

Abstract
mRNA steady state levels vary depending on environmental conditions. Regulation of the steady state accumulation levels of an mRNA ensures that the correct amount of protein is synthesized for the cell’s specific growth conditions. One approach for measuring mRNA decay rates is inhibiting transcription and subsequently monitoring the disappearance of the already present mRNA. The rate of mRNA decay can then be quantified, and an accurate half-life can be determined utilizing several techniques. In S. cerevisiae, protocols that measure mRNA half-lives have been developed and include inhibiting transcription of mRNA using strains that harbor a temperature sensitive allele of RNA polymerase II, rpb1-1. Other techniques for measuring mRNA half-lives include inhibiting transcription with transcriptional inhibitors such as thiolutin or 1,10-phenanthroline, or alternatively, by utilizing mRNAs that are under the control of a regulatable promoter such as the galactose inducible promoter and the TET-off system. Here, we describe measurement of S. cerevisiae mRNA decay rates using the temperature sensitive allele of RNA polymerase II. This technique can be used to measure mRNA decay rates of individual mRNAs or genome-wide.
Keywords: Cellular Biology, Issue 94, Saccharomyces cerevisiae, mRNA decay, mRNA stability, nonsense-mediated mRNA decay, mRNA half-life, transcription inhibition
Introduction
The transcription and decay of specific mRNA are crucial determinants of gene expression. The rate of synthesis and decay of specific mRNAs determines the steady-state level of that particular mRNA. The steady state levels of mRNAs govern the abundance of mRNAs and determine how much of each mRNA is available for protein synthesis. Measurements of mRNA half-lives are used extensively to determine the decay rate of mRNAs. Specific mRNAs decay at different rates that are related to features of the mRNA, the function of the protein encoded by the mRNA and the environmental conditions. Depending on the technique utilized to determine mRNA decay rates, decay rate measurements can be determined either globally or for individual transcripts. In the yeast S. cerevisiae, the techniques that are most commonly used to measure global mRNA decay rates include utilizing a yeast strain harboring the temperature sensitive allele of RNA polymerase II and chemical transcriptional inhibitors such as thiolutin and 1,10-phenanthroline 1-5. These methods can also be utilized to measure individual mRNA decay rates 4. Other methods can also be utilized to measure mRNA decay rates. These methods include approach to steady state labeling or utilization of mRNA molecules that are expressed from a regulated promoter that is expressed only in select conditions. Each of these techniques has certain advantages and limitations. The technique described here utilizes the temperature sensitive allele of RNA polymerase II. This method uses S. cerevisiae as the model, but can been modified and utilized in other systems using specific transcriptional inhibition techniques 6.
mRNA half-life measurements using the temperature sensitive allele of RNA polymerase II are extensively used for both genome-wide and individual measurements of mRNA decay 4-5. This technique requires the use of a specific yeast strain that harbors a temperature sensitive allele of RNA polymerase II, rpb1-11. The rationale for this technique is that exposure of the temperature sensitive yeast strain to the nonpermissive temperature inhibits mRNA synthesis. Subsequently, decay of the preexisting mRNA is monitored at different time points after transcription has been inhibited. The disappearance of the preexisting mRNA is monitored by extracting RNA from the yeast cells at different time points after transcription has been turned off. The time points at which the yeast cells are harvested are predetermined by a pilot experiment, and depend on the transcripts and system being investigated. Quicker time points are used for short-lived transcripts, while longer time points are used for longer lived transcripts. Afterwards, the decay of the mRNA is monitored by either northern blot analysis, quantitative PCR or RNAseq.
