear and cytoplasmic GFP values. These paired values are then used by Cell Profiler to calculate ratios, which inform as to each cell’s position in the cell cycle. Please click here to view a larger version of this figure.
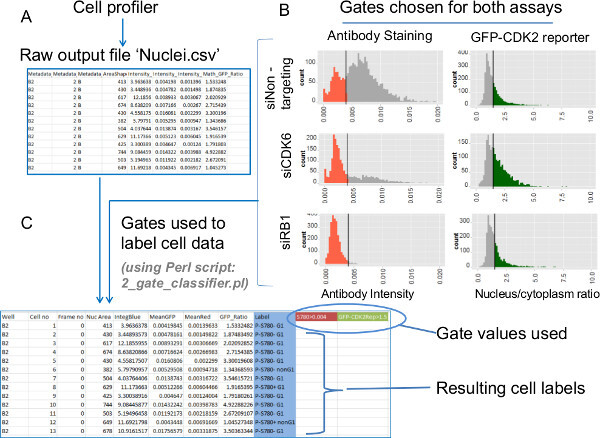 Figure 6: Data extraction - Processing raw individual cell data by imposing gates on assay values. Biological trends from the individual cell data for the antibody staining and GFP-CDK2 reporter assays are extracted using gated data. Histograms of the raw data enable identification of suitable gate values. These are then imposed with a Perl script. (A) The end product of analyzing the image files with the provided settings for Cell Profiler are comma-separated-value (.csv) files. These files contain individual cell data relating to each of the different sub-cellular segments. The file ‘Nuclei.csv’ contains all the selected measurements relating to the use of the Nuclei mask. These measurements include nuclear antibody intensity, nuclear DNA intensity and the GFP ratio (nucleus/cytoplasm). (B) Histograms of nuclear antibody intensity (left) and GFP-CDK2 reporter ratios (right) plotted from individual cell data for each siRNA knockdown condition. The bars on the displayed histograms show the desired gate positions for these assays. Colored data on the histograms indicate the gated subpopulations. (C) The gates for the two assays illustrated in B are applied to the raw data using the Perl script ‘2_gate_classifier.pl’. The script creates a modified copy of the original Cell Profiler output (Nuclei.csv) file to assist subsequent plotting. The two gate values are recorded in the new file (highlighted in color here) and a new ‘Label’ column is added. The labels bin each cell into one of four possible subgroups based on the two gated assay values for each cell. These labels are used in subsequent plots which feature calculations of the contributions of each subpopulation as well as the cross-referencing of additional parameters generated in Cell Profiler. Please click here to view a larger version of this figure.
Figure 6: Data extraction - Processing raw individual cell data by imposing gates on assay values. Biological trends from the individual cell data for the antibody staining and GFP-CDK2 reporter assays are extracted using gated data. Histograms of the raw data enable identification of suitable gate values. These are then imposed with a Perl script. (A) The end product of analyzing the image files with the provided settings for Cell Profiler are comma-separated-value (.csv) files. These files contain individual cell data relating to each of the different sub-cellular segments. The file ‘Nuclei.csv’ contains all the selected measurements relating to the use of the Nuclei mask. These measurements include nuclear antibody intensity, nuclear DNA intensity and the GFP ratio (nucleus/cytoplasm). (B) Histograms of nuclear antibody intensity (left) and GFP-CDK2 reporter ratios (right) plotted from individual cell data for each siRNA knockdown condition. The bars on the displayed histograms show the desired gate positions for these assays. Colored data on the histograms indicate the gated subpopulations. (C) The gates for the two assays illustrated in B are applied to the raw data using the Perl script ‘2_gate_classifier.pl’. The script creates a modified copy of the original Cell Profiler output (Nuclei.csv) file to assist subsequent plotting. The two gate values are recorded in the new file (highlighted in color here) and a new ‘Label’ column is added. The labels bin each cell into one of four possible subgroups based on the two gated assay values for each cell. These labels are used in subsequent plots which feature calculations of the contributions of each subpopulation as well as the cross-referencing of additional parameters generated in Cell Profiler. Please click here to view a larger version of this figure.
