

Abstract
The ability of malignant cells to evade the immune system, characterized by tumor escape from both innate and adaptive immune responses, is now accepted as an important hallmark of cancer. Our research on breast cancer focuses on the active role that tumor infiltrating lymphocytes play in tumor progression and patient outcome. Toward this goal, we developed a methodology for the rapid isolation of intact lymphoid cells from normal and abnormal tissues in an effort to evaluate them proximate to their native state. Homogenates prepared using a mechanical dissociator show both increased viability and cell recovery while preserving surface receptor expression compared to enzyme-digested tissues. Furthermore, enzymatic digestion of the remaining insoluble material did not recover additional CD45+ cells indicating that quantitative and qualitative measurements in the primary homogenate likely genuinely reflect infiltrating subpopulations in the tissue fragment. The lymphoid cells in these homogenates can be easily characterized using immunological (phenotype, proliferation, etc.) or molecular (DNA, RNA and/or protein) approaches. CD45+ cells can also be used for subpopulation purification, in vitro expansion or cryopreservation. An additional benefit of this approach is that the primary tissue supernatant from the homogenates can be used to characterize and compare cytokines, chemokines, immunoglobulins and antigens present in normal and malignant tissues. This protocol functions extremely well for human breast tissues and should be applicable to a wide variety of normal and abnormal tissues.
Keywords: Immunology, Issue 94, Tumor immunology, tumor infiltrating lymphocytes, CD45+, breast cancer, fresh tissue homogenate, non-enzymatic dissociation, primary tissue supernatant
Introduction
The tumor microenvironment is composed of various cell types with numerous studies showing they each play distinct and important roles in tumorigenesis1,2. These include, but are not limited to, infiltrating immune cells, stromal cells, endothelial cells and tumor cells3. Ex vivo studies of tumor infiltrating lymphocytes (TIL; CD45+ cells or leukocytes, which are predominantly lymphocytes in breast tumors) from fresh human tissue samples is made difficult by their low frequency, the small sample sizes often available for research and the potential for loss of viability during extraction. Because immune cells infiltrating tumors are usually present as passengers rather than permanent residents in general they are easier to release from the tissue matrix.
Dissociating tumor tissue while maintaining cellular integrity is technically challenging and has traditionally been performed using a combination of mechanical and enzymatic steps to prepare single cell suspensions4-8. This approach involves lengthy incubation periods and is associated with a significant reduction in cell viability as well as the loss of cell surface receptors by enzymatic cleavage. High quality flow cytometric studies characterizing TIL in the tumor microenvironment as well as clean purifications of CD45+ subpopulations by flow cytometry or antibody-coated beads are more difficult to achieve from enzyme-digested tumor tissue. In addition, the supernatant (SN) from the resulting tumor homogenate is not amenable to further analysis including quantification of secreted proteins (cytokines, chemokines, immunoglobulins or tumor antigens) or experimental treatment of normal cells, because of the potential for protein degradation in the enzymatic digests.
In our search for a method to prepare single cell homogenates from breast tissues [including tumor, non-adjacent non-tumor (NANT) and normal (from mammary reductions) breast tissues] without enzymatic digestion, we tested a variety of mechanical homogenization techniques. Homogenates prepared using a mechanical dissociator had increased cell viability (2-fold) and total cell recovery (2-fold) while preserving surface receptor expression. Enzymatic digestion of the remaining insoluble material did not recover additional CD45+ cells suggesting they were all released in the initial homogenate. Thus, this rapid and simple approach allows both qualitative and quantitative assessment of the CD45+ subpopulations present in various normal and malignant human tissues. An added advantage of this approach is that the SN from the initial homogenate (primary tissue SN) can be collected and stored for further analysis or experimentation.
Protocol
NOTE: All specimens were acquired using a protocol approved by the Medical Ethics Committee of the Institute Jules Bordet with written informed consent obtained from each patient.
1. Preparation of the Tissue Homogenate
Dissect resected tissues (malignant and normal tissue resected from the operating room) are in the pathology lab by trained personnel for immediate pickup. Tumor, NANT (taken the furthest distance from the tumor as possible) and normal tissue fragments are routinely processed within 1 - 3 hr of surgical excision in a BSL2 laboratory using standard biosafety procedures for human tissues. A flow chart of the protocol is illustrated in Figure 1.
Weigh all tissue fragments (normal, NANT and tumor) and measure the length, width, and height (length x width x height). This is an important step for the subsequent normalization of cell subpopulations, extracted RNA, etc. NOTE: The range of sample size is 100 to 10,000 mm3 with no fat if possible.
- Imprint the tumor fragment on a glass slide for H&E staining to verify that the tissue is actually part of the tumor.
- Do this by pressing a glass microscope slide on the tumor fragment and applying gentle pressure with your fingers for a few seconds.