Measurement of mRNA half-lives using a temperature sensitive allele of RNA polymerase II has its advantages. First, this technique is easy and straight forward. Second, once the yeast strain is acquired or generated in the laboratory the mRNA half-life measurements can be determined in different growth conditions; enabling determination of environmental influence on mRNA decay. Third, mRNA decay rates can be monitored genome-wide. Use of other transcription inhibition techniques also has advantages and limitations. For example, use of an inducible promoter requires subcloning to generate an mRNA that is under the control of the regulated promoter. Thiolutin is not readily available and is expensive when available. In addition, thiolutin’s mode of action is not completely understood and it has been reported to affect other cellular processes including inhibiting mRNA decay 7. Alternatively, 1,10-phenanthroline, is more readily available. Furthermore, all of the techniques used to inhibit transcription can perturb cellular function and can affect different mRNAs in distinct ways. An investigator needs to determine the most appropriate method to use in their experimental conditions to attain the most reliable results. To determine which method is most suitable for their application, a researcher needs to identify the transcripts and function of the proteins encoded by the transcripts being investigated. The most reliable mRNA decay rate measurements are those that are determined using multiple techniques and show the same decay rate. No single technique is always the best, and the most appropriate technique depends on the specific situation.
Numerous studies in S. cerevisiae have measured mRNA decay rates in various conditions and genetic backgrounds. The conditions that mRNA decay rates are measured in depend on the specific experiment being investigated. Measuring mRNA decay rates in different cellular environments determines whether the conditions being examined preferentially affect the decay rates of specific mRNAs. The decay rates of mRNAs can also vary depending on the yeast strain being used. For example, mRNA decay rates can be determined in wild-type yeast cells and yeast cells with a nonfunctional nonsense-mediated mRNA degradation (NMD) pathway. This mRNA degradation pathway is found in all eukaryotic organisms that have been examined so far and it triggers the degradation of mRNAs that prematurely terminate translation 8. NMD was initially identified as a pathway that degrades mRNAs with premature termination codons or nonsense codons, but is now recognized as a pathway that also regulates the expression of non-nonsense containing natural mRNAs. mRNAs that are targets of the pathway are rapidly degraded in yeast cells with a functional NMD pathway and stabilized in yeast cells with a nonfunctional NMD pathway. Thus, the half-lives of mRNAs that are direct targets of this pathway are shorter in wild-type yeast cells compared to yeast cells with a nonfunctional NMD pathway.
Protocol
1. Growth of Yeast Cells
Select the appropriate yeast strains to be utilized for the mRNA decay rate measurements. To inhibit transcription using the temperature sensitive allele of RNA polymerase II, use yeast strains harboring the rpb1-1 mutation 1. Obtain this yeast strain from a laboratory that already has one, or generate it in the laboratory using standard techniques if a specific genetic background is required9.
Using sterile technique, prepare yeast media using standard procedures 10. If no selection is required, prepare rich media such as YPD. Alternatively, prepare selective media if the yeast cells are transformed with plasmids and selection is required. Autoclave the media.
- Grow the yeast cells at 28 °C, which is the permissive temperature for this yeast strain. Do this in two steps:
- For the first O/N, grow the yeast cells in 5 ml of growth media to saturation.
- Set up the second O/N at the end of the second day. For the second O/N, inoculate different amounts of yeast cells from the first O/N into 100 to 150 ml of growth media (ranging from 100 µl to 1 ml, 2 ml or more, depending on when the yeast cells need to be harvested the following day). Do this step to ensure that one of the cultures is at the correct OD600 the following day.
2. Harvest the Yeast Cells
Prepare to harvest the yeast cells . The timing for this step is critical; label all the required tubes and gather all required equipment and materials in one place. Preheat a water bath to 39 °C and preheat two 15 ml test tubes of the growth media at 28 °C and at 60 °C. NOTE: The 15 ml growth media volume can be varied depending on the number of time points the cells are to be harvested.
Harvest the yeast cells when they reach an OD600 of 0.4 to 0.7. Transfer the cells to 4 - 6 sterile 50 ml screw cap bottles. Centrifuge at 7,500 x g in a high speed centrifuge for 5 min at RT.
Discard the supernatant and resuspend the pellets in the 15 ml growth media that was equilibrating at 28 °C. Pool the yeast cells in one sterile 250 ml flask and equilibrate them for ~5 min at the 28 °C growth temperature.
After the yeast cells have equilibrated at the growth temperature, immediately add the 15 ml of the growth media equilibrated at 60 °C to raise the temperature to 39 °C (the nonpermissive temperature) and immediately place the flask in the 39 °C water bath. Make sure that the culture medium remains in the 39 °C water bath throughout the remaining cell harvesting steps to ensure that transcription is turned off.