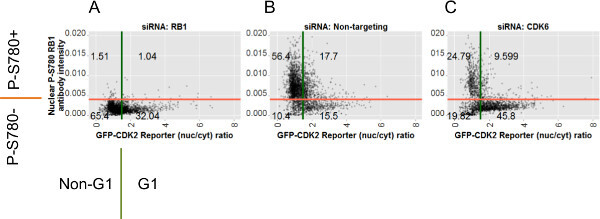 Figure 7: Scatter plots for each siRNA condition depicting raw data for individual cells and gate positions. Scatter plots of individual cell data from all images for the siRNA conditions indicated: (A) siRB1; (B) siNon-targeting negative control; (C) siCDK6. Plotted against the Y-axes are values of nuclear fluorescence from anti-P-S780 RB1 staining. Plotted against the X-axes are the corresponding ratio values calculated from the GFP-CDK2 reporter. The red and green bars indicate the positions of the gates for the P-S780 RB1 gate and the GFP-CDK2 reporter gates, respectively. The two gates divide the cells into four subpopulations and the numbers over the resulting quadrants are the percent number of cells from each of these. Annotations around the axes for A indicate the four possible label-elements applied to each cell by the 2_gate_classifier.pl Perl script. These labels are shown in relation to their respective assay gate and are used in the R-script (analysis.r) to generate the plots in Figures 6, 7 and 8. Please click here to view a larger version of this figure.
Figure 7: Scatter plots for each siRNA condition depicting raw data for individual cells and gate positions. Scatter plots of individual cell data from all images for the siRNA conditions indicated: (A) siRB1; (B) siNon-targeting negative control; (C) siCDK6. Plotted against the Y-axes are values of nuclear fluorescence from anti-P-S780 RB1 staining. Plotted against the X-axes are the corresponding ratio values calculated from the GFP-CDK2 reporter. The red and green bars indicate the positions of the gates for the P-S780 RB1 gate and the GFP-CDK2 reporter gates, respectively. The two gates divide the cells into four subpopulations and the numbers over the resulting quadrants are the percent number of cells from each of these. Annotations around the axes for A indicate the four possible label-elements applied to each cell by the 2_gate_classifier.pl Perl script. These labels are shown in relation to their respective assay gate and are used in the R-script (analysis.r) to generate the plots in Figures 6, 7 and 8. Please click here to view a larger version of this figure.
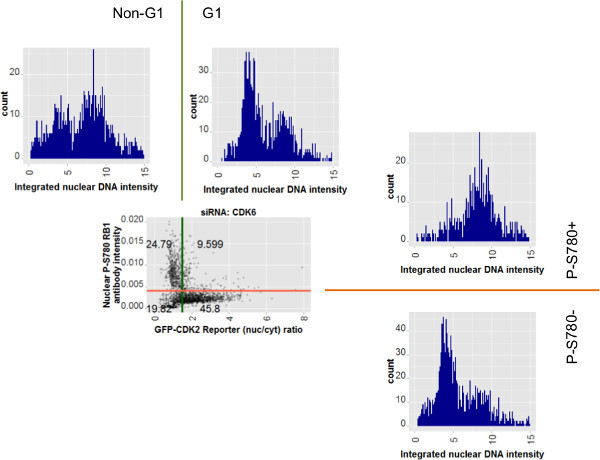 Figure 8: Cell subpopulations defined by the two G1 transit assays show 2N and 4N DNA profiles in keeping with assay outcome. The scatter plot of data for the siCDK6 cells is repeated from Figure 7C. Surrounding the scatter plot are histograms for integrated nuclear DNA intensity relating to subsets of the population. Those above the scatterplot relate to the GFP-CDK2 reporter assay. Those to the right of the scatterplot relate to nuclear phospho-RB1 antibody measurements alone. The colored gate lines are extended to show their relation to the histograms. Gate labels by which the cell data were selected for these additional plots are also shown. Cells with loss of RB1 phosphorylated on serine 780 (P-S780-) or those with a high GFP-CDK2 reporter nuclear to cytoplasmic ratio (indicating low CDK2 activity) show predominantly 2N-like DNA profiles, whereas their opposite counterparts for each respective assay show a distribution of 2N and 4N, characteristic of a mixed, post-G1 phase population of cells. Please click here to view a larger version of this figure.
Figure 8: Cell subpopulations defined by the two G1 transit assays show 2N and 4N DNA profiles in keeping with assay outcome. The scatter plot of data for the siCDK6 cells is repeated from Figure 7C. Surrounding the scatter plot are histograms for integrated nuclear DNA intensity relating to subsets of the population. Those above the scatterplot relate to the GFP-CDK2 reporter assay. Those to the right of the scatterplot relate to nuclear phospho-RB1 antibody measurements alone. The colored gate lines are extended to show their relation to the histograms. Gate labels by which the cell data were selected for these additional plots are also shown. Cells with loss of RB1 phosphorylated on serine 780 (P-S780-) or those with a high GFP-CDK2 reporter nuclear to cytoplasmic ratio (indicating low CDK2 activity) show predominantly 2N-like DNA profiles, whereas their opposite counterparts for each respective assay show a distribution of 2N and 4N, characteristic of a mixed, post-G1 phase population of cells. Please click here to view a larger version of this figure.
