

Abstract
Background
Genetic amplification of HER2 drives tumorigenesis and cancer progression in a subset of patients with gastric cancer (GC), and treatment with trastuzumab, a humanized HER2-neutralizing antibody, improves the overall survival rate of HER2-positive patients. However, a considerable portion of the patients does not respond to trastuzumab and the molecular mechanisms underlying the intrinsic resistance to anti-HER2 therapy in GC is not fully understood.
Methods
We performed whole-transcriptome sequencing on 21 HER2-positive tumor specimens from Chinese GC patients. Whole genome sequencing was performed on the three samples with HER2 fusion to discover the DNA integration structure. A multicolor FISH assay for HER2 split screening was conducted to confirm HER2 fusion and IHC (HercepTest™) was used to detect the membranous expression of HER2. Fusion cDNA were transfected into NIH/3T3 cells and generate stable cell line by lentivirus. The expression of exogenous HER2 fusion proteins and pHER2 were examined by western blot analysis. In vitro efficacy studies were also conducted by PD assay and softagar assay in cell line expression wild type and fusion HER2. T-DM1 was used to assess its binding to NIH/3T3 cells ectopically expressing wild-type and fusion HER2. Finally, the anti-tumor efficacy of trastuzumab was tested in NIH/3 T3 xenografts expressing the HER2 fusion variants.
Results
We identified three new HER2 fusions with ZNF207, MDK, or NOS2 in 21 HER2-amplified GC samples (14%; 3/21). Two of the fusions, ZNF207-HER2, and MDK-HER2, which are oncogenic, lead to aberrant activation of HER2 kinase. Treatment with trastuzumab inhibited tumor growth significantly in xenografts expressing MDK-HER2 fusion. In contrast, trastuzumab had no effect on the growth of xenografts expressing ZNF207-HER2 fusion, due to its inability to bind to trastuzumab.
Conclusions
Our results provide the molecular basis of a novel resistance mechanism to trastuzumab-based anti-HER2 therapy, supporting additional molecule stratification within HER2-positive GC patients for more effective therapy options.
Electronic supplementary material
The online version of this article (doi:10.1186/s12967-015-0476-2) contains supplementary material, which is available to authorized users.
Keywords: HER2, Fusion-gene, Gastric cancer, Trastuzumab, Lapatinib
Background
Gastric cancer (GC), as the second leading cause of cancer deaths worldwide, accounted for 989,600 new cases and 738,000 deaths globally in 2011 [1]; more than 50% of GC cases occur in Eastern Asia [2]. The conventional treatments for GC include surgery, radiotherapy and chemotherapy [3], which have limited efficacy because most GC patients are in the advanced stages when diagnosed; the five-year survival rate for patients with stage III/IV GC is around 10% [4]. Trastuzumab, a neutralization antibody of HER2, was recently approved for the treatment of a subset of advanced GC patients whose tumors are clinically defined as HER2-positive.
HER2 gene amplification was initially discovered as an oncogene in breast cancer (BC), which led to the development of HER2-targeted therapeutics for treating HER2-positive BC [5]. These drugs include trastuzumab; lapatinib, a small-molecular inhibitor of HER2 kinase; pertuzumab, an antibody-blocking heterodimerization of HER2 with HER3; and trastuzumab emtansine (T-DM1), which is trastuzumab conjugated with the antimitotic agent emtansine (DM1). The clinical application of these targeted agents dramatically changed the landscape of BC therapy and exemplified a new era of personalized medicine associated with companion molecular diagnosis for patient selection [6-8]. In addition to BC, HER2 amplification and overexpression was also found in about 20% of GC patients [9]. The anti-tumor activity of trastuzumab as a single agent or in combination with cytotoxic agents has been demonstrated in several HER2-positive human GC cell lines in vitro and in GC xenografts in vivo [10-12]. The preclinical efficacy translated into positive clinical trials in which a survival improvement was achieved in HER2-positive metastatic GC patients treated with trastuzumab plus cytotoxic agents [9,13]. These results led to the approval of trastuzumab as the first molecular targeted therapy for treating GC.
Despite the clinical benefits of trastuzumab in the treatment of patients with HER2-positive GC or BC [13,14], approximately 30-40% of HER2-positive tumors are insensitive to the treatment. Significant efforts to understand the resistance to anti-HER2 therapy in BC cases have recently been made, resulting in a diverse array of resistance mechanisms and clinical strategies to overcome the resistance [15]. However, there is little understanding of the resistance mechanism to anti-HER2 therapy in GC. Therefore, we used a next-generation sequencing (NGS) approach to elucidate molecular insights in HER2-positive GC. In this study, for the first time, we report three HER2 gene fusions in HER2-positive GC in Chinese patients, and we characterize their oncogenic properties and sensitivity to anti-HER2 agents.
Methods
Human primary tumor samples
Specimens were collected during surgery from Chinese GC patients with postoperative pathological confirmation. The study was carried out at Peking University Cancer Hospital and Institute, and Shanghai Renji Hospital (2007 ~ 2010). Written informed consent was provided by each patient, and the study was approved by the ethics committees of the hospitals.
RNA-seq for transcriptome analysis
Total RNA was extracted using TRIzol (Life Technologies). All RNA samples showed RNA integrity numbers >7 (Agilent 2100 bioanalyzer).
Total RNA quality and concentration was measured using an RNA Pico chip on a Bioanalyzer 2100 (Agilent). Normalized starting quantities of total RNA were used to prepare Illumina sequencing libraries with a TruSeq™ RNA sample preparation kit (Illumina). The library preparation was performed according to the manufacturer’s instructions. The cDNA libraries were placed on an Illumina c-Bot for paired-end (PE) cluster generation, according to the protocol outlined in the Illumina HiSeq Analysis User Guide. The template cDNA libraries (1.5 μg) were hybridized to a flow cell, amplified, linearized, and denatured to create a flow cell with ssDNA ready for sequencing. Each flow cell was sequenced on an Illumina HiSeq2000 sequencing system. After a 100-cycle PE sequencing run, the bases and quality values were generated for each read with the current Illumina pipeline.
Detection of fusion transcripts
We sequenced each tumor sample up to an average of about 150× coverage. Fusion transcripts were detected using FusionMap software [16]. Fusions supported by at least three reads were selected as candidates and subjected to RT-PCR and Sanger sequencing confirmation.
Quantification of mRNA expression level
Human gene expression quantification was measured according to sequenced fragments (reads) per kilobase of exon per million fragments mapped to the human genome (FPKM):
N: number of reads mapped in gene
L: gene length (bp) (intron excluded)
R: number of raw reads
RT-PCR and Sanger sequencing
First strand cDNA synthesis was performed with 0.5 μg total RNA using a High Capacity cDNA Reverse Transcription kit (Life Technologies) according to the manufacturer’s instructions. PCR was performed in a 25-μL reaction mix containing 1× AmpliTaq Gold® 360 Master Mix (Life Technologies), 200 μM of each primer, and 2 μL of cDNA. The PCR cycling conditions were: 10-min incubation at 95°C, followed by 40 cycles of 94°C for 30 s, 60°C for 30 s, 72°C for 60 s, and a final incubation at 72°C for 10 min. The resulting PCR products were digested with ExoSAP-IT reagent (Affymetrix, Cleveland, OH) and then sequenced in forward and reverse directions with a BigDye Terminator Kit (Life Technologies) and an ABI 3730XL DNA analyzer (Life Technologies), following the manufacturer’s instructions. The sequencing data were analyzed for mutations after assembly and quality calling with SeqScape sequence analysis software (version 2.5; Life Technologies). The RT-PCR primers used for fusion gene confirmation were: 1) ZNF207/HER2, Forward: 5'-CTGAAGCCGTGGTGCTGGTATTGTA-3', Reverse: 5'-TGGGCATGTAGGAGAGGTCAGGTTT-3'; 2) MDK/HER2, Forward: 5'-GTTTGAGAACTGGGGTGCGTGTGAT-3', Reverse: 5'-AGACCATAGCACACT CGGGCACA-3'; 3) NOS2/HER2, Forward: 5’-CAAGCCCCACAGTGAAGAACATCTG-3', Reverse: 5'-TGCTGGAGGTAGAGTGGTGAACAGG-3'.
