

Abstract
Studies in the mammalian neocortex have enabled unprecedented resolution of cortical structure, activity, and response to neurodegenerative insults by repeated, time-lapse in vivo imaging in live rodents. These studies were made possible by straightforward surgical procedures, which enabled optical access for a prolonged period of time without repeat surgical procedures. In contrast, analogous studies of the spinal cord have been previously limited to only a few imaging sessions, each of which required an invasive surgery. As previously described, we have developed a spinal chamber that enables continuous optical access for upwards of 8 weeks, preserves mechanical stability of the spinal column, is easily stabilized externally during imaging, and requires only a single surgery. Here, the design of the spinal chamber with its associated surgical implements is reviewed and the surgical procedure is demonstrated in detail. Briefly, this video will demonstrate the preparation of the surgical area and mouse for surgery, exposure of the spinal vertebra and appropriate tissue debridement, the delivery of the implant and vertebral clamping, the completion of the chamber, the removal of the delivery system, sealing of the skin, and finally, post-operative care. The procedure for chronic in vivo imaging using nonlinear microscopy will also be demonstrated. Finally, outcomes, limitations, typical variability, and a guide for troubleshooting are discussed.
Keywords: Neuroscience, Issue 94, spinal cord, in vivo microscopy, multiphoton microscopy, animal surgery, fluorescence microscopy, biomedical optics
Introduction
Time-lapse in vivo microscopy in intact organisms enables the direct visualization of complex biological processes that are inaccessible to traditional single-time point analysis, such as immunohistochemistry. Specifically, multi-photon microscopy (MPM)1 allows for imaging in scattering tissue, such as the rodent neocortex, where imaging up to2,3 and in excess of4 1 mm has been achieved. When combined with surgical preparations5-7 in which a single procedure allows optical access to the brain for weeks to months, these microscopy approaches have been used to study dynamic processes in the brain in healthy and diseased states8-11. In addition, protocols have been developed12,13 that provide for in vivo imaging in awake (i.e., non anesthetized) animals, allowing for cellular-resolution functional imaging during behavioral assays. These protocols have been used for comparisons of correlated neuronal activity14, astrocyte calcium signaling15 in anesthetized and awake animals, the identification of task-specific neuronal clusters16, and the ability of neurons to discriminate object location upon whisker stimulation17.
Given the potential of this approach to elucidate healthy and pathological mechanisms, time-lapse in vivo imaging was applied to the mouse spinal cord (SC), allowing for the identification of acute axonal degeneration (AAD) as a disease mechanism18. Subsequent studies investigated effects of peripheral lesions on dorsal root ganglia (DRG) axon regeneration19, the role of blood vessels in axon regeneration20, glial chemotaxis in response to injury21, T-cell migration in experimental autoimmune encephalomyelitis (EAE) 22, activity of microglia23,24 and astrocytes25 in response to amyotrophic lateral sclerosis (ALS), the role of STAT-3 in axonal sprouting after SC injury (SCI) 26, and a mechanism of axon loss and recovery in EAE27. Despite the success of these approaches, all these studies were limited to either a single imaging session, thereby limiting studies to short-term dynamics, or else required repeated surgical openings of the animal at every imaging time point, limiting the number of time points accessible and increasing the likelihood of confounding experimental artifacts. Protocols for these surgeries have been published previously28,29.
Recently, we published a technique30 for the implantation of a chronic spinal chamber that enabled time-lapse MPM imaging in the mouse SC over multiple weeks without the need for repeat surgeries. Briefly, this surgical preparation included performing laminectomy in the lower thoracic spine and the implantation of a four-part chamber. The chamber included three custom-machined stainless steel parts that clamped the vertebrae surrounding the laminectomy, and a glass coverslip placed over the SC and secured with silicone elastomer. This technique allowed for routine imaging out to more than 5 weeks postoperatively in healthy and injured states without the need for repeat surgeries. The number of imaging time points was limited only by the frequency at which the animal can tolerate anesthetic induction. Imaging lifetime was limited by the growth of a dense, fibrous tissue over the surface of the SC. In addition, we verified that the surgical implant had no long-term effect on motor function.
Since our initial publication, alternative approaches also enabling long-term imaging in the SC have been described elsewhere31-33. This protocol demonstrates our procedure for implanting the spinal chamber we developed.
Protocol
NOTE: The care and experimental manipulation of all mice in this paper was approved by the Cornell University Institutional Animal Care and Use Committee.
1. Setting up for Surgery
Sterilize all required tools using an autoclave or approved sterilization procedures. At a minimum, include a scalpel, scissors, vanna scissors, forceps, jeweler’s screwdriver, cotton swabs, a set of retractors, the implant (Figure 1A) and its delivery system (Figure 1D, E).