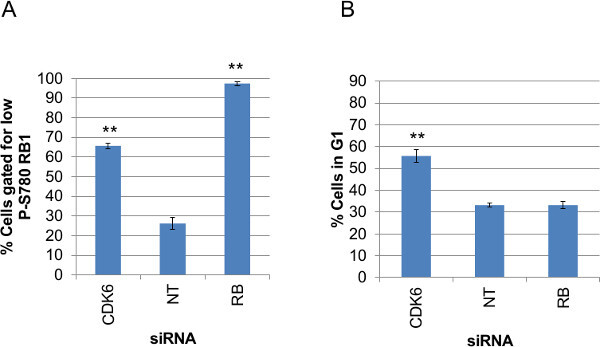 Figure 9: Summary plots of gated assay values for each siRNA condition. Summary data plots of gated (A) P-S780 RB1 data and (B) GFP-CDK2 data from triplicate wells for each siRNA knockdown condition. Values were calculated from the raw Cell Profiler output (Nuclei.csv) using the Perl scripts, ‘antibody_fluorescence_summary.pl’ (A) or ‘G1assay_summary.pl’ (B). The values plotted are means of the percent cells within the gate applied to each assay. Bars indicate standard errors calculated from triplicate wells. Unpaired, homoscedastic T-Test P values for each knockdown condition compared to non-targeting siRNA are shown above the plotted data where P < 0.001 (**) and P < 0.05 (*). Please click here to view a larger version of this figure.
Figure 9: Summary plots of gated assay values for each siRNA condition. Summary data plots of gated (A) P-S780 RB1 data and (B) GFP-CDK2 data from triplicate wells for each siRNA knockdown condition. Values were calculated from the raw Cell Profiler output (Nuclei.csv) using the Perl scripts, ‘antibody_fluorescence_summary.pl’ (A) or ‘G1assay_summary.pl’ (B). The values plotted are means of the percent cells within the gate applied to each assay. Bars indicate standard errors calculated from triplicate wells. Unpaired, homoscedastic T-Test P values for each knockdown condition compared to non-targeting siRNA are shown above the plotted data where P < 0.001 (**) and P < 0.05 (*). Please click here to view a larger version of this figure.
Figure S1. Setting up Cell Profiler software for image analysis. (A) Screenshot of Cell Profiler before any image analysis settings are entered. (B) Screenshot of Cell Profiler after the algorithm details contained in ‘3_channels_pipeline.cppipe’ have been loaded. The highlighted tab in the upper left corner indicates that this screen shows the parameters for the ‘LoadImages’ stage of the analysis. Clicking on the other parts of the list below this will reveal the details for the subsequent steps in the analysis. (C) Screenshot of Cell Profiler with details for Input Folder and Output Folder entered. (D) Screenshot of Cell Profiler after the ‘Analyze images’ button has been clicked to begin analysis. Superimposed are three new windows illustrating the algorithmically-produced masks generated by the software from the images under analysis. These windows are accessed by clicking the ‘eye’ icons to the open position next to the relevant steps in the analysis, in the upper left corner of the main Cell Profiler window. These views help the user to verify whether the settings generating the confetti-colored masks agree with the accompanying, original, greyscale data.
Figure S2. Use of Perl and RStudio to gate individual cell data and plot the resulting cell subpopulations. (A) The right panel shows the folder chosen to receive the output .csv files (green icons) from the Cell Profiler analysis. The Perl scripts provided with the manuscript (blue icons) are copied into this folder. Highlighted is the ‘2_gate_classifier.pl’ Perl script, which has been double-clicked with the mouse to produce the dialogue box in the left panel. Shown are the prompts and corresponding typed answers necessary to gate the individual cell data from the ‘Nuclei.csv’ file. (B) Screenshot of RStudio immediately after loading the ‘analysis.R’ script. Highlighted are the commands to upload the gated data from A into the software prior to plotting (note details in lines 5 and 6 will need to be adjusted according to where the gated data is located on the computer used for analysis). (C) Screenshot of RStudio once data has been uploaded. (D) Screenshot of RStudio showing highlighted the block of code required to produce the plot shown in the lower right window. Codes for each plot are separated by blank lines and grouped by type of plot.
| siRNA target | Plate well addresses |
| Non-targeting (NT) | E5, F5, G5 |
| Retinoblastoma (RB) | E7, F7, G7 |
| Cyclin dependent kinase 6 (CDK6) | B2, C2, D2 |
Table 1: Well addresses and corresponding siRNA conditions used in the example data set.
