

Abstract
Flow cytometry has been extensively used to define cell populations in immunology, hematology and oncology. Here, we provide a detailed description of protocols for flow cytometric analysis of the cluster of differentiation (CD) surface antigens and intracellular antigens in neural cell types. Our step-by-step description of the methodological procedures include: the harvesting of neural in vitro cultures, an optional carboxyfluorescein succinimidyl ester (CFSE)-labeling step, followed by surface antigen staining with conjugated CD antibodies (e.g., CD24, CD54), and subsequent intracellar antigen detection via primary/secondary antibodies or fluorescently labeled Fab fragments (Zenon labeling). The video demonstrates the most critical steps. Moreover, principles of experimental planning, the inclusion of critical controls, and fundamentals of flow cytometric analysis (identification of target population and exclusion of debris; gating strategy; compensation for spectral overlap) are briefly explained in order to enable neurobiologists with limited prior knowledge or specific training in flow cytometry to assess its utility and to better exploit this powerful methodology.
Keywords: Neuroscience, Issue 94, CD markers, surface antigens, intracellular antigens, flow cytometry, neurons, glial cells, neural stem cells, fluorescence-activated cell sorting (FACS), CD24, CD54, CFSE
Introduction
Flow cytometry has been extensively exploited in immunology, hematology and oncology to define cell populations via intrinsic scatter properties, cell surface antigen expression, and other fluorescence parameters1-3. Our insights into blood lineage development and disease are a result to a significant degree of the continuous refinement of this methodology after its initial implementation4,5. Increased awareness of the quantitative and overall analytic potential of flow cytometry has recently encouraged its more widespread use in stem cell research and may enable similarly profound progress in a shorter time frame6. However, the application of flow cytometry to specifically analyze and isolate neural populations has long been perceived as challenging. In contrast to hematopoietic cells that naturally exist in suspension, neural cell types are typically harvested from excessively complex sources that may include glia and various other surrounding cells as well as an intricate network of process-bearing neurons. Consequently, neurobiology has yet to implement the versatility of flow cytometry to its complete potential in daily research routines. However, as long as a viable single cell suspension can be generated (and protocols have been devised and optimized for that purpose7), flow cytometry and fluorescence-activated cell sorting (FACS) can be considered a valuable element of the analytical repertoire in neurobiology8-11.
 Figure 1. Principle of flow cytometric analysis and components of a flow cytometer. Flow cytometers comprise three main systems: fluidics, optics and electronics. A streamlined flow of cells in suspension (prepared from primary tissue or in vitro culture) is accomplished by the sheath fluid via hydrodynamic focusing, restricting the sample to its center core. The optical parts are composed of lasers that illuminate the stream of cells and optical filters that direct the signal to the appropriate detectors. The light signals detected are converted to electronic signals, subsequently processed by a computer and visualized on a monitor for data analysis and gating. Please click here to view a larger version of this figure.
Figure 1. Principle of flow cytometric analysis and components of a flow cytometer. Flow cytometers comprise three main systems: fluidics, optics and electronics. A streamlined flow of cells in suspension (prepared from primary tissue or in vitro culture) is accomplished by the sheath fluid via hydrodynamic focusing, restricting the sample to its center core. The optical parts are composed of lasers that illuminate the stream of cells and optical filters that direct the signal to the appropriate detectors. The light signals detected are converted to electronic signals, subsequently processed by a computer and visualized on a monitor for data analysis and gating. Please click here to view a larger version of this figure.
Users of flow cytometric methods profit from at least a basic understanding of the underlying fundamentals including a cytometer’s building blocks (for review see12,13; also see Figure 1). A laser beam intersects with a hydrodynamically focused fluidic stream that contains the cells in suspension, which in turn pass through the laser beam in ‘single file’ one after another. The interception of a cell (or any other particle, for that matter) with the laser results in the scattering of light from this interrogation point. Scattered light can be detected in continuation of the laser direction (forward scatter, associated with the size of the particle), as well as perpendicular to its direction (side scatter; reflecting the granulosity of the particle/cell). These aforementioned scatter properties do not require specific labeling, which is why an unlabeled sample (or also cellular debris, air bubbles, etc.) will generate a signal (event) on the bivariate forward scatter versus side scatter plot commonly used for initial gating. By using the appropriate lasers and filters specific for the corresponding excitation and emission spectra, a cell can be analyzed for its positivity, level of intensity, or absence of fluorescent markers. The majority of flow cytometric applications have focused on characterization via cell surface antigens. Unlike the hematopoietic lineage, the neural lineage has remained less extensively defined according to surface epitope expression patterns5. One advantage of exploiting surface antigens is that live cells can be subjected to cell sorting paradigms such as FACS. In contrast, intracellular antigen staining requires fixation and permeabilization steps to mediate the epitope-antibody interaction, precluding downstream applications that require viable cells. Of note, such approaches still allow for numerous quantitative assays14 as well as downstream analyses for RNA and protein expression15. Hematology, immunology and oncology have often used more than a dozen markers in conjunction to define particular subpopulations16. Additionally, mass cytometry or CyTOF can now be used to analyze up to 30 parameters simultaneously17,18.
For neural stem cell applications as well as primary cultures14,19,20 the heterogeneity of cells in vitro is a common phenomenon21-23. The cells not representing the target population of interest embody a potentially confounding factor for experimental readout24,25. Conveniently, the different cellular subsets present within a heterogeneous cell suspension bear distinct (known or yet to be deciphered) antigen expression profiles, which can be utilized to define these various populations. Flow cytometry can hence play a crucial role in resolving cellular heterogeneity and, thereby, facilitate biomedical applications (in vitro assays, cell therapy) and optimize quantitative readout by focusing on the most relevant subset24,26. Various surface antigen combinations have been identified over the past few years to allow the quantitation and isolation of specific neural cell types. This includes CD133 for the enrichment of neural stem cells27, a combination of the CD15/CD24/CD29 surface antigens for the isolation of NSC, differentiated neuron and neural crest cells28 or CD15/CD24/CD44/CD184/CD271 to isolate neural and glial subsets25, among other signatures29,30. Beyond neurons, glial markers include A2B531, CD4425, NG232 and GLAST33. A recent publication has exploited the midbrain floorplate precursor marker CORIN34,35 to enrich for dopaminergic precursors in Parkinson cell transplantation paradigms36. CD molecules are not only markers, but functionally relevant mediators of cell-cell interactions and of a cell’s ability to respond to cues from extracellular matrix molecules and growth factors37. One strategy of further enhancing the arsenal of combinatorial CD antigens to characterize neural lineage development is to use known intracellular markers to screen for and define CD antigen combinations for a particular cell type of interest. We have recently exploited such an approach and identified CD49f–/CD200high combinatorial expression patterns as a novel approach for enriching neuronal subsets from neurally differentiated induced pluripotent stem cell culture systems38. Here, we include and discuss the latter protocol (and optional variations thereof) in which surface staining and intracellular staining can be used simultaneously for defining neural cell subpopulations by flow cytometry.