Wipe down all surfaces, including any stereoscope knobs, with 70% ethanol and/or suitable disinfectants.
Create a sterile field by overlaying the custom stereotax (Figure 1D) and surrounding benchtop with sterile drapes. A heating pad (preferably feedback controlled) should be placed on top of the stereotax and under the sterile drapes.
2. Preparing the Mouse for Surgery
Anesthetize a mouse under 5% isoflurane/oxygen in an induction chamber until breathing rate slows to approximately 1 breath/sec.
Transfer mouse to a staging area away from the sterile field, using a nose cone to continue isoflurane exposure. Reduce isoflurane to 2%. Apply eye salve to prevent corneal drying.
Administer glycopyrrolate (an anticholinergic drug) at 0.05 mg/100 g mouse intramuscularly into the hindlimbs using 29 - 30 G needles and 3/10 cc syringe, delivering approximately 50% of the injection volume per limb. NOTE: Glycopyrrolate serves to prevent fluid buildup in the lungs while the mouse is under anesthesia.
Using electrical clippers, shave a portion of the dorsal aspect of the mouse, approximately 3 cm long by 2 cm wide and centered on the thoracic arch. Clean any trimmed hair well away from the shaved area.
Administer the following injections subcutaneously and distal to the shaved patch using 29 - 30 G needles and 3/10 cc syringes: 1 ml/100 g mouse 5% glucose in saline (for hydration), 5 mg/kg mouse ketoprofen (an NSAID for analgesia), 0.2 mg/kg mouse dexamethasone (an antiinflammatory steroid to reduce inflammation).
Using cotton swabs, perform three alternating washes of iodine and 70% ethanol, waiting 1 min in between washes.
Administer 100 µl of 0.125 % bupivacaine (a peripheral nerve block) subcutaneously using 29 - 30 G needles and 3/10 cc syringe to the center of the shaved patch to reduce pain at the incision site.
Transfer mouse to the custom stereotax (Figure 1D), insert rectal thermometer, and wait approximately 10 min for bupivicaine to take effect.
3. Expose and Clean the Dorsal Laminae of 3 Adjacent Thoracic Vertebrae
Using the scalpel blade, create a small (5 mm or less) incision above the vertebrae of interest (Figure 2A).
Using either forceps or by inserting the tip of surgical scissors and spreading the handles, increase the size of the opening in the skin by blunt dissection.
Using a scalpel or surgical scissors, gently dissociate connective fascia between the skin and underlying muscle.
Use retractors to hold back the skin, allowing for as large a working area as possible (Figure 2B). Take care not to increase the load on the chest and thereby obstruct breathing.
Using a pair of forceps, grip the rostral-most vertebra that will be exposed through the overlying muscle. Using a scalpel, cut tissue away on either side of the dorsal process from all 3 vertebrae (Figure 2c).
Using any combination of a scalpel blade, cotton swabs, and/or bone scraper, remove all overlying tissue from the dorsal laminae (Figure 2D).
Using surgical scissors, carefully cut away tendons attached to the vertebrae on both lateral edges of the spinal column (Figure 2E).
Using the scalpel, gently remove as much tissue from the lateral edges of the vertebral column as possible in order to provide a clean clamping surface.
Gently debride the vertebrae of any remaining soft tissue using a bone scraper and/or cotton swab. Trim away any incongruous tissue elsewhere using surgical scissors (Figure 2F). NOTE: It is essential for both clamping and performing the dorsal laminectomy that the vertebrae are cleanly exposed. A cauterizer may be useful in controlling subsequent bleeding.
Shift retractors to hold back the tissue surrounding the vertebral column in addition to the skin (Figure 2F).
Attach the implant side bars to the prongs on the delivery posts and load these into the post-holders (as in Figure 1E).
Clamp the 3 vertebrae using the side bars, applying just enough pressure to hold securely (Figure 2G, H). Use forceps to help ensure a level clamping of the 3 vertebrae. Note that several attempts may be necessary. Ensure that the side bars are both level and parallel prior to performing the laminectomy.
Carefully clean and dry the dorsal surface of the clamped vertebrae using cotton swabs.
4. Perform a Dorsal Laminectomy
Insert the tips of the vannas scissors into the epidural space of the medial clamped vertebra and squeeze the handles lightly; if the bone is dry, it should crack along the length of the dorsal lamina. Repeat this procedure on the contralateral side to release the lamina.
Gently gripping the loose lamina by the dorsal process with forceps, carefully cut away any connective tissue at the rostral and caudal ends of the lamina and lift it away.