Harvest the cells at different times after placing the flask in the 39 °C water bath. For the first time point, harvest the cells immediately after placing the flask at 39 °C. Harvest a 3 ml aliquot and distribute the 3 ml into two, 1.5 ml microcentrifuge tubes. Pellet the cells for 10 sec in a mini centrifuge or picofuge with rapid deceleration and pour out the supernatant.
Immediately freeze the pellet in a dry ice/ethanol bath or in liquid nitrogen. Designate the first time point the cells are harvested at as the zero time point.
Experimentally determine the time points the cells are harvested at thereafter by a pilot experiment, depending on the mRNAs of interest. Normally, harvest yeast cells at the following time points: 0, 3, 6, 9, 12, 18, 25 and 35 min (Figure 2B). If the mRNA half-life cannot be determined using the above mentioned time points, adjust these time points. For example, for mRNAs with anticipated short half-lives, use shorter times points, (i.e., 0, 1, 2, 3, 4, 6, 9, 12 and 18 min) and for mRNAs with anticipated long half-lives, extend the time points.
After the cells are harvested, store them in a -80 °C freezer until RNA extraction is carried out.
3. Extract RNA from the Yeast Cells
For the RNA extraction portion of the protocol, use RNase free techniques to prevent degradation of the RNA by RNases. Prepare RNase free solutions, glassware and plastic ware to be used in the extraction of the RNA.
Extract the RNA from the yeast cells according to standard protocols. Typically, use the hot phenol method 12. Alternatively, use kits that can be used to extract RNA from yeast cells.
Determine the quantity and purity of the RNA, normally by measuring the absorbance at A260 and A280 of 2 µl of RNA using a Nanodrop. Determine the concentration of the RNA from the A260. Based on the concentration, dilute the RNA to 1 µg/µl using DEPC treated water. NOTE: The ratio of the A260/A280 provides information on the purity of the RNA. An RNA sample is pure if the A260/A280 ratio is 2.0 ± 0.1.
4. Northern Blot Analysis
NOTE: Use northern blots to quantify mRNA levels and obtain information on the size of the transcripts. In addition, use northern blots to detect mRNAs that produce multiple isoforms of the same mRNA.
Prepare a 1.0% agarose-formaldehyde gel. Run equal amounts of the sample RNA on the agarose formaldehyde gel by electrophoresis according to standard protocols 12. Run a RNA ladder alongside the RNA samples and use it to determine the sizes of the RNA that are detected on the northern blot. Before transferring the RNA separated on the agarose formaldehyde gel to a membrane, cut the lane with the RNA ladder from the gel and visualize using ethidium bromide. NOTE: Alternatively, the whole gel can be stained with ethidium bromide before transfer of the RNA.
After the RNA samples have migrated to an appropriate distance on the gel, transfer the RNA to a membrane using RNase free techniques. Use one of several protocols available to transfer RNA to membranes 12-13. To transfer the RNA to a membrane, cut the membrane to approximately the same size as the gel. Transfer the RNA according to standard protocols 12.
UV cross-link the RNA to the membrane using a UV crosslinker following the manufacturer’s instructions. Alternatively, bake the membrane in an oven set to 80 °C for 1 hr. Store the membrane at -20 °C in a plastic bag indefinitely until it is hybridized to specific probes. NOTE: The dry membrane can also be stored at RT between filter papers.
5. Hybridize Probes Complementary to the RNA of Interest to the Membrane
NOTE: One way to detect mRNA on the membrane is to hybridize 32P labeled DNA probes. CAUTION: Researchers working with 32P need to use protective procedures to prevent contamination. Follow institutional guidelines on use of radioactive material.
Prepare the DNA probe by labeling 25 - 50 ng of the DNA to be hybridized to the membrane according to standard protocols 12. Generate the DNA fragment by PCR or plasmid digestion. Determine the specific activity of the DNA probe by counting 1 µl of the radiolabelled DNA probe after the unincorporated nucleotides are removed 12.