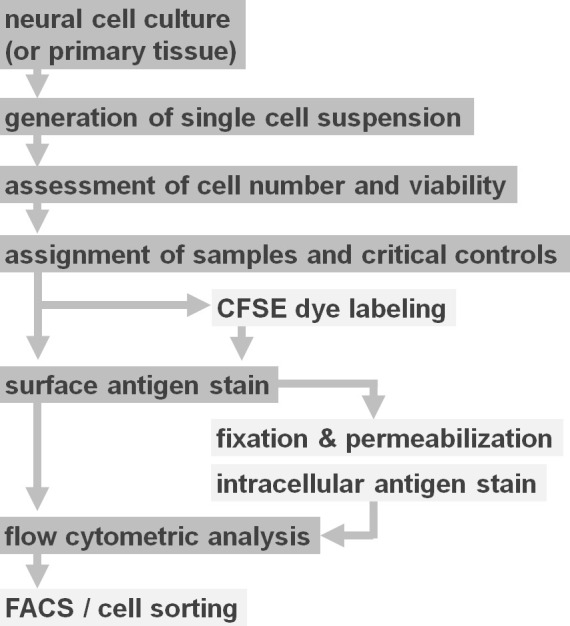 Figure 2. Flow diagram of experimental protocol options. The figure depicts a schematic representation of the major steps involved in the protocol. Optional steps (CFSE dye or intracellular antigen labeling) are indicated by light grey boxes. After harvesting, it is essential to assess the viability and cell number of neural cell suspensions prior to cell surface staining. Positive as well as negative controls need to be included in addition to the samples of interest. Samples can be analyzed by flow cytometric analysis and/or used in cell sorting paradigms. Please click here to view a larger version of this figure.
Figure 2. Flow diagram of experimental protocol options. The figure depicts a schematic representation of the major steps involved in the protocol. Optional steps (CFSE dye or intracellular antigen labeling) are indicated by light grey boxes. After harvesting, it is essential to assess the viability and cell number of neural cell suspensions prior to cell surface staining. Positive as well as negative controls need to be included in addition to the samples of interest. Samples can be analyzed by flow cytometric analysis and/or used in cell sorting paradigms. Please click here to view a larger version of this figure.
While we have previously used primary antibody in combination with secondary antibody for intracellular staining38, we now introduce non-covalent labeling of the primary antibody via fluorescent Fab fragments (Zenon labeling) as a slight variation, thereby reducing the steps of cell manipulation39. Moreover, as a further example of the protocol’s versatility, we employ an optional labeling of one experimental subset by carboxyfluorescein succinimidyl ester (CFSE) prior to surface antigen staining. Such CFSE pre-labeling enables the immediate direct comparison of two cell lines or experimental conditions (CFSE-labeled vs. unlabeled) within a single sample tube, reducing variance or subtle differences in incubation time and saving antibody. CFSE is an established fluorescent dye that is commonly used for cell tracking40, in proliferation41,42 and barcoding experiments43,44. Finally, while actual sorting steps (FACS, immunomagnetic cell separation or immunopanning) are not part of this protocol, in principle, the harvesting and labeling procedures described here do yield samples that can be subjected to surface antigen- or intracellular labeling-based sorting applications15 25,28.
With this article, we aim to: summarize a viable surface antigen staining protocol25,28, summarize a protocol for detection of intracellular targets as well as combined surface and intracellular antigen analysis38, present an intracellular CFSE dye labeling step41,45 as an experimental option for comparative analyses of neural cell populations, and summarize approaches to flow cytometric analysis (appropriate controls13,46, gating strategy and data presentation47).
Protocol
1. Neural Cell Harvesting
- Assessment by microscope:
- Prior to initiating an experiment, check the status of the culture with bright-field or phase contrast microscopy. NOTE: While primary neural tissue obtained from dissections is, in principle, equally amenable to flow cytometric analysis14,28, please note that the protocol’s focus is on cells obtained from in vitro neural cell systems.
- Harvesting cells7:
- Gently wash the dish/flask of adherent cells with Mg2+/ Ca2+ free phosphate-buffered saline (PBS) at RT (e.g., 5 ml for T75 flask, 3 ml per well for 6-well plate, or 10 ml for 10 cm dish). NOTE: The PBS used throughout the protocol is Mg2+/ Ca2+ free.
- For the video example, use a T75 flask of SH-SY5Y neuroblastoma cells at 80% confluency. Apply additional washing steps in cases where considerable debris is present in the dish.
- Consider washes with serum albumin-containing PBS48, Percoll, or Ficoll49 centrifugation gradients and/or commercially available beads50,51. Removal of myelin and other lipid or other contaminants is critical, particularly when adult primary tissue sources are being used.
- Add pre-warmed (37 °C) trypsin replacement at an appropriate volume that covers the entire surface of the tissue culture vessel. NOTE: Alternatively, consider Accutase or other enzymatic digestion options. This critical step may negatively affect surface epitope expression (see 7).
- Incubate the dish/flask at 37 °C for 2 - 5 min (depending on the cell type) to allow cells to detach. Gently tap the tissue culture vessel or flush with a serological pipet to dislodge the cells. Avoid over digestion (as this can cause cell loss and clotting at later steps).
- Quench the trypsin replacement by adding twice the volume of flow buffer (2% FBS in PBS) and collect the cells in a 15 ml conical tube.
- Gently triturate the cell suspension using a microliter pipet (100 - 1,000 µl) or a 5 ml serological pipet to prepare a single cell suspension.