Using 0.9 % saline and/or artificial cerebral spinal fluid (ACSF), thoroughly wash away any blood overlying the spinal cord. Use cotton swabs to absorb excess fluid.
Using vannas scissors, carefully trim the lateral edges of the bone back to the side bars of the implant. Again, wash away and control bleeding using cotton swabs and saline/ACSF.
In the event that a portion of the periosteum has detached and overlies the spinal cord, gently remove this tissue using cotton swabs and/or a 29 - 30 G needle. Take care not to injure the spinal cord in this portion of the procedure. Do not confuse the loose periosteum with the tightly wrapped dura mater.
Carefully, seal the remaining exposed edges of the bone with one or both of dental acrylic and cyanoacrylate (Figure 2I). Take special care not to allow the glues to flow onto the exposed spinal cord.
5. Seal the Chamber
Position the top plate carefully so that the 4 slots line up with the 4 holes on the side bars and the opening overlies the exposed spinal cord (Figure 1B and 2J).
Using the jeweler’s screwdriver, carefully start the first screw, stabilizing the shaft of the screwdriver with another pair of forceps. Do not tighten the screw at this stage, but insert it so that the first few threads are engaged. Repeat this process for the other 3 screws. NOTE: It is useful to use titanium or other non magnetizable forceps to help position the screws at this stage.
With all 4 screws in place, tighten each screw alternately by the same amount in small increments until screws are firmly secured (Figure 2K). Note that it is not unusual for the top plate to flex during this process. Take care not to tighten the screws too much, as the screw head can break and excessive downward pressure will dislodge the clamp and/or result in spinal cord injury.
Use a silicone elastomer to fill the space between the spinal cord and the glass window. Because it does not produce acetic acid during curing, use KwikSil brand elastomer and mixing tips. Apply a liberal portion of the silicone to the exposed spinal cord. Take care to get rid of any silicone containing air bubbles from the mixing tip prior to application to the spinal cord.
Working quickly, gently press a 5 mm #0 coverslip into the inset in the top plate. Apply enough pressure to gradually squeeze the liquid elastomer to surround the spinal cord, but not result in spinal cord compression (Figure 2L). Setting time for the elastomer is approximately 90 sec, so make sure the coverslip is applied rapidly.
In the event of a failed sealing, wait until the elastomer has set, gently remove it from the implant with forceps, and repeat 5.4 - 5.5.
Cover the edges of exuded elastomer with dental acrylic/cyanoacrylate and adhere to the surrounding bone as much as possible to prevent the elastomer from shifting over the surface of the spinal cord.
Remove retractors and pull the skin up around the edge of the implant. Using cyanoacrylate, adhere the skin to the edges of the implant (Figure 2M, N).
Insert #0-80 - 1/4” set screws into the wings of the top plate (Figure 1C and 2O).
Using dental acrylic, seal any gaps in the screw slots of the top plate (Figure 2P).
6. Postoperative Care
Remove animal from anesthesia and place in a large clean cage. Ensure that the cage is sufficiently large that the chamber will not snag on anything (such as the v-shaped dip for a water bottle and/or food) during the course of normal locomotion throughout the cage. NOTE: A standard rat cage is ideal.
Place the cage on a heated surface while the animal recovers. Do not return the mouse to the company of other animals until it has fully recovered.
Check animal behavior within 1 - 2 hr postoperation for signs of pain or distress and implant integrity. NOTE: Under normal conditions, the animal should be able to ambulate with modest perturbations to movement.
Continue to check animal every 8 - 12 hr for the first 72 hr, measuring weight and monitoring behavior. Check that the animals are active and mobile during the night. Wet food in the form of ground chow mixed with water or nutritional gels, chow pellets, and drinking water should be provided at ground level. Additional hydration by subcutaneous or intraperiotoneal injections should be provided as necessary.
Administer 5 mg/kg mouse ketoprofen and 0.2 mg/kg mouse dexamethasone subcutaneously at 24 and 48 hr time points, postoperatively. Additionally, 0.005-0.010 mg/100 g mouse should be administered every 8 hr by subcutaneous injection for 72 hr .
7. In Vivo Imaging
Anesthetize a mouse under 5% isoflurane/oxygen in an induction chamber until breathing rate slows to approximately 1 breath/sec.
Transfer mouse to the custom stereotax (Figure 1D) with feedback controlled heating pad and insert rectal thermometer. Reduce isoflurane to 2%.
Administer glycopyrrolate 0.05 mg/100 g mouse intramuscularly into the hindlimbs using 29 - 30 G needles and 3/10 cc syringe, delivering approximately 50% of the injection volume per limb.