Whole genome sequencing
DNA was extracted from the frozen tissues using a Puregene DNA extraction kit (Qiagen) and quantified using a PicoGreen fluorescence assay (Qubit; Invitrogen). To conduct whole genome sequencing, 2 μg of DNA were required for each sample. After electrophoresis, DNA fragments of the desired length were gel purified. Adapter ligation and DNA cluster preparation were performed and subjected to Illumina Hiseq2000 sequencing. Two paired-end libraries with an insert size of 500 bp were prepared for all samples, after which four lanes from each library were subjected to whole genome sequencing. Raw image files were processed by Illumina Pipeline for base calling with default parameters, and the sequences of each individual were generated as 90-bp paired-end reads. Raw sequence data was mapped to the reference human genome (hg19) using Bowtie 2. The total mapping rate was >90%, and the average coverage was about 30×. Unmapped reads were then used to conduct genomic fusion detection with FusionMap [16].
Analysis of HER2, BRAF, KRAS, or PI3K mutations
HER2, BRAF, KRAS, and PIK3CA gene mutations from the RNAseq data were analyzed using ArrayStudio software (http://www.omicsoft.com/array-studio.php). Allele frequencies below 10% were removed in case of potential false positive. The mutation status was further confirmed by the whole genome sequencing data.
Immunohistochemistry (IHC)
The primary antibodies used to detect the cytoplasmic domain of HER2 were purchased from Merck and Abcam, and the antibody used to detect the external domain of HER2 was purchased from Abnova. All of the collected tissues were fixed in FFPE blocks. Xenograft and cell-block sections were cut at 3 μm and human sections were cut at 4 μm for the HER2 IHC study. Paraffin sections were dewaxed and rehydrated in a Leica XL autostainer. Following antigen retrieval, the sections were incubated with 10 min of endogenous peroxidase block (DAKO), 60 min of primary antibodies, 30 min of EnVision System-HRP labeled polymer anti-mouse (DAKO), and 10 min of diaminobenzidine substrate (DAKO K3468), in that order. Finally, the sections were counter-stained, dehydrated, cleared, and mounted with coverslips in a Leica XL autostainer workstation. A HercepTest™ (DAKO) was used to detect the membranous expression of HER2, following standard procedures. Each slide was evaluated and scored on a 0–3 scale, following uniform guidelines developed for GC HER2 scoring from ToGA trials [13].
Fluorescent in situ hybridization (FISH)
A multicolor FISH assay for HER2 split screening was conducted via a dual-probe FISH break-apart test. The N-terminal and C-terminal probes for HER2 were generated internally by directly labeling BAC (N-terminal: RP11-98 J2; C-terminal: RP11-1044P23) DNA respectively with Green-dUTP (ENZO, Cat # 02 N32-050) and Red-dUTP (ENZO, Cat #02 N34-050). A CEP17 spectrum aqua probe (Vysis, Cat #32-131017) for the centromeric region of chromosome 17 was used as an internal control of the HER2 break-apart probes. Multicolor FISH was also used to confirm HER2 gene fusion with certain partner genes. The ZNF207 and NOS2 FISH probes were generated internally by directly labeling BAC (ZNF207: RP11-55 J8; NOS2: RP11-696H14) DNA with gold 525-dUTP (ENZO, Cat #ENZ42843). The FISH assays were performed as previous reported [17]. Briefly, the assay was run on 4-μm dewaxed and dehydrated FFPE TMAs. A SPoT-Light tissue pretreatment Kit (Invitrogen, Cat #00-8401) was used for the pretreatment (boiled in reagent 1 for ~18 minutes, then coated with reagent 2 for ~14 minutes, minor time adjustments were made for individual samples). The sections and probes were co-denaturated at 79°C for 6 minutes and then hybridized at 37°C for 48 hours. After a quick post wash off process (0.3% NP40/2xSSC at 75.5°C for 2 minutes, twice in 2 × SSC at room temperature for 2 minutes), the sections were mounted with 0.3 μg/ml DAPI (Vector, Cat #H-1200) and stored at 4°C, avoiding light for at least 30 minutes prior to observation. The FISH signals were observed using a fluorescence microscope equipped with the appropriate filters, allowing visualization of the intense red/green/gold signals of the target genes, the intense aqua centromere signals, and the blue counterstained nuclei. A minimum 100 nuclei were scored for each sample. Only nuclei with a minimum of two green and two red signals were scored. In the break-apart assay, fused N-terminal (green) and C-terminal (red) signals represent a normal HER2 gene. HER2 amplification status was defined as a ratio of fused HER2 signals to CEP17 (aqua) ≥2. The following situations indicated the presence of HER2-involved fusion: 1) broken apart: more than one set of broken-apart N-terminal and C-terminal signals in ≥10% tumor cells; 2) N-terminal deletion: more C-terminal signals in addition to fused and/or broken-apart signals in ≥30% tumor cells. Then, a multicolor FISH assay was performed on HER2 broken apart or N-terminal-deleted positive cases. The HER2 fusion was confirmed when HER2 C-terminal red signals co-localized with a certain partner gene’s gold signals.
Vector construction, cell culture, transduction, and transformation studies
ZNF207-HER2 and MDK-HER2 fusion cDNA were synthesized (Additional file 1: Files S1 and S2) at Generay (Shanghai, China). The products were subcloned into PLVX lentiviral vector (Sunbio, China). The integrity of the inserted cDNA was verified by Sanger sequencing of the constructs. Lentiviruses expressing ZNF207-HER2 and MDK-HER2 fusions were produced according to the manufacturer’s instructions. NIH/3 T3 fibroblast cells were infected with lentiviruses expressing empty vector, and ZNF207-HER2 and MDK-HER2 fusions were treated with puromycin (2 μg/ml) for two weeks. NIH/3 T3-resistant cells were seeded on 96-well plates (2000 cells/well) in 0.33% agar in complete medium. The expression of exogenous HER2, ZNF207-HER2, and MDK-HER2 proteins and phosphorylation of HER2 were examined by immune blot analysis. The overnight cell cultures in liquid or soft agar medium were treated with lapatinib, T-DM1 (synthesized at ChemPartner, Shanghai, China) for 3 and 14 days, respectively. The cell growth rate was measured by MTS assay according to the manufacturer’s instructions (Promega).
Immunoblot analysis
Total cellular extracts from the cell lines were prepared in an SDS lysis buffer supplemented with protease inhibitors and phosphatase inhibitors (Sigma). Protein samples were fractionated by SDS-PAGE and blotted onto polyvinylidene difluoride membranes (Millipore). After incubation with the indicated antibodies at 4°C overnight, the blots were detected with the relevant horseradish peroxidase-conjugated anti-mouse or anti-rabbit IgG antibody and enhanced chemilluminescence (GE Healthcare). The antibody information used in the Western blot assays are included were as follows: pHER2 (Y1221/1222) (CST, Cat #2243, diluted 1:1000); HER2 (CST, Cat #2165, diluted 1:1000); pErk1/2 (T202/Y204) (CST, Cat #4376, diluted 1:1000); Erk1/2 (CST, Cat #9102, diluted 1:1000); pAKT (Ser473) (CST, Cat# 9271, diluted 1:1000); and AKT (CST, Cat #9272, diluted 1:1000).