- Centrifuge the cells at 220 x g for 5 min at 25 °C. Carefully aspirate the supernatant leaving the pellet behind.
- Re-suspend the pellet in an appropriate volume of flow buffer, depending on the size of the pellet (e.g., for one confluent T75 flask of SH-SY5Y cells the typical yield is at least 10 x 106 cells, in which case the cells are resuspended in 5 ml of flow buffer). NOTE: If larger chunks or clotting are observed, filter through a 30 - 100 µm mesh.
- Cell counting52:
- Transfer a small aliquot of the cell suspension to a microcentrifuge tube and dilute at a defined ratio in a volume of trypan blue or an alternative viability dye before transfer to a hemocytometer or automated cell counting system.
- Dilute the cell suspension to a concentration of 1 x 106 viable cells/ml by adding the appropriate volume of flow buffer or PBS with 0.1% BSA (if proceeding with CFSE labeling). NOTE: Propidium iodide, 7-aminoactinomycin D, annexin V and commercially available fixable viability assay kits represent alternative options in order to assess the viability of cells. Also, apoptosis assays using caspase-3 fluorescence as described previously53 may be used. Fluorescent channels will be “occupied” by these reagents which may limit the options for subsequent steps if included in all samples.
2. Intracellular Dye Labeling Using CFSE (Figure 3)
Dilute the CFSE to a desired stock concentration that can be easily used. NOTE: For these experiments a stock concentration of 0.01 mM was used. Determine the optimal working concentration of CFSE empirically.
Add 10 µl of 0.01 mM CFSE solution per ml of cells (Section 1.3.2, concentration of 1 x 106 cells/ml in PBS + 0.1% BSA) for a final concentration of 0.1 µM. Briefly vortex to mix well. NOTE: CFSE concentrations used here are about ten-fold lower than the ones commonly applied in proliferation assays, hence cell toxicity of the dye is minimal. We do not observe negative effects on cell viability.
Incubate for 5 min at RT with constant shaking (200 rpm). Protect from light.
Quench the dye by adding 5 volumes of flow buffer to the tubes. Centrifuge at 94 x g for 5 min at RT.
Discard the supernatant leaving the pellet behind. Re-suspend the cells with 5 volumes of flow buffer.
Centrifuge at 94 x g for 5 min at RT. Discard the supernatant and re-suspend the cells in flow buffer at a concentration of 1 x 106 cells/ml.
Add an equal number of unstained cells of interest to the stained cell suspension. Proceed to surface antigen staining protocol (Section 3).
 Figure 3. Detection of differential CD surface antigen expression between two cell lines via CFSE dye labeling. SH-SY5Y neuroblastoma cells are pre-labeled with CFSE for subsequent identification in comparison to unlabeled BJ fibroblasts. Co-staining of the mixed sample (right panels) with surface markers CD24 or CD54 (both conjugated to APC) demonstrates that cell lines are easily distinguishable due to the CFSE staining (arrows = SH-SY5Y; arrowheads = BJ fibroblasts). The majority of SH-SY5Y cells express CD24 but not CD54 (ICAM-1). In contrast, BJ fibroblasts (CFSE-negative) are positive for CD54 but largely negative for CD24. Please click here to view a larger version of this figure.
Figure 3. Detection of differential CD surface antigen expression between two cell lines via CFSE dye labeling. SH-SY5Y neuroblastoma cells are pre-labeled with CFSE for subsequent identification in comparison to unlabeled BJ fibroblasts. Co-staining of the mixed sample (right panels) with surface markers CD24 or CD54 (both conjugated to APC) demonstrates that cell lines are easily distinguishable due to the CFSE staining (arrows = SH-SY5Y; arrowheads = BJ fibroblasts). The majority of SH-SY5Y cells express CD24 but not CD54 (ICAM-1). In contrast, BJ fibroblasts (CFSE-negative) are positive for CD54 but largely negative for CD24. Please click here to view a larger version of this figure.
3. Cell Surface Staining
Label tubes including samples and critical controls (see Table 1).
| Tube no. | Sample name | Antigen-fluorophore | Dilution |
| 1 | Unstained cells | - | |
| 2 | Single stained | CD24-APC | 1:50 |
| 3 | Single stained | TUJ1-Alexa fluor 488nm | 1:2,000 |
| 4 | Double stained | CD24-APC | 1:50 |
| TUJ1-Alexa fluor 488nm | 1:2,000 | ||
| 5 | Single stained | Secondary only: Alexa fluor 488nm | 1:2,000 |
Table 1. List of tubes to be included in a typical flow cytometry experiment. The table shows a minimal set of sample tubes required for a co-staining experiment described in this video article. An ideal experiment needs to include all necessary controls (negative, positive as well as compensation controls) for accurate interpretation of the results obtained.
Add 100 µl of cell suspension (from Section 2.7 or 1.3.2) to each 1.5 ml microcentrifuge tube. NOTE: Make sure a minimum of 0.1 x 106 cells are present per 100 µl of cell suspension.
Add fluorophore conjugated antibody to the sample at an appropriate dilution. NOTE: Determine working dilution for each antibody prior to the experiment. See Table 2 for a list of neural surface antigens.
| Antigen | Cell Type | Reference |
| CD15 | Neural stem cells | [28, 67] |
| CD24 | Neuronal cells | [28, 68] |
| CD29 | Neural stem cells | [28, 69, 70] |
| CD44 | Glial cells | [25] |
| CD49f | Neural stem cells | [38] |
| CD56 (NCAM) | Neuronal cells | [71] |
| CD133 | Neural stem cells | [27] |
| CD184 | Neural stem cells and glial cells | [25] |
| CD200 | Neuronal cells | [38] |
| CD271 | Neural crest stem cells | [25] |
| A2B5 | Glial cells | [31] |
| CORIN | Dopaminergic precursors | [35, 36] |
| FORSE1 | Neural stem cells (NSC) | [72 ] |
| GLAST | Glial cells | [33] |
| NG2 | Glial cells | [32] |
Table 2. Selection of neural surface antigens. This table provides a list of surface epitopes found to be expressed by various neural cell types to exemplify the increasing panel of surface antigens used to characterize the neural lineage. Note that this selection is far from being complete and that most of these markers are also expressed by a range of other neural and non-neural cells. Consequently, combinations of several markers will be required to better define and isolate the indicated neural subsets.