Administer 1 ml/100 g mouse 5% glucose (for hydration) subcutaneously in saline once per hr.
Insert the threaded chamber holders into the post-holders used for the surgery (Figure 1F). Adjust the height and twist the posts onto the set screws in the top plate.
With both holders attached, elevate the holders in the post-holders until the chest can freely expand upon inspiration. NOTE: Both the firm attachment of the holders and the free expansion of the chest significantly reduce motion artifacts due to breathing.
Perform imaging as desired.
When imaging is complete, lower posts, twist off chamber holders, remove rectal thermometer, and place mouse on a heated surface to recover. Do not return the mouse to the company of other animals until it has fully recovered.
Representative Results
By following the procedure above, each stage of the surgery should mimic the results for each stage of the surgery outlined in Figure 2. Special care must be taken when applying the silicone elastomer to eliminate air bubbles under the glass or at least ensure that they are located at the periphery of the area of interest (Figure 2L). For an experienced surgeon, surgical complications requiring euthanasia either during surgery or within the first 24 hr postsurgery occur in approximately 20% of operations. These complications most frequently arise from excessive soft tissue hemorrhaging or failure to securely attach the chamber to the vertebral column.
In successfully implanted chambers, the window typically remains optically transparent for multiple weeks (Figure 3). Despite best efforts, an opaque, fibrous, neoplastic growth often forms within a few days, resulting in partial obscuring of the window (Figure 3C - F, black dashed line). Fluorescent structures are difficult to see using 2PEF microscopy underneath this fibrous tissue. We find a bimodal distribution of results, with approximately 50% of successfully implanted windows remaining clear for more than 5 weeks postoperatively, and approximately 50% becoming obfuscated within 1 - 3 weeks. For windows that remain clear, most places within the viewing window experience only a modest degree of neovascularization in or above the dura (Figure 3D - F, green asterisks), which does not hinder 2PEF imaging. Our previous study30 showed an increase in microglia but not astrocyte density under the window and adjacent vertebrae, indicating mild inflammation analogous to that seen in cranial window preparations5.
In transgenic mice expressing yellow fluorescent protein (YFP) in a subset of dorsal root ganglia (DRG) neurons (YFPH line; Jackson Labs), axons were clearly distinguishable for weeks post-operatively (Figure 4A and b) and as long as four months in one animal (data not shown). Blood vessels (Figure 4A and B) were made visible by retro-orbitally injecting fluorescent Texas Red labeled dextran into the blood plasma. Microglia were also visualized in mice expressing GFP in a CX3CR1 knock-in mouse (CX3CR1-GFP; Jackson Labs) crossed to the YFPH line (Figure 4C). Contrast and resolution deteriorated modestly over time (Figure 4C) due to the formation of a fibrous overgrowth described previously30. We routinely imaged for 3 hr in a single session without mortality and as often as every 12 hr. Session duration and frequency is limited by the tolerance of the animal for anesthetic induction, concomitant with the ethical considerations accompanying pain and distress to laboratory animals.
 Figure1. A custom surgical suite facilitates chamber delivery during implantation and stabilizes the chamber during subsequent imaging sessions. Two side-bars and a top plate (A) are assembled (B) using small screws into the spinal chamber (C). A surgical table with extendable posts (D) allows for the delivery and clamping of the side-bars to the vertebrae using holding pins (E). Subsequent imaging sessions instead use internally threaded posts to attach to the chamber via the set screws on the wings of the implant (F). Panel b of this figure has been modified from Farrar et al., Nature Methods, 201230 with permission from Nature Publishing Group. Please click here to view a larger version of this figure.
Figure1. A custom surgical suite facilitates chamber delivery during implantation and stabilizes the chamber during subsequent imaging sessions. Two side-bars and a top plate (A) are assembled (B) using small screws into the spinal chamber (C). A surgical table with extendable posts (D) allows for the delivery and clamping of the side-bars to the vertebrae using holding pins (E). Subsequent imaging sessions instead use internally threaded posts to attach to the chamber via the set screws on the wings of the implant (F). Panel b of this figure has been modified from Farrar et al., Nature Methods, 201230 with permission from Nature Publishing Group. Please click here to view a larger version of this figure.