Preheat prehybridization/hybridization buffer at 42 °C. Use ~10 ml of the prehybridization/hybridization buffer per 100 cm2 of the membrane 12.
Hybridize 1 - 5 x 106 cpm of radiolabeled DNA probe per ml of hybridization buffer to the membrane O/N in a hybridization oven to detect the mRNA of interest 12. Make sure that during the hybridization the hybridization oven rotation is in a direction that causes the entire membrane to be exposed to the hybridization solution, to ensure proper hybridization.
Wash the membrane two times at RT for 15 min with 50 ml of 2x SSPE and one time at 65 °C with 50 ml of 2x SSPE/2% SDS for 15 min 12. Wrap the membrane in plastic wrap to ensure that it does not dry out or contaminate the Phosphor screen.
Using a Geiger counter, estimate the intensity of the radioactivity on the membrane. NOTE: This estimate ensures that the membrane is exposed to the Phosphor screen for the appropriate amount of time. Use longer exposure times for membranes with low amounts of radioactivity.
For northern blots, use a loading control to confirm that equal amounts of RNA are loaded on each spot. To do this, strip the membrane and reprobe it with an RNA that is not affected by the process being examined. Strip the membrane by placing it in a glass tray containing 200 ml of boiling stripping solution (0.1% SDS/0.01 X SSC) for 2 min. Pour out the stripping solution and repeat the procedure five times 12. NOTE: An example of an RNA used as a loading control is SCR1. SCR1 is an RNA polymerase III transcript that is stable and abundant. SCR1 RNA abundance should not change significantly during short periods of RNA polymerase II inhibition.
6. Quantify the RNA that is Bound to the Membrane
NOTE: To quantify the amount of radioactivity on membrane, expose the membrane to a Phosphor screen. After the appropriate exposure time, scan the Phosphor screen using a phosphorimager.
Scan the Phosphor screen using the instructions provided by the manufacturer of the phosphorimager.
Quantify the mRNA at each time point. Quantify mRNA levels using ImageQuant software or related software. Normalize the mRNA at each time point to the loading control. If SCR1 was used as the loading control, normalize the half-life northerns to SCR1.
The half-life of the mRNA is calculated by dividing the amount of mRNA remaining at each time point by the amount of mRNA present at time point 0 (the initial time point). Graph the percent mRNA remaining versus time on a semilogarithmic plot (Figure 2C). Calculate mRNA half-lives by the following equation: t1/2 = -0.693/k (negative 0.693)
k is the slope of the best fit line and t1/2 is the mRNA half-life.
Representative Results
The ability of this protocol to accurately measure mRNA decay rates depends on inhibition of transcription, the harvesting of yeast cells at the appropriate time points, and utilization of RNase free techniques while extracting RNA and northern blotting. Probing for two control mRNAs known to be unstable and stable, respectively, provides confidence that the experiment worked. For example, this can be accomplished by probing with a probe that detects both the CYH2 pre-mRNA and mRNA. Figure 2B shows the disappearance of the CYH2 pre-mRNA in wild-type and nmd mutant yeast strains at different time points after transcription inhibition. The CYH2 pre-mRNA is degraded faster in yeast cells with a functional NMD pathway (UPF1) relative to yeast cells with a nonfunctional NMD pathway (upf1).