Incubate for 30 min on an orbital shaker (200 rpm) in the dark.
- Flow buffer wash
- Add 1 ml of flow buffer to the tubes. Centrifuge at 380 x g for 4 min at 4 °C.
- Discard the supernatant leaving the pellet behind.
Repeat the wash step again.
After the second wash, decant the supernatant and re-suspend the cells in flow buffer to a final volume of 100 µl.
Use sample for flow cytometric analysis. Alternatively, proceed with Section 4 and 5. NOTE: If cells are to be sorted and put back into culture post-FACS (i.e., viable cell suspension without fixation or permeabilization), apply aseptic techniques during the harvest, staining and analytical steps.
4. Fixation and Permeabilization38
- Fixation using paraformaldehyde (PFA):
- Prepare fixation buffer containing 2% PFA in PBS. NOTE: PFA is harmful to humans and the environment. Use appropriate personal protective equipment and discard waste in accordance with local regulations.
- Add 500 µl of fixation buffer to 100 µl of cell suspension.
- Incubate the tubes at RT for 15 min on an orbital shaker (100 rpm) in the dark.
- PBS wash:
- Add 1 ml of PBS to the tube. Centrifuge at 380 x g for 3 min at 4 °C.
- Discard/ decant the supernatant leaving approximately 100 µl in the tube.
- Permeabilization with Tween-20:
- Prepare permeabilization buffer containing 0.7% Tween-20 in PBS.
- Add 500 µl of permeabilization buffer to 100 µl of cell suspension.
- Incubate the tubes at RT for 15 min on an orbital shaker (100 rpm) in the dark.
Wash the cells once with PBS (as described in Section 4.2) and remove the supernatant from the tubes completely, leaving only the pellet behind.
5. Intracellular Antigen Staining38 (Figure 4)
- Preparation of primary antibody solutions:
- Dilute the primary antibodies in dilution buffer containing 1% bovine serum albumin, 10% serum (e.g., normal donkey or goat serum) and 0.5% Tween-20 in PBS. NOTE: Choose the serum to be used depending on the species the secondary antibodies were raised in.
- Alternatively, use Zenon fluorescein labeling of the primary antibody according to the manufacturer’s instructions.
- Prepare 1 µg of primary antibody in PBS at an appropriate dilution (total volume ≤ 20 µl).
- Add 5 µl of Zenon fluorescein IgG labeling reagent (Component A) to the antibody solution.
- Incubate the mixture for 5 min at RT.
- Add 5 µl of Zenon blocking reagent (Component B) to the reaction mixture.
- Incubate the mixture for 5 min at RT. Apply the antibody to the sample within 30 min.
- Primary antibody staining:
- Add 100 µl of the primary antibody solution to the cell pellet and gently triturate to mix.
- Alternatively, add Zenon fluorescein labeled antibody to the cell suspension at the appropriate dilution.
- Incubate the tubes at RT for 30 min on an orbital shaker (200 rpm), protected from light. Wash the cells once with PBS (as describes in Section 4.2) and remove the supernatant from the tubes completely, leaving only the pellet behind.
- Secondary antibody staining (not required for the Zenon fluorescein labeled antibodies):
- Dilute the secondary antibodies in PBS at an appropriate concentration.
- Add 100 µl of the secondary antibody solution to the cell pellet and gently triturate to mix. Incubate the tubes at RT for 30 min on a shaker (200 rpm) in the dark.
Wash the samples two times with PBS (as described in Section 4.2).
Wash once with flow buffer (see Section 3.5).
Re-suspend the cells in approximately 150 µl of flow buffer and analyze on the flow cytometer.
 Figure 4. Co-staining of surface and intracellular proteins. Flow cytometry data exemplifies a comparison between primary + secondary antibody-based versus Zenon fluorescein-based intracellular staining in combination with surface staining. Exclusive positivity on the y-axis (upper left quadrant) and the x-axis (lower right quadrant) shows cells stained for MAP2 and CD24, respectively. After co-staining, shared MAP2 and CD24 expression can be seen in the upper right quadrant (right panels). Comparison of using Alexa fluor 488 (top; AF488) versus Zenon fluorescein (bottom) for MAP2-labeling yields similar results. Please click here to view a larger version of this figure.
Figure 4. Co-staining of surface and intracellular proteins. Flow cytometry data exemplifies a comparison between primary + secondary antibody-based versus Zenon fluorescein-based intracellular staining in combination with surface staining. Exclusive positivity on the y-axis (upper left quadrant) and the x-axis (lower right quadrant) shows cells stained for MAP2 and CD24, respectively. After co-staining, shared MAP2 and CD24 expression can be seen in the upper right quadrant (right panels). Comparison of using Alexa fluor 488 (top; AF488) versus Zenon fluorescein (bottom) for MAP2-labeling yields similar results. Please click here to view a larger version of this figure.
6. Flow Cytometric Analysis
Conduct flow cytometric analysis immediately after the completion of the staining protocol using a flow cytometer with appropriate filters for signal detection. Use 488 nm blue and 640 nm red laser with FL-1 (533/30), FL-2 (585/40), and FL-4 (675/25) bandpass filters.
Set up primary gates based on the forward and side scatter excluding debris and dead cells.
Set fluorescence gates for surface and intracellular antigen to ≤0.5% based on the unstained samples and compensation for spectral overlap using single stained controls.
Representative Results
The protocol presented here allows for versatile experimental approaches (Figure 2). In its shortest version (steps 1, 3 and 6), it can be considered a guide for simple staining of surface antigens. In its more complex form, a number of co-labeling paradigms with a range of intracellular antigens can be pursued (optional steps 2 and/or 4 to 5). Moreover, the CFSE-labeling step exemplifies its utility for cell labeling/tracking of subpopulations in cell-cell interaction studies or flow cytometric barcoding experiments (step 2). Viable cell populations (obtained from steps 2 and/or 3) are amenable to cell sorting paradigms (FACS) and downstream analysis including post-sort cell culture. On the other hand, the combined intracellular and surface antigen staining (steps 3 to 5) provides analytical readout opportunities in its own right. For example, identifying co-localization of certain CD antigens on particular subsets of neural cell populations (applied by us in Turaç et al.38 to identify CD49f–/CD200high signatures for neuronal FACS). These and other example neural markers are provided in Table 2, of which CD15, CD29, CD49f and FORSE1, are primarily associated with neural stem cells, and CD24 and CD56 are abundant on neuronal cells. While few of these markers are exclusive, different staining intensities and multivariate expression analysis can help to identify specific marker sets for analysis and isolation of particular neural subpopulations.