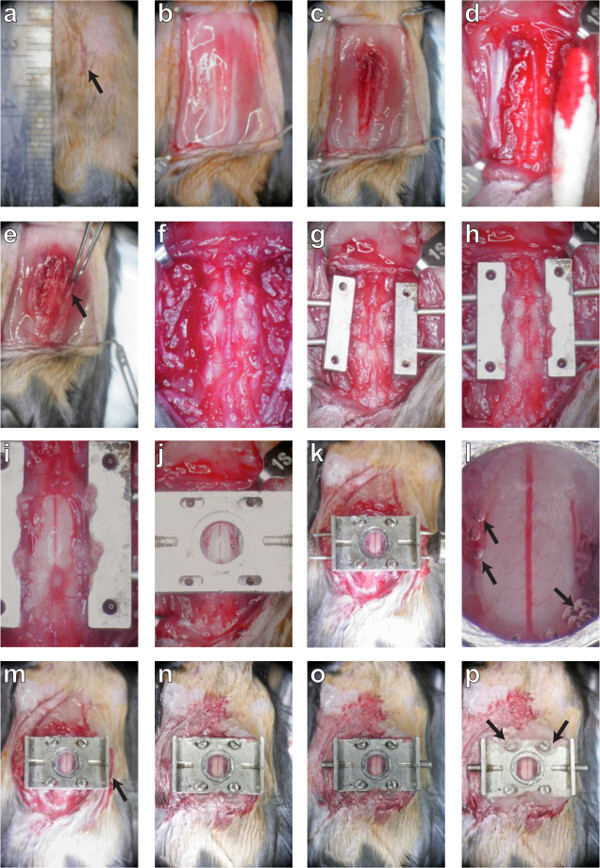 Figure 2. A dorsal laminectomy is performed and a chronic imaging chamber is implanted to provide optical access to the spinal cord. The procedure is described in montage. First, the skin (panels A, B; protocol sections 3.1 - 3.4) and muscles (C; 3.5) are retracted to expose the three underlying vertebrae. The tissue overlying these three vertebrae is removed (D, E, F; 3.6 - 3.10) and the lateral processes are scraped clean before they are clamped on both sides (G, H; 3.12). Carefully, the dorsal lamina of the central vertebra is removed and the edges of the bone sealed (I; 4. 6). A top plate is applied (J; 5.1) and screws fastened into position (K; 5.3) to secure the chamber before sealing the chamber with silicone elastomer and cover glass (L; 5.5). After the delivery pins are removed (M; 5.8), the skin is glued to the sides of the chamber (M, N; 5.8) and the set screws are inserted into the wings (O; 5.9). Finally, the screw slots are sealed with dental acrylic (P; 5.10) and the mouse is placed in a clean cage to recover. Please click here to view a larger version of this figure.
Figure 2. A dorsal laminectomy is performed and a chronic imaging chamber is implanted to provide optical access to the spinal cord. The procedure is described in montage. First, the skin (panels A, B; protocol sections 3.1 - 3.4) and muscles (C; 3.5) are retracted to expose the three underlying vertebrae. The tissue overlying these three vertebrae is removed (D, E, F; 3.6 - 3.10) and the lateral processes are scraped clean before they are clamped on both sides (G, H; 3.12). Carefully, the dorsal lamina of the central vertebra is removed and the edges of the bone sealed (I; 4. 6). A top plate is applied (J; 5.1) and screws fastened into position (K; 5.3) to secure the chamber before sealing the chamber with silicone elastomer and cover glass (L; 5.5). After the delivery pins are removed (M; 5.8), the skin is glued to the sides of the chamber (M, N; 5.8) and the set screws are inserted into the wings (O; 5.9). Finally, the screw slots are sealed with dental acrylic (P; 5.10) and the mouse is placed in a clean cage to recover. Please click here to view a larger version of this figure.
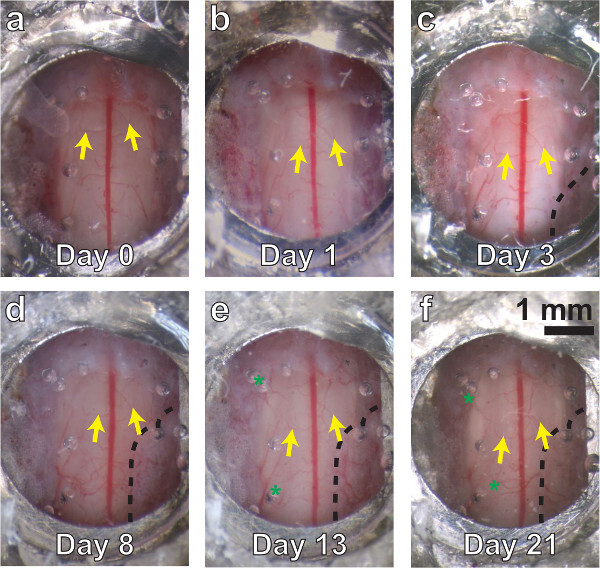
Figure 3. A chronic spinal chamber remains optically transparent over multiple weeks. White light images of the spinal cord, ranging from several minutes (A) to three weeks postoperatively (F). Vascular landmarks are identifiable at all time points (yellow arrows). Little change is seen at one day postsurgery (B). Dense fibrous overgrowth (C - F, bounded by dashed line on lower right) is present as early as three days and results in partial obfuscation of the imaging area. Mild neovascularization (D, E, F, green asterisks) is also present, but does not obscure the original vasculature in white light or 2PEF imaging.Please click here to view a larger version of this figure.