Trastuzumab emtansine (T-DM1) receptor binding assay
Approximately 5 × 105 NIH/3T3 cells expressing vector control, wild-type HER2, ZNF207-HER2, or MDK-HER2 were collected using enzyme-free cell dissociation buffer (Invitrogen). After blocking with 10% donkey serum for 30 minutes at 4°C, the cells were incubated with 10 μg/mL T-DM1 (ChemPartner) at 4°C for one hour. The cells were rinsed three times with wash buffer (0.5% BSA in PBS) and further incubated with 10 ug/mL Alex488 labeled donkey anti-human IgG (Jackson immunology) at 4°C for one hour. After rinsing five times with wash buffer, the mean intensity of the fluorescence was detected by FACSCanto (BD). The receptor binding was qualitatively evaluated by the peak shift in the histogram.
In vivo efficacy study in xenograft models
6- to 8-week-old female nude (nu/nu) mice (Vital River, Beijing, China) were used for in vivo efficacy studies. All experiments using immunodeficient mice were carried out in accordance with the guidelines approved by the Institutional Animal Care and Use Committees. NIH/3 T3 cells expressing ZNF207-HER2, MDK-HER2, or a control vector were inoculated subcutaneously into female nude mice. Tumor-bearing mice with tumors ranging 100–200 mm3 in size were selected randomly and placed in groups according to their tumor volume and body weight (eight animals per group) for treatment. Trastuzumab (15 mg/kg) was administrated by intravenous injection twice a week. The xenograft tumors were measured in two perpendicular diameters with a caliper, and tumor volumes (TV) were calculated using the formula TV = (length × width2)/2. Percentage of tumor growth inhibition (%TGI) was calculated using the formula [1-(change of tumor volume in treatment group/change of tumor volume in control group)] × 100, and was used to evaluate anti-tumor efficacy. Student’s t tests were used to compare the TGI of the treatment group with that of the control group. Statistical tests were two sided, with P < 0.05 considered significant.
Results
Novel HER2 fusion genes identified in HER2-positive gastric cancer
To uncover the genetic aberrations that might confer resistance to trastuzumab treatment in GC, we performed whole-transcriptome sequencing (RNAseq) on 21 HER2-positive tumor specimens from Chinese GC patients whose tumors were surgically removed and who were treatment naïve, using the HiSeq2000 system (Illumina). A number of candidate fusion transcripts were identified with more than three chimerical reads, which were subsequently followed by RT-PCR/Sanger sequencing confirmation. This led to the identification of three HER2 (chr17q12) in-frame fusion transcripts with 5’ partners of ZNF207 (chr17q11.2), MDK (chr11p11.2), or NOS2 (chr17q11.2) in three HER2-positive GC samples of GC196, 431-9540474 T, and GC334, respectively (Figure 1A, B and Table 1).
Figure 1.
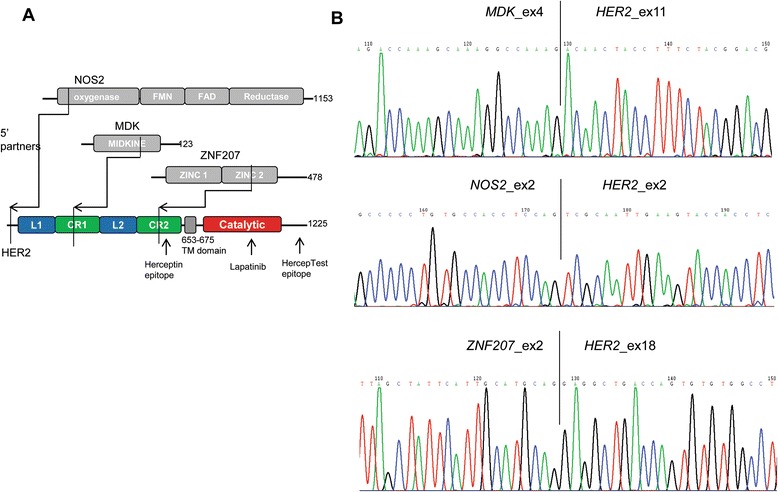
Identification of HER2 fusions in gastric cancer. A. Schematic illustration of the wild-type HER2 protein and the three fusions identified in this study. The breakpoints for each fusion are indicated by arrows. B. Sanger sequencing of the fusion junctions.
Table 1.
A summary of patients with gastric cancer harboring HER2 fusions
| Samples | Country | Sex | Age | Fusions* | TNM | Pathological type |
|---|---|---|---|---|---|---|
| GC196 | China | M | 76 | ZNF207_ex2/HER2_ex18 | T3N1M0 | Mixed |
| 431-9540474 T | China | M | 63 | MDK_ex4/HER2_ex11 | T2N2M0 | - |
| GC334 | China | F | 62 | NOS2_ex2/HER2_ex2 | T3N2M0 | Diffused |
*The amplification of MDK-HER2 was determined by aCGH, whereas the other two HER2 fusion amplifications were defined by FISH and IHC assays.
To understand the genomic alterations of these fusion transcripts, tumor DNA samples from the three corresponding GC patients were analyzed by whole genome sequencing. Consistent with the data from the RNAseq analysis, three HER2 fusion genes—ZNF207 (exon 1–2)/HER2 (exon 18–30), MDK (exon 1–4)/HER2 (exon 11–30), and NOS2 (exon 1–2)/HER2 (exon 2–30)—were further confirmed in these tumor DNA samples (Figure 2). These results demonstrate the existence of three novel HER2 gene fusions in this cohort of HER2-positiv GC patients.
Figure 2.
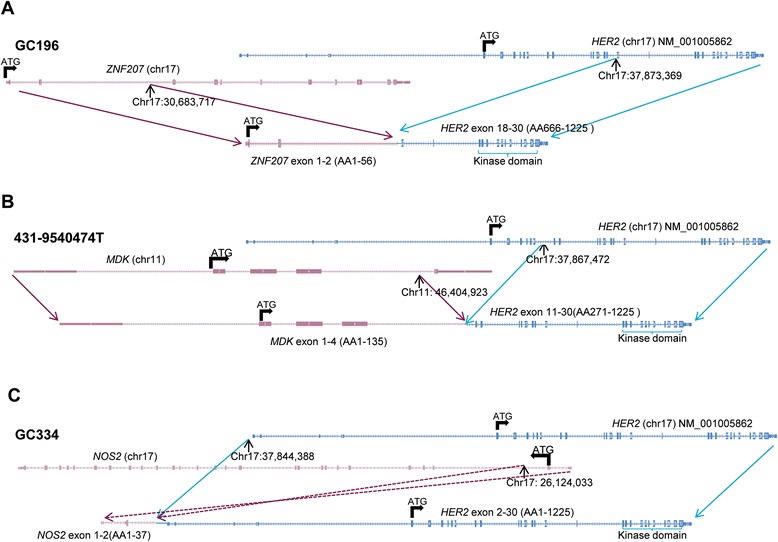
Schematic of genomic fusion structure of the HER2 fusion. A. Genomic fusion structure for sample GC196 harboring ZNF207-HER2 fusion B. Genomic fusion structure for sample 431-9540474 T harboring MDK-HER2 fusion C. Genomic fusion structure for sample GC334 harboring NOS2-HER2 fusion. The genomic structure was discovered by whole genome sequencing.
Amplification and overexpression of the HER2 fusions in GC
In the GC196 sample, over 100 reads were detected for ZNF207 genomic sequences only composing the 5’ partner and HER2 sequences only composing the 3’ partner of the ZNF207-HER2 fusion; low reads (~20) were captured for the genomic sequences outside the fusion gene. In the GC334 specimen, in addition to the high reads (>1800) of the fusion partners of NOS2-HER2 fusion that were detected, an average of 500 reads was also captured for the entire genomic sequence of wild-type HER2 gene.