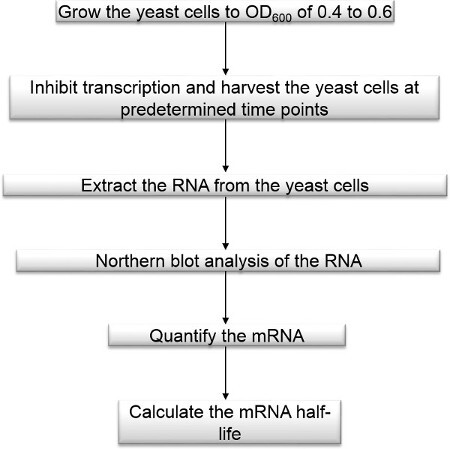 Figure 1. Method flowchart. Measurement of mRNA decay rate flowchart
Figure 1. Method flowchart. Measurement of mRNA decay rate flowchart
 Figure 2. mRNA half-life of CYH2-pre-mRNA and mRNA. (A) Schematic representation of the CYH2 pre-mRNA and CYH2 mRNA. CYH2 pre-mRNA is inefficiently spliced and transported to the cytoplasm where it is degraded by the NMD pathway. The CYH2 mRNA is not degraded by the NMD pathway because it lacks the intron containing the premature termination codon (PTC). (B) Half-life northern blots of RNA extracted from wild-type (UPF1) and nmd mutant strains (upf1). The time points after transcription inhibition are listed above the northern blots. The blots were probed with radiolabelled CYH2 DNA. (C) A graph of % CYH2 pre-mRNA remaining versus time in wild-type (UPF1) and nmd mutant strains (upf1). This graph shows that the CYH2 pre-mRNA is degraded at a faster rate in wild-type (UPF1) than in nmd mutant yeast strains (upf1).
Figure 2. mRNA half-life of CYH2-pre-mRNA and mRNA. (A) Schematic representation of the CYH2 pre-mRNA and CYH2 mRNA. CYH2 pre-mRNA is inefficiently spliced and transported to the cytoplasm where it is degraded by the NMD pathway. The CYH2 mRNA is not degraded by the NMD pathway because it lacks the intron containing the premature termination codon (PTC). (B) Half-life northern blots of RNA extracted from wild-type (UPF1) and nmd mutant strains (upf1). The time points after transcription inhibition are listed above the northern blots. The blots were probed with radiolabelled CYH2 DNA. (C) A graph of % CYH2 pre-mRNA remaining versus time in wild-type (UPF1) and nmd mutant strains (upf1). This graph shows that the CYH2 pre-mRNA is degraded at a faster rate in wild-type (UPF1) than in nmd mutant yeast strains (upf1).
Discussion
Inhibition of mRNA synthesis and monitoring mRNA turnover in the absence of new synthesis is a method that is frequently used to measure mRNA decay rates. In S. cerevisiae, measurement of mRNA decay rates by inhibiting transcription using the temperature sensitive allele of RNA polymerase II is one of the most frequently used methods. This method specifically inhibits RNA polymerase II. The most critical steps for determination of mRNA decay rates using this technique are: 1) Prior to harvesting the yeast cells, ensure that transcription has been shut off by maintaining the culture at 39°C; 2) During the RNA extraction and RNA gel electrophoresis steps of the protocol ensure that RNase free techniques are used; 3) Use a control RNA for normalization to ensure that equal amounts of RNA were loaded and that the experimental treatments are working as expected; 4) repeat the mRNA decay rate measurements at least three times to ensure reproducibility and accuracy of the half-life measurements.
rpb1-1 yeast strains are normally transferred to 37 °C to inhibit transcription. However, we have found that at 37 °C inhibition of transcription occurs ~3 min after the temperature shift. At 39 °C transcription inhibition is immediate 4, 11. However, the use of this technique to determine mRNA decay rates has some limitations. The primary limitation is that a special yeast strain is required. As previously stated, this yeast strain can either be obtained from other laboratories or generated in the laboratory if a specific yeast genetic background is required. Once the yeast strain is obtained, this technique is simple and straight forward. A second limitation is that the method entails exposing the yeast cells to heat shock to inhibit transcription. Heat stress can affect cellular processes including the decay rate of particular mRNAs. For example, the decay rate of those mRNAs that encode for proteins that are involved in stress response may be affected. Lastly, the utilization of a yeast strain with a mutation in RNA polymerase II can result in the production of alternative transcripts that behave differently from the normal transcripts.
As discussed in the introduction, other techniques are utilized to measure mRNA decay rates in S. cerevisiae. This includes inhibition of transcription using chemicals such as thiolutin and 1-10-phenanthroline. These techniques are advantageous in that they can be done using any yeast strain and mRNA decay rates can also be determined genome-wide or for individual endogenous transcripts. In addition, mRNA decay rate measurements can be done in various physiological conditions. The utility of these drugs is limited by the fact that they can also affect cellular processes and influence the decay rates of mRNA differentially. Additionally, thiolutin is not readily available and when available is expensive.