Figure 3 exemplifies typical results obtained from the combinatorial usage of intracellular dye and surface staining. The data show how two distinct cell populations can easily be distinguished in a mixed sample due to the prior labeling of one of the populations. Here, SH-SY5Y neuroblastoma cells were labeled with CFSE, and exhibit fluorescence in the green channel (FL1; y-axis) that is clearly separate from the unstained second population, in this case BJ fibroblasts. While well-established characteristic fibroblast (CD54) and neural (CD24) markers are shown (present on their respective target populations; FL2 or FL4 fluorescence, respectively; x-axis) analogous approaches can be exploited to screen for additional unidentified antigens. Furthermore, the effects of cell-cell interactions of two (or more) subpopulations that were labeled prior to co-culture experiments can be studied in this manner.
Figure 4 shows the unstained SH-SY5Y cells as well as samples stained for CD24-APC and/or the intracellular neuronal MAP2 antigen (FITC channel) after fixation and permeabilization. As the latter can affect scatter properties, background fluorescence and surface epitope staining, the plots serve to document maintained surface expression of neural CD24 after intracellular staining. Moreover, the co-localization of neuronal MAP2 staining on the CD24-positive fraction is shown. It is critical to ensure faithful detection of the surface antigen by comparison to CD24 surface expression on live cells. Moreover specificity of the intracellular detection over background, non-specific antibody binding and autofluorescence needs to be confirmed. Detection of intracellular antigens via Zenon fluorescence-based or primary + secondary antibody-based strategies yields comparable results, which provides a rationale for considering this more time-efficient approach (skipping step 5.3). While the protocol is robust and enables the combined detection of a range of surface and intracellular markers with limited background fluorescence, it is recommended to closely adhere to the incubation times and washing steps provided in the protocol. Moreover, it is advisable to analyze at least three independent experimental repeats and to note that labeling intensity and surface epitope stability can be marker-specific and may require further optimization. Once established for a particular target, despite lacking the ability to analyze morphological features, flow cytometric detection of surface and intracellular antigens or intracellular antigens alone is compatible with conventional fluorescence microscopy.
Discussion
The protocol presented here is well established for neural cell cultures derived from human stem cells, but can be equally applied to other neural cell sources including primary tissue or neural cell lines. In addition to embryonic sources, neural stem or progenitor cells can be extracted from the neurogenic regions of adult brain27. Moreover, flow cytometry and FACS can be exploited to quantify, analyze and isolate various cell populations including mature neurons54, astroglia33, microglial55, oligodendrocytes56, or endothelial cells57 from postnatal brain tissue.
Regardless of the source, optimal conditions for fixation and permeabilization need to be determined for each cell type experimentally, as excess of either of the steps could lead to a massive change of scatter properties affecting flow cytometric readout. Efficiency of the protocol also depends on the stability of the surface molecules post permeabilization. It should be noted that different surface epitopes can be affected by cell manipulation as part of the procedure to varying degrees. Also enzymatic harvesting with trypsin replacement or liberase-1, for instance, may affect a range of cell surface epitopes but to a lesser degree compared to the usage of Accutase or papain7.
Specific epitopes may be affected by the same treatment paradigm to a varying degree: while VCAM-1 (Vascular cell adhesion protein-1; CD106) can be affected by harvesting, dissociation, fixation and permeabilization in a quite sensitive manner, ICAM-1 (Intercellular Adhesion Molecule-1; CD54) may display a more robust expression pattern preserved throughout the procedure58.
While previously either surface25,28 or intracellular antigen labeling14 have been applied in neurobiology paradigms, approaches combining both methods38 can yield novel neural surface antigen candidates and provide additional cytometric readout options.
We routinely conduct the surface staining step prior to intracellular antigen staining and have used that strategy to expand our set of surface markers for neuropoiesis38 using a range of human neural cell lines. Titration of antibodies and incubation times will need to be optimized for the respective cell types, target epitopes and experimental paradigm. Similarly, the CFSE concentration and the amount of cells used per staining must be optimized for each cell line, as not all cell lines stain in an identical fashion with the same amount of CFSE concentration.
CFSE has a peak excitation of 494 nm and peak emission of 521 nm (green channel; FL-1 detector), hence any fluorescence spillover to other channels needs to be compensated to determine specific fluorescence emission. Alternative dyes such as Cell Trace Violet (CTV), Cell Proliferation Dye eFluor 670 (CPD), Horizon V550, Horizon V450 have been successfully tried and tested to be used instead of CFSE59,60.
In flow cytometry, it is critical to determine the actual specific fluorescent signal from background. Therefore, the most appropriate controls have to be carefully chosen, and may include using a non-neural or otherwise negative cell line to underline antibody specificity (so called internal negative controls38,46). Autofluorescence of each cell source must be taken into account. Isotype controls can be considered largely dispensable in most neural cell detection paradigms13,46.
Given the considerable variation observed in cell culture systems (particularly those derived from stem cells) and of the staining procedure, the inclusion of biological replicates is strongly recommended. It should be noted that usage of genetic reporters is another way to use neural flow cytometry61,62. Recently, Maroof et al. used an Nkx2.1-GFP reporter approach to sort distinct cortical-like neuronal subpopulations from human embryonic stem cells63.
An up-and-coming field is represented by the exploration of neural exosomes in biomedical areas as diverse as tumor biology and neurodegeneration64, and flow cytometry of exosomes or their isolation by bead-based assays provides important tools for this quest65. For related video protocol see66.
Disclosures
The authors declare no potential conflicts of interest.
Acknowledgments
Our research program is funded through the Emmy Noether-Program of the German Research Foundation (DFG), grant PR1132/3-1. Further support by the Müller-Fahnenberg Foundation of the University of Freiburg is gratefully acknowledged. This study was supported in part by the Excellence Initiative of the German Research Foundation (GSC-4, Spemann Graduate School).