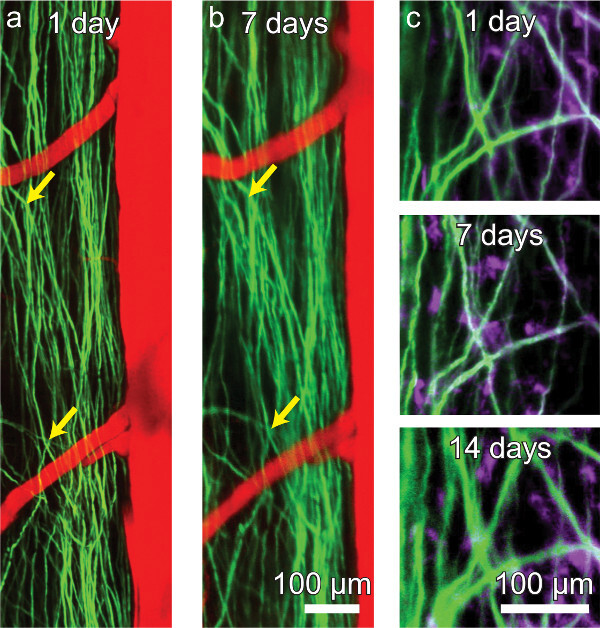
Figure 4. A chronic spinal chamber allows for repeated multi-photon imaging without the need for repeat surgeries. Repeated imaging of transgenic mice expressing YFP in a subset of DRG axons (green) in the dorsal funiculi of the spinal cord and vasculature (red) labeled by intravascular dye injection (A, B). The activity of microglia (mauve) can also be tracked over time in Thy1-YFP/CX3CR1-GFP double transgenic mice (C). While axons and microglia remain visible, contrast and resolution decline modestly over time (C). Please click here to view a larger version of this figure.
Discussion
The technique demonstrated here allows for repeated, time-lapse, in vivo imaging of the dorsal mouse SC out to many weeks post operatively without the need for subsequent surgical procedures. This procedure represents a substantial improvement over repeat-surgery imaging studies or over portmortem histological approaches, where information on cellular dynamics is lost. We have previously30 demonstrated the value of this technique for studying SCI pathology in vivo.
The maximum longitudinal extent of imaging was determined by the growth of a dense, fibrous tissue over the dorsal surface of the SC. Over time, this growth resulted in the loss of image contrast and resolution. This growth has also been seen in alternative surgical preparations31. Anecdotally, we have observed that this growth can be minimized by carefully washing the dorsal SC surface to remove blood products, sealing the surface of the cut bone with cyanoacrylate, sealing edges of the chamber well with silicone elastomer, and minimizing the interstitial space between the dorsal SC and the cover glass.
Recently, another approach similar to our own using a polycarbonate chamber has been demonstrated32. The use of polycarbonate is advantageous since it is compatible with X-ray and acoustic imaging modalities, which is not the case for our stainless steel parts. However, with the now ubiquitous technology of 3D printers, chamber parts can be fabricated from a wide variety of materials to suit specific needs. We have recently printed all our parts in a clear photopolymer.
One key disadvantage of a closed chamber is the inability to administer repeated dosages of drugs or exogenous dyes suitable for SC imaging34. However, by capitalizing on the mechanical stability of our present system, we have already used our present chamber to successfully anchor an intrathecal catheter connected to a subcutaneously implanted injection port, which allowed for drug delivery at multiple time points even in a closed chamber system (unpublished work). Furthermore, due to the modular nature of the top plate, future versions of this preparation will include a resealable chamber to allow for repeated application of both therapeutic agents and fluorescent labels. It is also possible to envision top plates with mounts for recording electrodes, optical inserts, and ports for drug delivery. Such additions would likely be more challenging to implement using the more minimalist system of Fenrich et al.31. In conclusion, our chamber provides a gateway platform upon which diverse experiments can be based.
Disclosures
The authors having nothing to disclose, and no competing financial interests.