Next, we assessed the relative mRNA expression levels of the three amplified fusion genes. Consistent with the whole genome sequencing data, high levels of mRNA expression of the three fusion variants were obtained in the three HER2-fusion-positive tumor specimens (GC196 FPKM 346, 431-9540474 T FPKM 1359, and GC334 FPKM 2805). In the tumor harboring ZNF207-HER2, the increased expression of HER2 mRNA was observed only after the fusion site, whereas the increased expression of ZNF207 mRNA was detected only before the fusion site, further confirming that the amplification of the ZNF207-HER2 fusion was a homogeneous event in the sample (Figure 3A). In contrast, in the tumor harboring MDK-HER2, overexpressed transcripts were detected for MDK-HER2 fusion, wild-type HER2, and wild-type MDK, consistent with the heterogeneity populations of amplified wild-type HER2 with MDK-HER2 (Figure 3B). A similar observation was made in the GC334 sample, in which overexpression of both NOS2-HER2 and wild-type HER2 were detected (Figure 3C).
Figure 3.
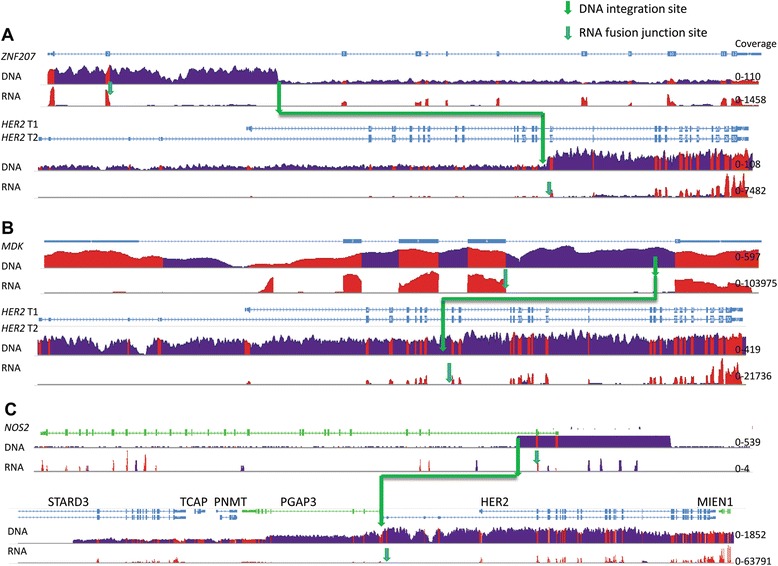
Illustration of DNA coverage and mRNA expression levels of HER2 and fusion partners. A. DNA coverage and mRNA expression levels of HER2 and ZNF207 in sample GC196. B. DNA coverage and mRNA expression levels of HER2 and MDK in sample 431-9540474 T C. DNA coverage and mRNA expression levels of HER2 region (including HER2 and nearby gene STARD3, TCAP, PNMT, PGAP3 and MIEN1) and fusion partner NOS2 in sample GC334. The DNA integration sites were marked in green arrows and the RNA fusion junction sites were marked in cyan arrows. Exon coverage was shown in red and intron coverage was shown in blue. The coverage range for each gene was given in the right side of the figure. Average coverage for WGS is 30X and average coverage for RNASeq is 150X.
Given that the tumor samples harboring the three fusions were all HER2-positive, we first developed a multicolor fluorescence in situ hybridization (FISH) assay to assess the genetic amplification status of the ZNF207-HER2 and NOS2-HER2 fusion genes, as well as to dissect their relationship to the wild-type HER2 gene in the GC196 and GC334 samples, respectively. Based on hematoxylin and eosin staining, both primary tumors with the two HER2 fusions were defined as adenocarcinoma by pathologists (Figure 4A). The multicolor FISH assay detected the N-terminal and C-terminal of the HER2 gene, along with the ZNF207 or NOS2 gene. As shown in Figure 4B, the ZNF207-HER2 fusion gene was amplified homogeneously without wild-type HER2 amplification in the GC196 sample. However, co-amplification of both NOS2-HER2 and wild-type HER2 were observed in the GC334 specimen, but in separate tumor cell populations (Figure 4C). The co-localization of 5’ partners and the C-terminus of HER2 gene by the FISH analysis not only further confirmed the genomic fusions of the involved genes, but also indicated the intratumoral heterogeneity within the HER2-amplified tumor. For the MDK-HER2 fusion, although a formalin-fixed and paraffin-embedded (FFPE) sample of 431-9540474 T was unavailable for the multicolor FISH analysis, the whole genome sequencing data suggested a mixture of tumor cell populations harboring gene amplifications of wild-type HER2, MDK-HER2, and wild-type MDK (Figure 3).
Figure 4.
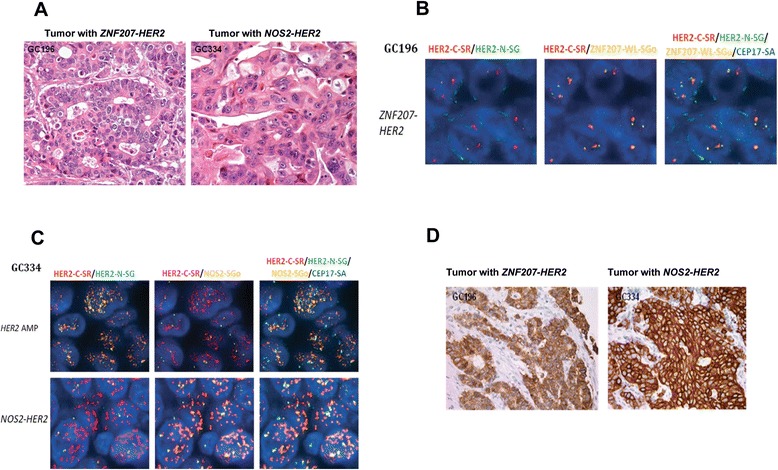
Amplification and over-expression of HER2 fusions in gastric tumors. A. Histological characterization of human gastric carcinomas. Representative images from H&E staining of GC196 and GC334 primary tumors. B. ZNF207-HER2 gene fusion in primary tumor GC196. Representative images of break-apart FISH on GC196 shows normal copy of HER2 N-terminal (green signals), coexistence of amplified HER2 C-terminal (red signals) and amplified ZNF207 (gold signals). Red and green signals represent C-terminal (HER2-C-SR) and N-terminal (HER2-N-SG) of HER2 gene, respectively; gold signals represent whole length of ZNF207 gene (ZNF207-WL-SGo); aqua signal represent CEP17 as internal control. Cell nuclei were counterstained with DAPI. C. NOS2-HER2 gene fusion in tumor GC334 primary tumor. Representative images of break-apart FISH on GC334 show NOS2-HER2 fusion with heterogeneity. The upper lane showed tumor cells with both HER2 N-terminal (green signals) and C-terminal (red signals) amplified and normal copy number of NOS2 (gold signals). The lower lane showed tumor cells with normal copy of HER2 N-terminal (green signals), coexistence of amplified HER2 C-terminal (red signals) and amplified NOS2 (gold signals). Red and green signals represent C-terminal and N-terminal of HER2 gene; gold signals represent NOS2 gene; aqua signal represent CEP17 as internal control. Cell nuclei were counterstained with DAPI. D. IHC analysis of HER2. Representative images showed HER-2 IHC strong membrane staining (+++) on GC196 and GC334 primary tumors.
Lastly, we performed immunohistochemistry (IHC) with a HercepTest™ in FFPE samples of GC196 and GC334. Strong staining (IHC 3+) was detected in both samples (Figure 4D), indicating an overexpression of the HER2 proteins. A summary of the HER2 fusion status was shown in Table 2.
Table 2.