Discussion
The workflow described constitutes a procedure for multiwell perturbance of cells using siRNA, subsequent marker detection and finally use of a series of software-supported steps to facilitate extraction of quantitative data from the resulting fluorescent microscopy images. The approach is focused on the delivery of nuclear and cytoplasmic intensity values for individual cells, which has broad practical application in many cell-based applications. The example data used here was generated in an siRNA screen setting in which two fluorescent assays for G1 phase cell cycle transit are tested and correlated back to a more direct biophysical measure of nuclear DNA content.
The use of a fluorescent DNA stain to image nuclear DNA is an indispensable step in the image segmentation process as it allows identification of individual cells and the resulting ‘Nuclei mask’ serves as the starting point to identify corresponding cytoplasmic regions. The GFP-tagged CDK2 reporter, which is stably expressed in the cells, gives a variable yet consistently higher than background signal in the cytoplasm by which this compartment can be delineated. The same analysis pipeline should be applicable to the analysis of protein translocation events using other suitable fluorescence-linked reporters and their response to perturbance. Also, substituting the GFP-CDK2 reporter with cytoplasm-specific fluorescent dyes would allow the alternative use of this algorithm to measure the dimensions of the cytoplasm and the relative sizes of the cells in the images.
Another design consideration in the image segmentation strategy described here is the use of Cell Profiler to deliver integrated intensity values for the DNA quantification. Integration of the intensity values for the nuclear DNA staining data allow for possible variations in nucleus size, and represents a close match for the quantification profiles seen for propidium iodide stained FACS data. However, integrated intensity may not provide an appropriate means to assess protein function where average concentration, exemplified by mean intensity of antigen fluorescence, is more biologically relevant than the integrated total amount of protein (and associated fluorescence) within a cell compartment. Therefore mean intensity values were used for the P-S780 RB1 and GFP data. The option to alter between the two modes (mean or integrated) of data assessment is found on the ‘ExportToSpreadsheet’ panel of the Cell Profiler software.
The analysis settings in the 3_channels_pipeline.cppipe file are optimized for the images in the example data set. Analysis of new images sets with this protocol will require that the file names adopt the naming convention described above (Figure 3). Also, sensitivity values to suit the brightness of nuclear DNA staining and thresholds for background intensities in the new image sets may need to be adjusted within the Cell Profiler settings. Given the key role the DNA staining holds for building the various image segmentation masks, the application of correct sensitivity settings for this channel is key to the successful analysis of new image data with the Cell Profiler software. The provided Cell Profiler settings file (3_channels_pipeline.cppipe) contains notes on the most frequently useful parameters for adapting the analysis to new data. These notes are in the text box at the top of the screen in the Cell Profiler main window and include guidance on changing the sensitivity settings and adjusting the number of channels to be analyzed. As indicted in Protocol section 2.8, to test settings for new image data it may be necessary to observe the image segmentation during image analysis by clicking open the ‘eye’ icons for each of the ‘Identify…Objects’ protocol steps (Figure S1D). In particular, visualization of the image data through ‘IdentifyPrimaryOjects’ will show if the Nuclei mask is correctly identified from images of the DNA staining. On the Cell Profiler software page for the ‘IdentifyPrimaryOjects’ module is the threshold correction factor. Trial and error adjustment of this value will fix most nuclear recognition errors. The values balance the DNA channel against the background intensity for each image. Threshold correction factor values hinge around 1, where greater than this is more stringent (good for clear images) and less than 1 is lenient (suited to images with less contrast between staining and background).
The raw output of individual cell data from Cell Profiler can be analyzed in varying ways to suit the needs of other studies. Shown here is the use of a Perl script to apply gates to two of the parameters measured per cell in order to assist extracting biological trends from the data and permit cross-referencing of the identified subpopulations with additional measurements. Although it is equally possible to include elements of gating within the framework of Cell Profiler, the alternative route used here provides greater flexibility and speed, specifically if large data sets need to be assessed. The slowest stage in the post-image acquisition phases of the current protocol is the running of the Cell Profiler software. Cell profiler here is run without imposing gates to produce an un-gated raw data set which can be reanalyzed with the subsequent Perl script more quickly and, if needed, iteratively with different gate values. Not all studies will know in advance the suitable gate values as this may vary with reagents on any given set of data, and potentially over time. It is, therefore, recommended to generate histograms depicting the raw data distribution obtained from Cell Profiler for positive controls and mock-perturbed cells in order to identify suitable gate values for the parameters of interest.