Acknowledgments
We thank Dr. Joseph R. Fetcho for his input throughout the development of the procedure.We would like to acknowledge funding from the US National Institutes of Health (R01 EB002019 to C.B.S and DP OD006411 to Joseph R. Fetcho) and the National Science and Research Council of Canada (to M.J.F.) for financial support.
References
- Denk W, Strickler JH, Webb WW. Two-photon laser scanning fluorescence microscopy. Science. 1990;248(4951):73–76. doi: 10.1126/science.2321027. [DOI] [PubMed] [Google Scholar]
- Theer P, Hasan MT, Denk W. Two-photon imaging to a depth of 1000 μm in living brains by use of a Ti: Al 2 O 3 regenerative amplifier. Optics Letters. 2003;28(12):1022–1024. doi: 10.1364/ol.28.001022. [DOI] [PubMed] [Google Scholar]
- Kobat D, Durst ME, Nishimura N, Wong AW, Schaffer CB, Xu C. Deep tissue multiphoton microscopy using longer wavelength excitation. Optics Express. 2009;17(16):13354–13364. doi: 10.1364/oe.17.013354. [DOI] [PubMed] [Google Scholar]
- Horton NG, Kobat D, Wang K. In Vivo, Deep Tissue Three-Photon Imaging at the 1700-nm Spectral Window. Biomedical Optics. 2012;7(3) [Google Scholar]
- Holtmaat A, et al. Long-term, high-resolution imaging in the mouse neocortex through a chronic cranial window. Nature Protocols. 2009;4(8):1128–1144. doi: 10.1038/nprot.2009.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang G, Pan F, Parkhurst CN, Grutzendler J, Gan WB. Thinned-skull cranial window technique for long-term imaging of the cortex in live mice. Nature Protocols. 2010;5(2):201–208. doi: 10.1038/nprot.2009.222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drew PJ, et al. Chronic optical access through a polished and reinforced thinned skull. Nature methods. 2010;7(12):981–984. doi: 10.1038/nmeth.1530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misgeld T, Kerschensteiner M. In vivo imaging of the diseased nervous system. Nature Reviews Neuroscience. 2006;7(6):449–463. doi: 10.1038/nrn1905. [DOI] [PubMed] [Google Scholar]
- Svoboda K, Yasuda R. Principles of two-photon excitation microscopy and its applications to neuroscience. Neuron. 2006;50(6):823–839. doi: 10.1016/j.neuron.2006.05.019. [DOI] [PubMed] [Google Scholar]
- Kerr JND, Denk W. Imaging in vivo: watching the brain in action. Nature Reviews Neuroscience. 2008;9(3):195–205. doi: 10.1038/nrn2338. [DOI] [PubMed] [Google Scholar]
- Lichtman JW, DENK W. The Big and the Small: Challenges of Imaging the Brain's Circuits. Science. 2011;334(6056):618–623. doi: 10.1126/science.1209168. [DOI] [PubMed] [Google Scholar]
- Dombeck DA, Khabbaz AN, Collman F, Adelman TL, Tank DW. Imaging Large-Scale Neural Activity with Cellular Resolution in Awake, Mobile Mice. Neuron. 2007;56(1):43–57. doi: 10.1016/j.neuron.2007.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wienisch M, Blauvelt DG, Sato TF, Murthy VN. Two-Photon Imaging of Neural Activity in Awake, Head-Restrained Mice. Neuromethods. 2011;67(18):45–60. [Google Scholar]
- Greenberg DS, Houweling AR, Kerr JND. Population imaging of ongoing neuronal activity in the visual cortex of awake rats) Nature Neuroscience. 2008;11(7):749–751. doi: 10.1038/nn.2140. [DOI] [PubMed] [Google Scholar]
- Thrane AS, Thrane RV, Zeppenfeld D, Lou N, Xu Q, Nagelhus EA, et al. General anesthesia selectively disrupts astrocyte calcium signaling in the awake mouse cortex. Proc Natl Acad Sci USA. 2012;109(46):18974–18979. doi: 10.1073/pnas.1209448109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dombeck DA, Graziano MS, Tank DW. Functional Clustering of Neurons in Motor Cortex Determined by Cellular Resolution Imaging in Awake Behaving Mice. Journal Of Neuroscience. 2009;29(44):13751–13760. doi: 10.1523/JNEUROSCI.2985-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connor DH, Peron SP, Huber D, Svoboda K. Neural activity in barrel cortex underlying vibrissa-based object localization in mice. Neuron. 2010;67(6):1048–1061. doi: 10.1016/j.neuron.2010.08.026. [DOI] [PubMed] [Google Scholar]
- Kerschensteiner M, Schwab ME, Lichtman JW, Misgeld T. In vivo imaging of axonal degeneration and regeneration in the injured spinal cord. Nature Medicine. 2005;11(5):572–577. doi: 10.1038/nm1229. [DOI] [PubMed] [Google Scholar]