A summary of the HER2 fusion status
| Sample ID | Fusion Type(RNA) | DNA structure | 5' fusion partner status | HER2 status | |||
|---|---|---|---|---|---|---|---|
| GC196 | ZNF207_exon2 /HER2_exon18 | ZNF207_intron2/HER2_intron17 | ZNF207 | mRNA expression | High | mRNA Expression | High |
| Gene copy | N Ter AMP | Gene Copy | C Ter AMP | ||||
| Protein level | - | Protein level | IHC 3+ | ||||
| 431-9540474 T | MDK_exon4 -/HER2_exon11 | MDK_intron4/HER2_intron10 | MDK | mRNA expression | High | mRNA Expression | High |
| Gene copy | tAMP | Gene Copy | tAMP | ||||
| Protein level | - | Protein level | - | ||||
| GC334 | NOS2_exon2 /HER2_exon2 | NOS2_intron2/HER2_5’UTR | NOS2 | mRNA expression | Low | mRNA Expression | High |
| Gene copy | N Ter AMP | Gene Copy | tAMP | ||||
| Protein level | - | Protein level | IHC 3+ | ||||
N Ter AMP: N terminal amplification.
C Ter AMP: C terminal amplification.
tAMP: total amplification.
Together, these data demonstrate the gene amplification of the HER2 fusions, which correlated with dis-regulation of both mRNA and protein expressions of the fusion variants. In addition, the HercepTest™ did not distinguish the fusion proteins from the wild-type HER2.
Oncogenic driver activity of the HER2 fusions
A number of genetic aberrations with oncogenic properties have been reported recently in GC cases [8]. Thus, we aimed to determine whether the HER2 fusions overlapped with the known oncogenic alterations. RNAseq and WGS data showed that the three HER2-fusion-positive GC patients were negative for HER3, BRAF, KRAS, PI3KCA, and HER2 mutations, and negative for amplifications of FGFR2 and cMET (data no shown), which are common genetic alterations identified in GC [18]. The mutually exclusive nature of HER2 fusions from these known oncogenic alterations suggests that the HER2 fusions are oncogenic drivers. In addition, sequence analysis revealed that proteins encoded by ZNF207-HER2 and MDK-HER2 fusion variants contained a partial extracellular domain, a transmembrane domain, and a full kinase domain of HER2 (Additional file 1: Files S1 and S2), whilst the predicated fusion protein of NOS2-HER2 would be a full-length HER2 protein as result of a stop codon that is introduced prior to the HER2 start codon (Additional file 1: File S3). Based on the sequence predication that NOS2-HER2 fusion encodes a full-length HER2 without NOS2, our efforts on function characterization of these HER2 fusion proteins were then focused on ZNF207-HER2 and MDK-HER2 fusions.
Given the presence of the HER2 dimerization domain [19,20], ZNF207-HER2 and MDK-HER2 fusion variants are likely to form homodimers in a manner similar to that of amplified wild-type HER2. Consistently, when MDK-HER2 and ZNF207-HER2 variants were ectopically expressed in NIH/3 T3 cells, the autophosphorylation sites Tyr1221/1222 at the C-terminus of HER2 involved in the activation of HER2 signaling were phosphorylated in a similar manner as wild-type HER2 [21] (Figure 5A). This result indicates an aberrant activation of HER2 kinase by the fusions with MDK or ZNF207. Downstream-signaling AKT was also phosphorylated by the fusion variants (Figure 5A). Phospho-HER2Y1221/1222 and phospho-AKTS473 were suppressed by Lapatinib (Figure 5A), thus predicting the sensitivity of GC harboring the HER2 fusions to the HER2 kinase inhibitor, such as Lapatinib.
Figure 5.
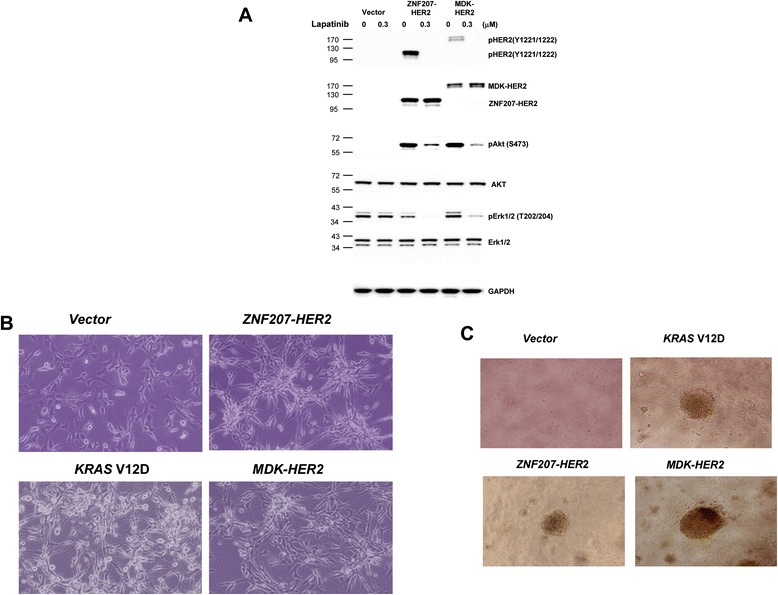
Transformation of NIH/3T3 cells by ZNF207-HER2 and MDK-HER2 fusions. A. Modulation of HER2 signaling in NIH/3 T3 cells expressing HER2 fusions. Cell lysates collected from cells stably expressing HER2 and HER2 fusions were subjected to western blotting analysis with antibodies against phospho and total HER2, Akt and Erk. B. Exogenous expression of ZNF207-HER2 and MDK-HER2 led to transformational morphological changes. Cells were photographed under a phase-contrast light microscope (×150) under identical conditions. C. Anchorage-independent growth of NIH/3 T3 cells expressing HER2 fusions. Cells were seeded in soft agar culture in 96-well plates for 14 days and colony formation was photographed under a phase-contrast light microscope (×150). The results are representative from three independent experiments.
In addition to the induction of phosphorylation of Tyr1221/1222, the exogenous expression of ZNF207-HER2 or MDK-HER2 fusions in NIH/3 T3 cells also induced transformational morphology (Figure 5B); anchorage-independent growth of the cells in vitro, which was comparable to the oncogenic phenotypes caused by mutant KRAS (V12D) (Figure 5C). Together, these results demonstrate the oncogenic properties of the ZNF207-HER2 and MDK-HER2 fusions in GC.
Different binding and responsiveness of the HER2 fusions to T-DM1 and trastuzumab
To determine whether the 5’ partners fused to a truncated HER2 extracellular domain affects their binding ability to trastuzumab in the two HER2 fusion variants, we used T-DM1 to assess its binding to NIH/3 T3 cells ectopically expressing wild-type HER2, MDK-HER2, or ZNF207-HER2. Our results clearly showed that under those conditions, the ZNF207-HER2 fusion lost its ability to bind to T-DM1, while MDK-HER2 bound to T-DM1 in a manner similar to that of wild-type HER2 (Figure 6A). Because the receptor binding assay was performed using the engineered cells, it might be argued that the impaired binding of ZNF207-HER2 could be due to improper cellular localization of the fusion protein. To address this issue, cellular localization of ectopically expressed MDK-HER2 or ZNF207-HER2 in the NIH/3 T3 cells was assessed by HercepTest™. As shown in Figure 6B, strong membrane staining (IHC 3+) was observed in the NIH/3 T3 cells expressing MDK-HER2 or ZNF207-HER2, similar to the results observed in the original primary tumor samples (Figure 4D), thus further supporting the inability of ZNF207-HER2 to bind to T-DM1.
Figure 6.