References
- Herzenberg LA, et al. The History and Future of the Fluorescence Activated Cell Sorter and Flow Cytometry: A View from. 2002;48(10):1819–1827. [PubMed] [Google Scholar]
- Chattopadhyay PK, Roederer M. Cytometry: today’s technology and tomorrow's horizons. Methods (San Diego, Calif) 2012;57(3):251–258. doi: 10.1016/j.ymeth.2012.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaye DL, Bray RA, Gebel HM, Harris WAC, Waller EK. Translational applications of flow cytometry in clinical practice) J. Immunol. 2012;188(10):4715–4719. doi: 10.4049/jimmunol.1290017. [DOI] [PubMed] [Google Scholar]
- Henel G, Schmitz J. Basic Theory and Clinical Applications of Flow Cytometry. Lab Med. 2007;38(7):428–436. [Google Scholar]
- Seita J, Weissman IL. Hematopoietic stem cell: self-renewal versus differentiation. Wiley Interdiscip. Rev. Syst. Biol. Med. 2010;2(6):640–653. doi: 10.1002/wsbm.86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulrich H, Bocsi J. Phenotypes of stem cells from diverse origin. Cytometry. A. 2010;77(1):6–10. doi: 10.1002/cyto.a.20844. [DOI] [PubMed] [Google Scholar]
- Panchision DM, et al. Optimized flow cytometric analysis of central nervous system tissue reveals novel functional relationships among cells expressing CD133, CD15, and CD24. Stem cells. 2007;25(6):1560–1570. doi: 10.1634/stemcells.2006-0260. [DOI] [PubMed] [Google Scholar]
- Meyer RA, Zaruba ME, McKhann GM. Flow cytometry of isolated cells from the brain. Anal. Quant. Cytol. 1980;2(1):66–74. [PubMed] [Google Scholar]
- Junger H, Junger WG. CNTF and GDNF, but not NT-4, support corticospinal motor neuron growth via direct mechanisms. Neuroreport. 1998;9(16):3749–3754. doi: 10.1097/00001756-199811160-00033. [DOI] [PubMed] [Google Scholar]
- McLaren FH, Svendsen CN, Vander Meide P, Joly E. Analysis of neural stem cells by flow cytometry: cellular differentiation modifies patterns of MHC expression. J. Neuroimmunol. 2001;112(1-2):35–46. doi: 10.1016/s0165-5728(00)00410-0. [DOI] [PubMed] [Google Scholar]
- Wang S, Roy NS, Benraiss A, Goldman SA. Promoter-based isolation and fluorescence-activated sorting of mitotic neuronal progenitor cells from the adult mammalian ependymal/subependymal. 2000;22(1-2):167–176. doi: 10.1159/000017437. [DOI] [PubMed] [Google Scholar]
- Tanke HJ, vander Keur M. Selection of defined cell types by flow-cytometric cell sorting. Trends Biotechnol. 1993;11(2):55–62. doi: 10.1016/0167-7799(93)90123-Q. [DOI] [PubMed] [Google Scholar]
- Baumgarth N, Roederer M. A practical approach to multicolor flow cytometry for immunophenotyping. J. Immunol. Methods. 2000;243(1-2):77–97. doi: 10.1016/s0022-1759(00)00229-5. [DOI] [PubMed] [Google Scholar]
- Sergent-Tanguy S, Chagneau C, Neveu I, Naveilhan P. Fluorescent activated cell sorting (FACS): a rapid and reliable method to estimate the number of neurons in a mixed population. J. Neurosci. Methods. 2003;129(1):73–79. doi: 10.1016/s0165-0270(03)00210-3. [DOI] [PubMed] [Google Scholar]
- Ernst A, et al. Neurogenesis in the striatum of the adult human brain. Cell. 2014;156(5):1072–1083. doi: 10.1016/j.cell.2014.01.044. [DOI] [PubMed] [Google Scholar]
- Perfetto SP, Chattopadhyay PK, Roederer M. Seventeen-colour flow cytometry: unravelling the immune system. Nat. Rev. Immunol. 2004;4(8):648–655. doi: 10.1038/nri1416. [DOI] [PubMed] [Google Scholar]
- Bandura DR, et al. Mass cytometry: technique for real time single cell multitarget immunoassay based on inductively coupled plasma time-of-flight mass spectrometry. Anal. Chem. 2009;81(16):6813–6822. doi: 10.1021/ac901049w. [DOI] [PubMed] [Google Scholar]
- Bendall SC, et al. Single-cell mass cytometry of differential immune and drug responses across a human hematopoietic continuum. Science. 2011;332(6030):687–696. doi: 10.1126/science.1198704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neveu I, Rémy S, Naveilhan P. The neuropeptide Y receptors, Y1 and Y2, are transiently and differentially expressed in the developing cerebellum. Neuroscience. 2002;113(4):767–777. doi: 10.1016/s0306-4522(02)00256-7. [DOI] [PubMed] [Google Scholar]
- Pruszak J, Just L, Isacson O, Nikkhah G. Isolation and culture of ventral mesencephalic precursor cells and dopaminergic neurons from rodent brains. Curr. Protoc. Stem Cell Biol. 2009;2(Unit 2D.5) doi: 10.1002/9780470151808.sc02d05s11. [DOI] [PubMed] [Google Scholar]
- Suslov ON, Kukekov VG, Ignatova TN, Steindler DA. Neural stem cell heterogeneity demonstrated by molecular phenotyping of clonal neurospheres. Proc. Natl. Acad. Sci. U.S.A. 2002;99(22):14506–14511. doi: 10.1073/pnas.212525299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bez A, et al. Neurosphere and neurosphere-forming cells: morphological and ultrastructural characterization. Brain Res. 2003;993(1-2):18–29. doi: 10.1016/j.brainres.2003.08.061. [DOI] [PubMed] [Google Scholar]
- Pruszak J, Isacson O. Molecular and cellular determinants for generating ES-cell derived dopamine neurons for cell therapy. Adv. Exp. Med. Biol. 2009;651:112–123. doi: 10.1007/978-1-4419-0322-8_11. [DOI] [PubMed] [Google Scholar]
- Carson CT, Aigner S, Gage FH. Stem cells: the good, bad and barely in control. Nat. Med. 2006;12(11):1237–1238. doi: 10.1038/nm1106-1237. [DOI] [PubMed] [Google Scholar]