- Fix the slide with isopropanol for 2 min followed by a washing step in water. Counterstain the tissue for 30 sec with Mayer's hematoxylin.
- Wash the slide in six baths of water. Stain in Phloxine B 2% for 15 sec.
- Wash in one bath of water followed by four baths of isopropanol and finish with one bath of water.
- Incubate in isopropanol for 1 min and drain. Clear in two baths of xylene.
- Mount with xylene based mounting medium. Examine for tumor cellularity (Figure 2). NOTE: Mainly, tumor cells stick to the imprinted slide - the stromal, lymphoid or adipose cells rarely remain, leaving spaces between the tumor cells (Figure 2).
Place the tissue fragment in a small culture dish containing 1 ml of the chemically defined, serum-free hematopoietic cell medium (hereafter referred to as medium) at room temperature and dice it into small pieces (~1 - 2 mm2) using a sterile scalpel.
Transfer everything (tissue fragments + medium) to a mechanical dissociator C tube.
Rinse the Petri dish and scalpel with 2 ml of medium using a Pasteur pipette and add this to the C tube (volume of medium for dissociation = 3 ml).
Use the mechanical dissociator program A.01 for C tubes (the most gentle program) to homogenize the tissue fragments into a single cell suspension. Place the C tube in the apparatus and run the program twice in succession (one cycle = 25 sec). NOTE: This homogenization procedure has been established and validated for human breast tissue, other tumor or tissue types may need to use a different program and should be tested first.
Remove the C tube from the apparatus and decant the homogenate directly into a 40 μm cell strainer seated on a 50 ml tube. Using the same Pasteur pipette as in step 1.6, transfer any liquid remaining in the C tube to the cell strainer.
Transfer the filtered liquid into a 15 ml tube using a 1 ml micropipette tip. Temporarily keep the cell strainer and its 50 ml tube.
Rinse the C tube with an additional 3 ml of medium and transfer this, again using the same Pasteur pipette as in step 1.6, to the cell strainer still seated on the 50 ml tube. Squeeze a maximum amount of the residual liquid trapped in the unhomogenized tissue into the 50 ml tube by gently moving it around the strainer with a clean Pasteur pipette or 1 ml tip that is subsequently thrown away to avoid contaminating the eluate.
Place the cell strainer upside down on the original C tube and rinse with 3 ml of medium so that the unhomogenized tissue drops back into the C tube.
Re-homogenize as in step 1.7 for two cycles of the A.01 program.
Pour this second homogenate through the cell strainer seated on the 50 ml tube, rinse the C tube again with 3 ml medium (as in step 1.10) and transfer with the Pasteur pipette to the cell strainer seated on the 50 ml tube again squeezing a maximum amount of liquid from the residual connective tissue trapped in the cell strainer.
At this point, a volume of ~2.5 ml is in the 15 ml tube and ~9 ml in the 50 ml tube.
2. Separation of the Tissue Supernatant and Cells
Centrifuge the homogenates in the 15 ml and 50 ml tubes for 15 min at 600 x g at room temperature.
Decant the SN from the 15 ml tube into a clean tube and temporarily store at 4 °C. This supernatant = primary tumor, NANT or normal tissue SN (final volume 2.5 ml) is subsequently clarified and aliquoted prior to storage at -80 °C for future analyses (see below).
Discard the supernatant from the 50 ml tube.
Gently resuspend both cell pellets in a final volume of 1 ml medium. Briefly, first gently break the cell pellet in both tubes (by tapping the tube on a hard surface). Resuspend the loose cell pellet in the 50 ml with 500 µl of medium and transfer this cell suspension to the 15 ml tube to resuspend the second pellet. Repeat this step once with the second 500 µl of medium for maximum recovery of cells.
Transfer 10 µl of the cell suspension to a small tube, mix with 10 µl of trypan blue (dilution 1:1) and count the number of viable cells using a hemocytometer. NOTE: At this point a fraction of the cell suspension can also be analyzed by flow cytometry to evaluate cell size, granularity and if desired a limited number of subpopulation markers for more precise assessment of the relative cell distribution in the homogenate prior to extensive analysis or experimentation. All analyses by flow cytometry incorporate CD45 labeling for normalization of subpopulations.
Pellet the cells by centrifugation at 300 x g for 10 min at room temperature. The cells from the tumor, NANT, or normal tissue are now ready for further purification or analysis. These additional steps are best when performed on the same day as surgery. NOTE: For flow cytometric analysis but not cell sorting the residual red blood cells should be lysed after antibody labeling by adding 0.4 ml of red blood cell lysis buffer to the cell pellet, immediately vortexing for 1 sec and incubating a minimum of 10 min at room temperature (protected from light) before analysis.
3. Clarification of the Tissue Supernatant
Centrifuge the 1.5 ml tubes with tissue SN at 15,000 x g for 15 min at 4 °C.