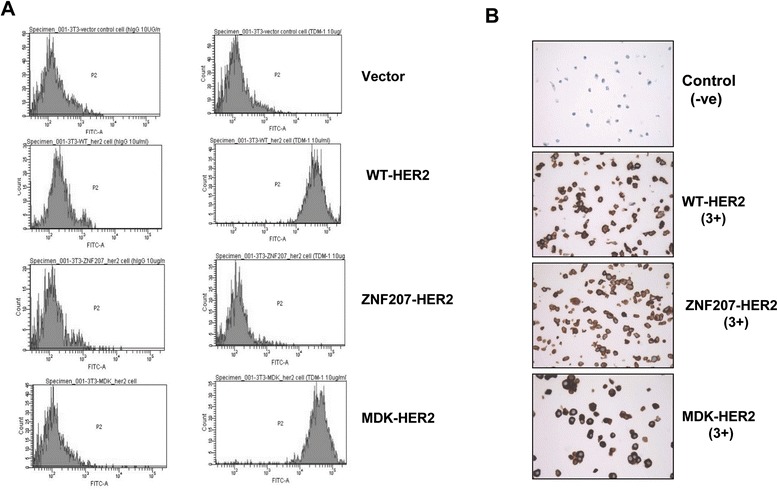
Binding of the HER2 fusions with T-DM1. A. Cells were incubated with T-DM1 at 4°C for 1 hr and the bound T-DM1 was measured by FACS with an Alex488 labeled secondary antibody. The results are representative from three independent experiments. B. IHC analysis of HER2 with HercepTest™).
The different binding capabilities of the HER2 fusions to T-DM1 predict different responsiveness of GC cells with the HER2 fusions to trastuzumab-based anti-HER2 therapies. T-DM1 was effective in inhibiting the growth of cells expressing wild-type HER2 or MDK-HER2 fusion, but this inhibitory effect was significantly impaired in the cells expressing the ZNF207-HER2 fusion (Figure 7A), indicating an intrinsic resistance mechanism to trastuzumab-based therapy. The resistance of ZNF207-HER2 to T-DM1 was further confirmed in in vivo efficacy study. We tested the anti-tumor efficacy of trastuzumab in NIH/3 T3 xenografts expressing the ZNF207-HER2 or MDK-HER2 fusion variant. As expected, the trastuzumab treatment resulted in significant tumor growth inhibition in the xenografts expressing the MDK-HER2 fusion (TGI = 67%) (Figures 7B), but it showed no efficacy in the xenografts expressing the ZNF207-HER2 fusion (TGI = 2%) (Figures 7C), thus supporting the resistant mechanism to trastuzumab in GC.
Figure 7.
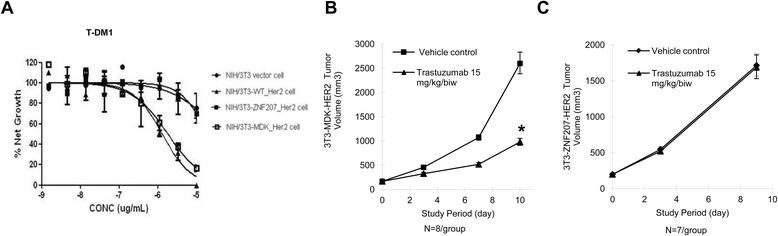
Response of NIH/3T3 cells expressing HER2 fusions to T-DM1 and trastuzumab. A. Response of NIH/3 T3 cells expressing HER2 fusions to T-DM1. Cells were treated with T-DM1 at indicated concentrations for 72 hrs and cell proliferation was measured by MTS assay. The error bars represent SD. B-C. Anti-tumor efficacy of trastuzumab in xenograft models expressing the HER2 fusions. Nude mice bearing NIH/3 T3 xenografts stably expressing the MDK-HER2 (B) or ZNF207-HER2 fusion variant (C), were treated with trastuzumab at 15 mg/kg biweekly and the tumor size was measured with a caliper. The error bars represent SEM and students’ T-tests were used to compare the growth rate in the treatment group with that in the control group *: P < 0.05.
In summary, our results clearly demonstrate that the ZNF-207-HER2 fusion does not respond to trastuzumab, due to the loss of its binding ability.
Discussion
Several studies have recently used NGS to understand the molecular basis of GC, and a number of previously unknown genetic alterations have been reported [22], including genetic fusions of HER2 in human GC cell lines. For example, two HER2 fusions were identified at the same time in human GC cell line MKN7 (HER2 positive): a fusion between CDK12 exon 12 and HER2 intron 4, and a second fusion between NEUROD2 exon 1 and HER2 exon 8 [22]. However, there was no direct evidence in that report that demonstrated the oncogenic driver of these HER2 fusions. In the current study, we performed a whole-transcriptome sequencing of 21 HER2-positive GC tumor samples taken from Chinese patients, and discovered three HER2 fusion transcripts due to HER2 gene fusions. Two of them, ZNF207-HER2 and MDK-HER2 were truncated in the N-terminal extracellular domains, but they remained intact in the kinase and transmembrane domains of HER2. The amplification and overexpression of the three HER2 gene fusions were detected in the primary GC samples by multicolor FISH and RNAseq or IHC-based HercepTest™ analysis. The ectopic expression of ZNF207-HER2 and MDK-HER2 in the NIH/3T3 cells led to a constitutive activation of HER2 and downstream signaling, and thus, cell transformation in vitro and tumorigenesis in vivo, demonstrating the oncogenic driver of the fusions. Furthermore, the xenografts ectopically expressing MDK-HER2 but not ZNF207-HER2 were sensitive to trastuzumab. Interestingly, the HER2 fusions were found to be mutually exclusive with mutations of PI3KCA, BRAF, KRAS, and HER3 and amplifications of FGFR2 and c-MET. Collectively, our data confirmed for the first time the presence of oncogenic HER2 arrangements in patients with GC. The ZNF207-HER2 fusion represents a novel intrinsic resistance mechanism to trastuzumab-based anti-HER2 therapy, due to the loss of binding ability to trastuzumab.
Despite the overall survival benefit achieved with trastuzumab in GC patients carrying HER2 amplification, a significant portion of the patients do not respond clinically to the treatment, and there is little understanding of the molecular mechanism underlying this intrinsic resistance. In contrast, the understanding of the mechanisms of both intrinsic and acquired resistance to HER2 inhibitors in HER2-positive BC is far more advanced [15,23]. For example, some intrinsic resistance mechanisms affect the ability of HER2 inhibitors to directly engage HER2 in BC; a truncated form of HER2, p95, lacking the trastuzumab binding region [24,25]; a splice variant that eliminates exon 16 (HER2-Δ16) in the extracellular domain of the HER2 receptor, preventing disruption of HER2 homodimers upon binding by trastuzumab [26]; In our study, we did not assess p95, but we did find HER2-Δ16 in a GC tumor sample, which naturally harbored HER2-Δ16. Surprisingly, its corresponding patient-derived gastric cancer xenograft (PDGCX), which retains HER2-Δ16 (data not shown), responded well to trastuzumab treatment, with a significant tumor regression being observed (data not shown). Given the lack of clinical evidence of association between HER2-Δ16 and resistance to trastuzumab in BC, as well as the lack of preclinical data on the anti-tumor efficacy of trastuzumab in xenografts carrying HER2-Δ16 [26], our data suggest that HER2-Δ16 was not a resistance mechanism to trastuzumab in GC when tested in the PDGCX model. Additional HER2-Δ16 positive PDGCX models are warranted to confirm this observation further.
Surprisingly, in addition to the HER2 fusions, we also found two recurrent in-frame BRAF fusion transcripts, BAIAP2L1-BRAF (data not shown), in another cohort of HER2-amplified GC patients. Although further work is needed to demonstrate its oncogenic activity and sensitivity to trastuzumab or BRAF inhibitor, the findings suggest that BAIAP2L1-BRAF could be a potential resistance mechanism to trastuzumab, as it maintains an intact BRAF kinase domain [27].