- Ylera B, Ertürk A, et al. Chronically CNS-Injured Adult Sensory Neurons Gain Regenerative Competence upon a Lesion of Their Peripheral Axon. Current Biology. 2009;19(11):930–936. doi: 10.1016/j.cub.2009.04.017. [DOI] [PubMed] [Google Scholar]
- Dray C, Rougon G, Debarbieux F. Quantitative analysis by in vivo imaging of the dynamics of vascular and axonal networks in injured mouse spinal cord. Proceedings of the National Academy of Sciences. 2009;106(23):9459. doi: 10.1073/pnas.0900222106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dibaj P, et al. NO mediates microglial response to acute spinal cord injury under ATP control in vivo. Glia. 2010;58(9):1133–1144. doi: 10.1002/glia.20993. [DOI] [PubMed] [Google Scholar]
- Kim JV, et al. Two-photon laser scanning microscopy imaging of intact spinal cord and cerebral cortex reveals requirement for CXCR6 and neuroinflammation in immune cell infiltration of cortical injury sites. Journal of Immunological Methods. 2010;352(1-2):89–100. doi: 10.1016/j.jim.2009.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dibaj P, et al. In Vivo imaging reveals distinct inflammatory activity of CNS microglia versus PNS macrophages in a mouse model for ALS. Plos One. 2011;6(3):17910. doi: 10.1371/journal.pone.0017910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davalos D, et al. Fibrinogen-induced perivascular microglial clustering is required for the development of axonal damage in neuroinflammation. Nature Communications. 2012;3:1227. doi: 10.1038/ncomms2230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dibaj P, Steffens H, Zschüntzsch J, Kirchhoff F, Schomburg ED, Neusch C. In vivo imaging reveals rapid morphological reactions of astrocytes towards focal lesions in an ALS mouse model. Neuroscience Letters. 2011;497(2):148–151. doi: 10.1016/j.neulet.2011.04.049. [DOI] [PubMed] [Google Scholar]
- Bareyre FM, Garzorz N, Lang C, Misgeld T, Buning H, Kerschensteiner M. In vivo imaging reveals a phase-specific role of STAT3 during central and peripheral nervous system axon regeneration. Proceedings of the National Academy of Sciences. 2011;108(15):6282–6287. doi: 10.1073/pnas.1015239108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikic I, et al. A reversible form of axon damage in experimental autoimmune encephalomyelitis and multiple sclerosis. Nature Medicine. 2011;17(4):495–499. doi: 10.1038/nm.2324. [DOI] [PubMed] [Google Scholar]
- Misgeld T, Nikic I, Kerschensteiner M. In vivo imaging of single axons in the mouse spinal cord. Nature Protocols. 2007;2(2):263–268. doi: 10.1038/nprot.2007.24. [DOI] [PubMed] [Google Scholar]
- Davalos D, Akassoglou K. In vivo Imaging of the Mouse Spinal Cord Using Two-photon Microscopy. J. Vis. Exp. 2012;59:2760. doi: 10.3791/2760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farrar MJ, Bernstein IM, Schlafer DH, Cleland TA, Fetcho JR, Schaffer CB. Chronic in vivo imaging in the mouse spinal cord using an implanted chamber. Nature. Methods. 2012;9(3):297–302. doi: 10.1038/nmeth.1856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fenrich KK, Weber P, Hocine M, Zalc M, Rougon G, Debarbieux F. Long-term in vivo imaging of normal and pathological mouse spinal cord with subcellular resolution using implanted glass windows. The Journal of Physiology. 2012;590(16):3665–3675. doi: 10.1113/jphysiol.2012.230532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Figley SA, et al. A Spinal Cord Window Chamber Model for In Vivo Longitudinal Multimodal Optical and Acoustic Imaging in a Murine Model. Plos One. 2013;8(3):58081. doi: 10.1371/journal.pone.0058081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fenrich KK, Weber P, Rougon G. Implanting Glass Spinal Cord Windows in Adult Mice with Experimental Autoimmune Encephalomyelitis. Journal of Visualized Experiments. 2013;82:50826. doi: 10.3791/50826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romanelli E, Sorbara CD, cacute IN, Dagkalis A, Misgeld T, Kerschensteiner M. Cellular, subcellular and functional in vivo labeling of the spinal cord using vital dyes. Nature Protocols. 2013;8(3):481–490. doi: 10.1038/nprot.2013.022. [DOI] [PubMed] [Google Scholar]