- Yuan SH, et al. Cell-surface marker signatures for the isolation of neural stem cells, glia and neurons derived from human pluripotent stem cells. PloS One. 2011;6(3):e17540. doi: 10.1371/journal.pone.0017540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy NS, Cleren C, Singh SK, Yang L, Beal MF, Goldman S. Functional engraftment of human ES cell-derived dopaminergic neurons enriched by coculture with telomerase-immortalized midbrain astrocytes. Nat. Med. 2006;12(11):1259–1268. doi: 10.1038/nm1495. [DOI] [PubMed] [Google Scholar]
- Uchida N, et al. Direct isolation of human central nervous system stem cells. Proc. Natl. Acad. Sci. U. S. A. 2000;97(26):14720–14725. doi: 10.1073/pnas.97.26.14720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pruszak J, Ludwig W, Blak A, Alavian K, Isacson O. CD15, CD24, and CD29 define a surface biomarker code for neural lineage differentiation of stem cells. Stem Cells. 2009;27(12):2928–2940. doi: 10.1002/stem.211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peh GS-L, Lang RJ, Pera MF, Hawes SM. CD133 expression by neural progenitors derived from human embryonic stem cells and its use for their prospective isolation. Stem Cells Dev. 2009;18(2):269–282. doi: 10.1089/scd.2008.0124. [DOI] [PubMed] [Google Scholar]
- Golebiewska A, Atkinson SP, Lako M, Armstrong L. Epigenetic landscaping during hESC differentiation to neural cells. Stem Cells. 2009;27(6):1298–1308. doi: 10.1002/stem.59. [DOI] [PubMed] [Google Scholar]
- Dietrich J, Noble M, Mayer-Proschel M. Characterization of A2B5+ glial precursor cells from cryopreserved human fetal brain progenitor cells. Glia. 2002;40(1):65–77. doi: 10.1002/glia.10116. [DOI] [PubMed] [Google Scholar]
- Nishiyama A. NG2 cells in the brain: a novel glial cell population. Hum. Cell. 2001;14(1):77–82. [PubMed] [Google Scholar]
- Jungblut M, et al. Isolation and characterization of living primary astroglial cells using the new GLAST-specific monoclonal antibody ACSA-1. Glia. 2012;60(6):894–907. doi: 10.1002/glia.22322. [DOI] [PubMed] [Google Scholar]
- Ono Y, et al. Differences in neurogenic potential in floor plate cells along an anteroposterior location: midbrain dopaminergic neurons originate from mesencephalic floor plate cells. Development. 2007;134(17):3213–3225. doi: 10.1242/dev.02879. [DOI] [PubMed] [Google Scholar]
- Chung S, et al. ES cell-derived renewable and functional midbrain dopaminergic progenitors. Proc. Natl. Acad. Sci. U.S.A. 2011;108(23):9703–9708. doi: 10.1073/pnas.1016443108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doi D, et al. Isolation of Human Induced Pluripotent Stem Cell-Derived Dopaminergic Progenitors by Cell Sorting for Successful Transplantation. Stem Cell Reports. 2014;2(3):337–350. doi: 10.1016/j.stemcr.2014.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solozobova V, Wyvekens N, Pruszak J. Lessons from the embryonic neural stem cell niche for neural lineage differentiation of pluripotent stem cells. Stem Cell Rev. 2012;8(3) doi: 10.1007/s12015-012-9381-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turaç G, et al. Combined flow cytometric analysis of surface and intracellular antigens reveals surface molecule markers of human neuropoiesis. PloS One. 2013;8(6):e68519. doi: 10.1371/journal.pone.0068519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchwalow IB, Böcker W. Chapter 2. Immunohistochemistry: Basics and Methods. 2010. pp. 9–17.
- Tario JD, et al. Optimized staining and proliferation modeling methods for cell division monitoring using cell tracking dyes. J. Vis. Exp. 2012. p. e4287. [DOI] [PMC free article] [PubMed]
- Lyons AB, Parish CR. Determination of lymphocyte division by flow cytometry. J. Immunol. Methods. 1994;171(1):131–137. doi: 10.1016/0022-1759(94)90236-4. [DOI] [PubMed] [Google Scholar]
- Hawkins ED, et al. Measuring lymphocyte proliferation, survival and differentiation using CFSE time-series data. Nat. Protoc. 2007;2(9):2057–2067. doi: 10.1038/nprot.2007.297. [DOI] [PubMed] [Google Scholar]
- Quah BJC, Parish CR. The use of carboxyfluorescein diacetate succinimidyl ester (CFSE) to monitor lymphocyte proliferation. J. Vis. Exp. 2010;44 doi: 10.3791/2259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sukhdeo K, et al. Multiplex flow cytometry barcoding and antibody arrays identify surface antigen profiles of primary and metastatic colon cancer cell lines. PloS One. 2013;8(1):e53015. doi: 10.1371/journal.pone.0053015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang L, et al. Daucosterol promotes the proliferation of neural stem cells. The J. Steroid Biochem. Mol. Biol. 2014;140:90–99. doi: 10.1016/j.jsbmb.2013.12.002. [DOI] [PubMed] [Google Scholar]
- Hulspas R, et al. Considerations for the control of background fluorescence in clinical flow cytometry. Cytometry. B. 2009;76(6):355–364. doi: 10.1002/cyto.b.20485. [DOI] [PubMed] [Google Scholar]
- Moloney M, Shreffler WG. Basic science for the practicing physician: flow cytometry and cell sorting. Annals of Allergy, Asthm., & Immunology: Official Publication of the American College of Allergy, Asthma., & Immunology. 2008;101(5):544–549. doi: 10.1016/S1081-1206(10)60295-5. [DOI] [PubMed] [Google Scholar]
- Siebzehnrubl FA, et al. Isolation and characterization of adult neural stem cells. Methods Mol. Biol. 2011;750:61–77. doi: 10.1007/978-1-61779-145-1_4. [DOI] [PubMed] [Google Scholar]
- Guez-Barber D, et al. FACS purification of immunolabeled cell types from adult rat brain) J. Neurosci. Methods. 2012;203(1):10–18. doi: 10.1016/j.jneumeth.2011.08.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tham C-S, Lin F-F, Rao TS, Yu N, Webb M. Microglial activation state and lysophospholipid acid receptor expression. Int. J. Dev. Neurosci. 2003;21(8):431–443. doi: 10.1016/j.ijdevneu.2003.09.003. [DOI] [PubMed] [Google Scholar]