The current clinical protocol for selecting HER2-positive patients is based on FISH positivity or an IHC (HercepTest™) score of 3+ (HercepTest™ score of 2+ needs further FISH confirmation). HER2 gene amplification is determined by the ratio between the numbers of signals from the hybridization of the HER2 gene probe (covers the whole HER2 gene) and the number of signals from the hybridization of the reference chromosome 17 centromere probe [28]. The antibody used in the HercepTest™ recognizes the HER2 epitope located at the HER2 intracellular site, which is also covered by all three HER2 fusions (Figure 1A). Therefore, the current HER2 tests cannot distinguish between the amplifications of the HER2 fusions and that of the wild-type. The use of multicolor FISH and RT-PCR assays to detect the HER2 fusions developed in this study demonstrated the feasibility of detecting HER2 fusions as clinical biomarkers in either FFPE or frozen surgical GC specimens. This finding warrants further clinical validation of the novel resistant mechanism of the ZNF207-HER2 fusion to trastuzumab-based therapy.
In addition to the antibody, a number of small-molecule HER2 kinase inhibitors are available: lapatinib, an approved agent for HER2-positive BC patients, and afatinib and neratinib, two irreversible kinase inhibitors, currently in phase III clinical trials for HER2-positive BC. The modulation of AKT signaling by lapatinib in the cells expressing the HER2 fusions (Figure 5A) suggests that the small-molecule inhibitor against HER2 kinase is a potential option for cancer patients with the HER2 fusions. This notion is further supported by our observations that NIH/3T3 cells expressing either ZNF207-HER2 or MDK-HER2 showed to be sensitivity to lapatinib, afatinib, and neratinib in vitro (data not shown).
Besides the functional and phonotypical heterogeneity, emerging evidences indicate that genetic heterogeneity among tumor cells contributes to the advantages for survival, proliferation, metastasis and resistance to anti-cancer therapies. Recently, Tajiri et al. examined 475 GC samples using multiple ligation-dependent probe amplification (MLPA) and FISH analysis and revealed intratumoral heterogeneity of HER2 amplification in 41% (21/51) of HER2-amplified tumors. The mutually exclusive co-amplification of HER2 with EGFR, FGFR2, FGFR2 and MET was also observed respectively in some of the tumors, suggesting the potential challenges for design of targeted-therapy approaches [29]. The homogenous expression of ZNF270-HER2 supports the driver role of this fusion gene, which is consistent with our experimental results. However, the advantage of co-amplification of MDK-HER2 and NOS2-HER2 with wild HER2 (intra-tumor heterogeneity) is yet to be uncovered. Further studies to explore the responsiveness of the HER2 fusions to combination of trastuzumab with pertuzumab or chemotherapies may lead to additional insights into the impact of the HER2 fusions to anti-HER2 therapies.
It is noteworthy that the HER2 positive GC samples used for this study were collected prior the introduction of trastuzumab to China, thus we lack evidence for clinical response of the tumors harboring HER2 fusions to trastuzuamb. Further studies on GC samples from patients treated with trastuzumab at different stages will help to confirm the effect of the HER2 fusions to trastuzmab therapy and their oncogenic property.
Although further large-scale clinical investigations are needed to understand the clinical prevalence in GC patients, our data on ZNF207-HER2, along with the discovery of two recurrent BAIAP2L1-BRAF, strongly indicate a large degree of molecular heterogeneity, even in the well-defined HER2-positive segment, representing potential de novo resistance to trastuzumab-based GC therapies. In addition, whether a similar mechanism would exist in BC also needs to be exploited.
Conclusions
In summary, we uncovered three previously unidentified HER2 fusion genes in GC patients whose tumors were clinically classified as HER2-positive. Our results suggest that these HER2 fusions are genetically amplified driver oncogenes that respond differently to the HER2-neutralizing antibody trastuzumab. The resistance of ZNF207-HER2 to trastuzumab and the existence of the recurrent BRAF fusion variants warrant molecular subtype diagnoses of HER2-positive GC patients for more effective personalized trastuzumab therapies and for future treatment options.
Acknowledgement
The authors thank Charles Liu, Peter Lu and Jingyan Ding for assistance in data generation, analysis and interpretation; Jie Zang for editing, review, revision of the manuscript.
This work was sponsored by AstraZeneca
Additional file
Gene fusion CDS sequence of ZNF207-HER2 fusion MDK-HER2 fusion and NOS2-HER2 fusion. File S1 . ZNF207-HER2 gene fusion CDS sequence. File S2. MDK-HER2 gene fusion CDS sequence. File S3. NOS2-HER2 gene fusion CDS sequence.
Footnotes
De-Hua Yu and Lili Tang contributed equally to this work.
Competing interests
We declare the following Competing Interests: DY, LT, HD, ZD, JF, XS, TZ, HF, LH, LX, QY, ZQ, GZ, YC, JZ, JW, XY, XZ and QJ are employees of AstraZeneca during this study. No other conflicts of interests to declare.
Authors’ contributions
DY was responsible for collecting and analyzing the data, and drafting the manuscript; LT, ZD, JF, XS, TZ, HF, LH, LX and QY for data generation; LZ, JJ, HC for sample collection and data interpretation; DH, ZQ, GZ, JZ, XY, XZ and JJ for related study design and data interpretation. QJ was responsible for overall study design, hypothesis generation, data interpretation, drafting and finalizing the manuscript. All authors have read and approved the final manuscript.
Contributor Information
De-Hua Yu, Email: yudehua@yahoo.com.
Lili Tang, Email: lily.tang@astrazeneca.com.
Hua Dong, Email: hua.dong@astrazeneca.com.
Zhengwei Dong, Email: harvey.dong@astrazeneca.com.
Lianhai Zhang, Email: zlhzlh@hotmail.com.
Jiangang Fu, Email: jiangang.fu@astrazeneca.com.
Xinying Su, Email: xinying.su@astrazeneca.com.
Tianwei Zhang, Email: tianwei.zhang@astrazeneca.com.
Haihua Fu, Email: haihua.fu@astrazeneca.com.
Lu Han, Email: luna.han@astrazeneca.com.
Liang Xie, Email: xieliang_02@yahoo.com.
Hao Chen, Email: hyschen@163.com.
Ziliang Qian, Email: zl_qian@aliyun.com.
Guanshan Zhu, Email: guanshan_zhu@hotmail.com.
Jia Wang, Email: jia.wang@astrazeneca.com.
Qingqing Ye, Email: yeqingqing616@163.com.
Jingchuan Zhang, Email: jingchuanzhang@yahoo.com.
Xiaolu Yin, Email: lucy.yin@astrazeneca.com.
Xiaolin Zhang, Email: xiaolin.zhang@astrazeneca.com.
Jiafu Ji, Email: jiafuj@hotmail.com.
Qunsheng Ji, Email: qsji18@yahoo.com.