After mastering this technique one will be able to determine mRNA decay rates of individual mRNAs or genome-wide. In addition, mRNA decay rates can be measured in different physiological conditions to examine whether different conditions affect mRNA decay rates differentially.
Disclosures
The authors declare that they have no competing financial interest.
Acknowledgments
Research in the author’s laboratory is supported by the Texas Higher Education Coordinating Board’s Norman Hackerman Advanced Research Program and start-up funds from Baylor University.
References
- Nonet M, Scafe C, Sexton J, Young R. Eukaryotic RNA polymerase conditional mutant that rapidly ceases mRNA synthesis. Mol. Cell. Biol. 1987;7:1602–1616. doi: 10.1128/mcb.7.5.1602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santiago TC, Purvis IJ, Bethany AJ, Brown AJ. The relationship between mRNA stability and length in Saccharomyces cerevisiae. Nucleic Acids Res. 1986;14(21):8347–8360. doi: 10.1093/nar/14.21.8347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jimenez A, Tipper DJ, Davies J. Mode of action of thiolutin, an inhibitor of macromolecular synthesis in Saccharomyces cerevisiae. Antimicrob Agents Chemother. 3(6):729–738. doi: 10.1128/aac.3.6.729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrick D, Parker R, Jacobson A. Identification and Comparison of stable and unstable mRNAs in Saccharomyces cerevisiae. Mol Cell Biol. 10(5):2269–2284. doi: 10.1128/mcb.10.5.2269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grigull J, Mnaimneh S, Pootoolal J, Robinson MD, Hughes TR. Genome-wide Analysis of mRNA Stability Using Transcription Inhibitors and Microarrays Reveals Postranscriptional Control of Ribosome Biogenesis Factors. Mol Cell Biol. 2004;24(12):5534–5547. doi: 10.1128/MCB.24.12.5534-5547.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tani H, Akimitsu N. Genome-wide technology for determining RNA stability in mammalian cells. RNA Biology. 2012;9(10):1233–1238. doi: 10.4161/rna.22036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelechano V, Perez-Ortin JE. The transcriptional inhibitor thiolutin blocks mRNA degradation in yeast. Yeast. 2008;25:85–92. doi: 10.1002/yea.1548. [DOI] [PubMed] [Google Scholar]
- Kervestin S, Jacobson A. NMD: a multifaceted response to premature translational termination. Nat Rev Mol Cell Biol. 2012;13:700–712. doi: 10.1038/nrm3454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolodziej PA, Woychik N, Liao SM, Young RA. RNA Polymerase II subunit composition, stoichiometry and phosphorylation. Mol Cell Biol. 1990;10(5):1270–1275. doi: 10.1128/mcb.10.5.1915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ausubel FM, Brent R, Kingston RE, Moore DD, Seidman JG, Smith JA, Struhl K. Current Protocols in Molecular Biology. John Wiley and Sons, Inc.; 1998. [Google Scholar]
- Kebaara BW, Nazarenus T, Taylor R, Atkin AL. Genetic background affects relative nonsense mRNA accumulation in wild-type and upf mutant yeast strains Current Genetics) 2003;43:171–177. doi: 10.1007/s00294-003-0386-3. [DOI] [PubMed] [Google Scholar]
- Kebaara BW, Baker KE, Patefield KD, Atkin AL. Analysis of Nonsense-Mediated mRNA Decay in Saccharomyces cerevisiae. Current Protocols in Cell Biology. 2012;27(27.3):27.3.1–27.3.39. doi: 10.1002/0471143030.cb2703s54. [DOI] [PubMed] [Google Scholar]
- Hu W, Coller J. Methods for measuring mRNA Decay Rate in Saccharomycescerevisiae. Methods in Enzymology. 2013;530:137–155. doi: 10.1016/B978-0-12-420037-1.00007-5. [DOI] [PubMed] [Google Scholar]