- Nguyen HX, Beck KD, Anderson AJ. Quantitative assessment of immune cells in the injured spinal cord tissue by flow cytometry: a novel use for a cell purification method. J. Vis. Exp. 2011. p. e2698. [DOI] [PMC free article] [PubMed]
- Marchenko S, Flanagan L. Counting human neural stem cells. J. Vis. Exp. 2007. p. 262. [DOI] [PMC free article] [PubMed]
- Brunlid G, Pruszak J, Holmes B, Isacson O, Sonntag KC. Immature and neurally differentiated mouse embryonic stem cells do not express a functional Fas/Fas ligand system. Stem Cells. 2007;25(10):2551–2558. doi: 10.1634/stemcells.2006-0745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brewer GJ. Isolation and culture of adult rat hippocampal neurons. J. Neurosci. Methods. 1997;71(2):143–155. doi: 10.1016/s0165-0270(96)00136-7. [DOI] [PubMed] [Google Scholar]
- Cardona AE, Huang D, Sasse ME, Ransohoff RM. Isolation of murine microglial cells for RNA analysis or flow cytometry. Nat. Protoc. 2006;1(4):1947–1951. doi: 10.1038/nprot.2006.327. [DOI] [PubMed] [Google Scholar]
- Nielsen JA, Maric D, Lau P, Barker JL, Hudson LD. Identification of a novel oligodendrocyte cell adhesion protein using gene expression profiling. J. Neurosci. 2006;26(39):9881–9891. doi: 10.1523/JNEUROSCI.2246-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daneman R, et al. The mouse blood-brain barrier transcriptome: a new resource for understanding the development and function of brain endothelial cells. PloS One. 2010;5(10):e13741. doi: 10.1371/journal.pone.0013741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gräbner R, Till U, Heller R. Flow cytometric determination of E-selectin, vascular cell adhesion molecule-1, and intercellular cell adhesion molecule-1 in formaldehyde-fixed endothelial cell monolayers. Cytometry. 2000;40(3):238–244. doi: 10.1002/1097-0320(20000701)40:3<238::aid-cyto9>3.0.co;2-3. [DOI] [PubMed] [Google Scholar]
- Quah BJC, Parish CR. New and improved methods for measuring lymphocyte proliferation in vitro and in vivo using CFSE-like fluorescent dyes. J. Immunol. Methods. 2012;379(1-2):1–14. doi: 10.1016/j.jim.2012.02.012. [DOI] [PubMed] [Google Scholar]
- Lathia JD, et al. High-throughput flow cytometry screening reveals a role for junctional adhesion molecule a as a cancer stem cell maintenance factor. Cell Rep. 2014;6(1):117–129. doi: 10.1016/j.celrep.2013.11.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganat YM, et al. Identification of embryonic stem cell-derived midbrain dopaminergic neurons for engraftment. J. Clin. Invest. 2012;122(8):2928–2939. doi: 10.1172/JCI58767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hedlund E, et al. Embryonic stem cell-derived Pitx3-enhanced green fluorescent protein midbrain dopamine neurons survive enrichment by fluorescence-activated cell sorting and function in an animal model of Parkinson’s disease. Stem Cells. 2008;26(6):1526–1536. doi: 10.1634/stemcells.2007-0996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maroof AM, et al. Directed differentiation and functional maturation of cortical interneurons from human embryonic stem cells. Cell Stem Cell. 2013;12(5):559–572. doi: 10.1016/j.stem.2013.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chivet M, Hemming F, Pernet-Gallay K, Fraboulet S, Sadoul R. Emerging role of neuronal exosomes in the central nervous system. Front. Physiol. 2012;3:145. doi: 10.3389/fphys.2012.00145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graner MW, et al. Proteomic and immunologic analyses of brain tumor exosomes. FASEB J. 2009;23(5):1541–1557. doi: 10.1096/fj.08-122184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eldh M, Lötvall J. Isolation and characterization of RNA-containing exosomes. J. Vis. Exp. 2012. p. e3037. [DOI] [PMC free article] [PubMed]
- Capela A, Temple S. LeX is expressed by principle progenitor cells in the embryonic nervous system, is secreted into their environment and binds Wnt-1. Dev. Biol. 2006;291(2):300–313. doi: 10.1016/j.ydbio.2005.12.030. [DOI] [PubMed] [Google Scholar]
- Nieoullon V, Belvindrah R, Rougon G, Chazal G. mCD24 regulates proliferation of neuronal committed precursors in the subventricular zone. Mol. Cell. Neurosci. 2005;28(3):462–474. doi: 10.1016/j.mcn.2004.10.007. [DOI] [PubMed] [Google Scholar]
- Nagato M, et al. Prospective characterization of neural stem cells by flow cytometry analysis using a combination of surface markers. J. Neurosci. Res. 2005;80(4):456–466. doi: 10.1002/jnr.20442. [DOI] [PubMed] [Google Scholar]
- Hall PE, Lathia JD, Miller NGA, Caldwell MA, French-Constant C. Integrins are markers of human neural stem cells. Stem Cells. 2006;24(9):2078–2084. doi: 10.1634/stemcells.2005-0595. [DOI] [PubMed] [Google Scholar]
- Hargus G, et al. Differentiated Parkinson patient-derived induced pluripotent stem cells grow in the adult rodent brain and reduce motor asymmetry in Parkinsonian rats. Proc. Natl. Acad. Sci. U.S.A. 2010;107(36):15921–15926. doi: 10.1073/pnas.1010209107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elkabetz Y, et al. Human ES cell-derived neural rosettes reveal a functionally distinct early neural stem cell stage. Genes Dev. 2008;22(2):152–165. doi: 10.1101/gad.1616208. [DOI] [PMC free article] [PubMed] [Google Scholar]