References
- 1.Parkin DM, Bray F, Ferlay J, Pisani P. Global cancer statistics, 2002. CA Cancer J Clin. 2005;55:74–108. doi: 10.3322/canjclin.55.2.74. [DOI] [PubMed] [Google Scholar]
- 2.Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA Cancer J Clin. 2011;61:69–90. doi: 10.3322/caac.20107. [DOI] [PubMed] [Google Scholar]
- 3.Macdonald JS, Smalley SR, Benedetti J, Hundahl SA, Estes NC, Stemmermann GN, et al. Chemoradiotherapy after surgery compared with surgery alone for adenocarcinoma of the stomach or gastroesophageal junction. N Engl J Med. 2001;345:725–30. doi: 10.1056/NEJMoa010187. [DOI] [PubMed] [Google Scholar]
- 4.Wagner AD, Unverzagt S, Grothe W, Kleber G, Grothey A, Haerting J, et al. Chemotherapy for advanced gastric cancer. The Cochrane database of systematic reviews. 2010;3:CD004064. doi: 10.1002/14651858.CD004064.pub3. [DOI] [PubMed] [Google Scholar]
- 5.Perez R, Crombet T, de Leon J, Moreno E. A view on EGFR-targeted therapies from the oncogene-addiction perspective. Front Pharmacol. 2013;4:53. doi: 10.3389/fphar.2013.00053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Paez JG, Janne PA, Lee JC, Tracy S, Greulich H, Gabriel S, et al. EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science. 2004;304:1497–500. doi: 10.1126/science.1099314. [DOI] [PubMed] [Google Scholar]
- 7.Brufsky AM, Mayer M, Rugo HS, Kaufman PA, Tan-Chiu E, Tripathy D, et al. Central nervous system metastases in patients with HER2-positive metastatic breast cancer: incidence, treatment, and survival in patients from registHER. Clin Cancer Res. 2011;17:4834–43. doi: 10.1158/1078-0432.CCR-10-2962. [DOI] [PubMed] [Google Scholar]
- 8.Wang K, Lim HY, Shi S, Lee J, Deng S, Xie T, et al. Genomic landscape of copy number aberrations enables the identification of oncogenic drivers in hepatocellular carcinoma. Hepatology. 2013;58(2):706–17. doi: 10.1002/hep.26402. [DOI] [PubMed] [Google Scholar]
- 9.Gravalos C, Jimeno A. HER2 in gastric cancer: a new prognostic factor and a novel therapeutic target. Ann Oncol. 2008;19:1523–9. doi: 10.1093/annonc/mdn169. [DOI] [PubMed] [Google Scholar]
- 10.Tanner M, Hollmen M, Junttila TT, Kapanen AI, Tommola S, Soini Y, et al. Amplification of HER-2 in gastric carcinoma: association with Topoisomerase IIalpha gene amplification, intestinal type, poor prognosis and sensitivity to trastuzumab. Ann Oncol. 2005;16:273–8. doi: 10.1093/annonc/mdi064. [DOI] [PubMed] [Google Scholar]
- 11.Matsui Y, Inomata M, Tojigamori M, Sonoda K, Shiraishi N, Kitano S. Suppression of tumor growth in human gastric cancer with HER2 overexpression by an anti-HER2 antibody in a murine model. Int J Oncol. 2005;27:681–5. [PubMed] [Google Scholar]
- 12.Fujimoto-Ouchi K, Sekiguchi F, Yasuno H, Moriya Y, Mori K, Tanaka Y. Antitumor activity of trastuzumab in combination with chemotherapy in human gastric cancer xenograft models. Cancer Chemother Pharmacol. 2007;59:795–805. doi: 10.1007/s00280-006-0337-z. [DOI] [PubMed] [Google Scholar]
- 13.Bang YJ, Van Cutsem E, Feyereislova A, Chung HC, Shen L, Sawaki A, et al. Trastuzumab in combination with chemotherapy versus chemotherapy alone for treatment of HER2-positive advanced gastric or gastro-oesophageal junction cancer (ToGA): a phase 3, open-label, randomised controlled trial. Lancet. 2010;376:687–97. doi: 10.1016/S0140-6736(10)61121-X. [DOI] [PubMed] [Google Scholar]
- 14.Slamon DJ, Leyland-Jones B, Shak S, Fuchs H, Paton V, Bajamonde A, et al. Use of chemotherapy plus a monoclonal antibody against HER2 for metastatic breast cancer that overexpresses HER2. N Engl J Med. 2001;344:783–92. doi: 10.1056/NEJM200103153441101. [DOI] [PubMed] [Google Scholar]
- 15.Stern HM. Improving treatment of HER2-positive cancers: opportunities and challenges. Sci Transl Med. 2012;4:127rv122. [DOI] [PubMed]
- 16.Ge H, Liu K, Juan T, Fang F, Newman M, Hoeck W. FusionMap: detecting fusion genes from next-generation sequencing data at base-pair resolution. Bioinformatics. 2011;27:1922–8. doi: 10.1093/bioinformatics/btr310. [DOI] [PubMed] [Google Scholar]
- 17.Xie L, Su X, Zhang L, Yin X, Tang L, Zhang X, et al. FGFR2 gene amplification in gastric cancer predicts sensitivity to the selective FGFR inhibitor AZD4547. Clin Cancer Res. 2013;19:2572–83. doi: 10.1158/1078-0432.CCR-12-3898. [DOI] [PubMed] [Google Scholar]
- 18.Cho JY. Molecular diagnosis for personalized target therapy in gastric cancer. Journal of gastric cancer. 2013;13:129–35. doi: 10.5230/jgc.2013.13.3.129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fleishman SJ, Schlessinger J, Ben-Tal N. A putative molecular-activation switch in the transmembrane domain of erbB2. Proc Natl Acad Sci U S A. 2002;99:15937–40. doi: 10.1073/pnas.252640799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Matsushita C, Tamagaki H, Miyazawa Y, Aimoto S, Smith SO, Sato T. Transmembrane helix orientation influences membrane binding of the intracellular juxtamembrane domain in Neu receptor peptides. Proc Natl Acad Sci U S A. 2013;110:1646–51. doi: 10.1073/pnas.1215207110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schulze WX, Deng L, Mann M. Phosphotyrosine interactome of the ErbB-receptor kinase family. Mol Syst Biol. 2005;1:2005.0008. [DOI] [PMC free article] [PubMed]
- 22.Zang ZJ, Ong CK, Cutcutache I, Yu W, Zhang SL, Huang D, et al. Genetic and structural variation in the gastric cancer kinome revealed through targeted deep sequencing. Cancer Res. 2011;71:29–39. doi: 10.1158/0008-5472.CAN-10-1749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rexer BN, Arteaga CL. Intrinsic and acquired resistance to HER2-targeted therapies in HER2 gene-amplified breast cancer: mechanisms and clinical implications. Crit Rev Oncog. 2012;17:1–16. doi: 10.1615/CritRevOncog.v17.i1.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Scaltriti M, Rojo F, Ocana A, Anido J, Guzman M, Cortes J, et al. Expression of p95HER2, a truncated form of the HER2 receptor, and response to anti-HER2 therapies in breast cancer. J Natl Cancer Inst. 2007;99:628–38. doi: 10.1093/jnci/djk134. [DOI] [PubMed] [Google Scholar]
- 25.Anido J, Scaltriti M, Bech Serra JJ, Santiago Josefat B, Todo FR, Baselga J, et al. Biosynthesis of tumorigenic HER2 C-terminal fragments by alternative initiation of translation. EMBO J. 2006;25:3234–44. doi: 10.1038/sj.emboj.7601191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mitra D, Brumlik MJ, Okamgba SU, Zhu Y, Duplessis TT, Parvani JG, et al. An oncogenic isoform of HER2 associated with locally disseminated breast cancer and trastuzumab resistance. Mol Cancer Ther. 2009;8:2152–62. doi: 10.1158/1535-7163.MCT-09-0295. [DOI] [PubMed] [Google Scholar]
- 27.Subbiah V, Westin SN, Wang K, Araujo D, Wang WL, Miller VA, et al. Targeted therapy by combined inhibition of the RAF and mTOR kinases in malignant spindle cell neoplasm harboring the KIAA1549-BRAF fusion protein. J Hematol Oncol. 2014;7:8. doi: 10.1186/1756-8722-7-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Allison M. The HER2 testing conundrum. Nat Biotechnol. 2010;28:117–9. doi: 10.1038/nbt0210-117. [DOI] [PubMed] [Google Scholar]
- 29.Tajiri R, Ooi A, Fujimura T, Dobashi Y, Oyama T, Nakamura R, et al. Intratumoral heterogeneous amplification of ERBB2 and subclonal genetic diversity in gastric cancers revealed by multiple ligation-dependent probe amplification and fluorescence in situ hybridization. Hum Pathol. 2014;45:725–34. doi: 10.1016/j.humpath.2013.11.004. [DOI] [PubMed] [Google Scholar]