The Perl scripts are written to accept a rigidly defined column structure of data from Cell Profiler and may stop working if a user modifies the number of parameters output by Cell Profiler using the ‘ExportToSpreadsheet’ settings. To help implement modification of the settings notes are included within the Perl script files. To see these view the script in a text editor, preferably a programmer’s text editor set to color code Perl elements (e.g., http://www.activestate.com/komodo-edit). These notes indicate where to adjust the script to adapt to changes in data format. Similar to the Perl scripts, the R-code file provided (analysis.r), containing the instructions for plotting the figures from the image analysis data, can be read in a text editor or RStudio software to see additional notes on use and adaptation. These notes can be supplemented with details on regular expressions and Perl12 and the ggplot213 package for R, both of which form the basis for how the data is read, annotated and plotted, respectively.
New studies using fluorescence microscopy as well as raw data deposited with open source publications are amenable to methods of analysis such as those described here. The very nature of high-content data lends itself to recursive analysis with different analytic emphases depending on the research interests of any given observer. Although the questions that can be asked of the data are limited by the probes originally used, image data can often be meaningfully reanalyzed beyond the scope of the studies which generated them.
Disclosures
The authors have nothing to disclose
Acknowledgments
This work was supported by grants CRUK 15043 and CRUK 14251.
We thank Daniel Wetterskog and Ka Kei Ho for technical assistance and critical reading of the manuscript.
References
- Carpenter AE, et al. CellProfiler: image analysis software for identifying and quantifying cell phenotypes. Genome Biol. 2006;7(10):100. doi: 10.1186/gb-2006-7-10-r100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan A, Eldaly H, Rajpoot N. A gamma-gaussian mixture model for detection of mitotic cells in breast cancer histopathology images. J Pathol Inform. 2013;4:11. doi: 10.4103/2153-3539.112696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selzer P, Beibel M, Gubler H, Parker CN, Gabriel D. Comparison of multivariate data analysis strategies for high-content screening. J Biomol Screen. 2011;16(3):338–347. doi: 10.1177/1087057110395390. [DOI] [PubMed] [Google Scholar]
- Lyman SK, et al. High content, high-throughput analysis of cell cycle perturbations induced by the HSP90 inhibitor XL888. PLoS One. 2011;6(3):e17692. doi: 10.1371/journal.pone.0017692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richardson E, Stockwell SR, Li H, Aherne W, Cuomo ME, Mittnacht S. Mechanism-based screen establishes signalling framework for DNA damage-associated G1 checkpoint response. PLoS One. 2012;7(2):e17692. doi: 10.1371/journal.pone.0031627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heynen-Genel S, Pache L, Chanda SK, Rosen J. Functional genomic and high-content screening for target discovery and deconvolution. Expert Opin Drug Discov. 2012;7(10):955–968. doi: 10.1517/17460441.2012.711311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krausz E. High-content siRNA screening. Mol Biosyst. 2007;3(4):232–240. doi: 10.1039/b616187c. [DOI] [PubMed] [Google Scholar]
- Gu J, Xia X, et al. Cell Cycle-dependent Regulation of a Human DNA Helicase That Localizes in. DNA Damage Foci. Mol Biol Cell. 2004;15(7):3320–3332. doi: 10.1091/mbc.E04-03-0227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mittnacht S. Control of pRB phosphorylation. Curr Opin Genet Dev. 1998;8(1):21–27. doi: 10.1016/s0959-437x(98)80057-9. [DOI] [PubMed] [Google Scholar]
- Mittnacht S. The retinoblastoma protein--from bench to bedside. Eur J Cell Biol. 2005;84(2-3):97–107. doi: 10.1016/j.ejcb.2004.12.012. [DOI] [PubMed] [Google Scholar]
- Nybo K. GFP imaging in fixed cells. BioTechniques. 2012;52(6):359–360. doi: 10.2144/000113872. [DOI] [PubMed] [Google Scholar]
- Schwartz RL, Foy BD, Phoenix T. Learning Perl - Making Easy Things Easy and Hard Things Possible. 6th ed. O'Reilly Media; 2011. pp. 1–363. [Google Scholar]
- Wickham H. ggplot2: Elegant Graphics for Data Analysis. New York: Springer; 2009. pp. 978–970. [Google Scholar]