Carefully remove the supernatant without touching or disturbing the pellet. Transfer to a clean tube (or tubes) depending upon the number and volume of aliquots desired.
Store the supernatant at -80 °C for future use.
4. Patient Blood
Collect a blood sample from each patient as a control by venipuncture into heparinized tubes the day prior to surgery. Centrifuge the blood at 400 x g for 10 min at 20 ºC with the brake off to obtain the plasma and a buffy coat.
Remove the plasma and clarify it by centrifugation at 10,000 x g for 15 min at 20 ºC. Aliquot and store plasma at -80 °C for future use. Dilute the buffy coat in medium
Separate the mononuclear cells using standard ficoll-hypaque gradient centrifugation prior to immediate analysis, subpopulation isolation, DNA/RNA/protein extraction or cryopreservation.
5. Flow Cytometry
Label cells according to manufacturer’s instructions at 4 °C, protected from light. Lyse red blood cells in cell suspensions from tissue fragments after antibody labeling by adding 0.4 ml of cell lysis buffer to the cell pellet.
Vortex immediately for 1 sec and incubate a minimum of 10 min at room temperature, protected from light. Pass the labeled cells in a flow cytometer for data acquisition without washing.
Representative Results
Enzymatic digestion of tissue fragments with either commercially available tissue dissociation solutions or various laboratory mixtures of collagenase, DNase and/or hyaluronidase inhibitors, cleave a wide variety of receptors on the surface of cells. Our studies, initially focused on CD4+ T cells infiltrating breast tumors, were quickly presented with a major technical problem due to cleavage of surface CD4 receptors using standard enzymatic digestion protocols4-8. We tested a variety of collagenase enzymes with or without DNase and hyaluronidase inhibitors, finding that collagenase I and II completely removed CD4 from the cell surface, despite high lymphocyte viability (Figure 3A). A moderate effect on CD4 was observed with collagenase IV at short incubation times (1 - 2 hr) with lower CD4 antibody labeling detected in digested lymph node and tumor tissue compared to undigested blood and bone marrow from the same patient (Figure 3B; note that due to limitations on the tissue we receive we were unable to compare digested and undigested lymph node and tumor tissue). This loss of CD4 was confirmed to be a technical byproduct of collagenase IV digestion by comparing undigested and digested CD4+ T cells isolated from healthy donor blood (data not shown). While these CD4lo T cells can be isolated from collagenase IV-digested tissue fragments using magnetic beads, the purified populations frequently contain varying amounts of contamination by other cells from the tumor microenvironment and thus were suboptimal for our experimental purposes.
In a continuous search for a better approach, in 2008 we tested a new mechanical dissociator without using enzymes. This apparatus produced breast tissue homogenates rapidly and with surface receptor integrity maintained9. Representative flow cytometry dot plots of the tumor cell homogenates following dissociation and labeling with specific antibodies against epithelial cells (EpCAM) and leukocytes (CD45) are shown in Figure 3C. These images are typical routine observations for breast tumor homogenates. This protocol does not significantly alter the viability of CD45+ cells (4% dead cells); however, there is a considerable loss of viable EpCAM+ cells (38% dead cells in tumor shown; varies between tumors). Despite this loss, the viable EpCAM+ and CD45+ subsets can be segregated on the basis of size and structure into large epithelial cells (= tumor cells), small epithelial cells and large CD45+ cells and small CD45+ cells (the majority of cells, which are principally lymphocytes; Figure 3C).
The significant increase in viable CD45+ cells obtained using this approach permits the acquisition of reproducible multicolor flow cytometry data (using up to ten colors) to more fully characterize the TIL subsets in breast cancer. Representative flow cytometry dot plots of the major TIL subsets are shown in Figure 4, with our recent experiments demonstrating that it is also possible to detect and quantify minor T and B cells subsets infiltrating the tumor10-12, including intracellular staining for canonical T and B cell transcription factors12. Gating strategies for lymphocytes are based on viable CD45+ cells identified with a viability stain (note: tumors can vary greatly in the amount of cellular debris and dead epithelial cells depending on the BC subtype, grade, etc.). This approach has been successfully used to purify lymphocyte subpopulations from the homogenate with magnetic beads or cell sorting for gene expression analysis using microarrays or qRT-PCR9. Soluble mediators in the SN, including cytokines and immunoglobulins, have been quantified using a bead-based immunoassay with the resulting data normalized to the size of the tissue fragment. A comparison of cytokine expression (a Th1/Th2/Th7/Th9/Th17 pool) or total immunoglobulins (IgA, IgE, IgG, IgM) in BC SN with SN from normal breast tissue reveals increases in both associated with the tumor tissue (Figure 5). These primary tissue SN’s have also been effectively analyzed for antigens10-12 and used to experimentally treat lymphocytes from healthy donors, thereby reproducing the TIL phenotype in normal cells9.
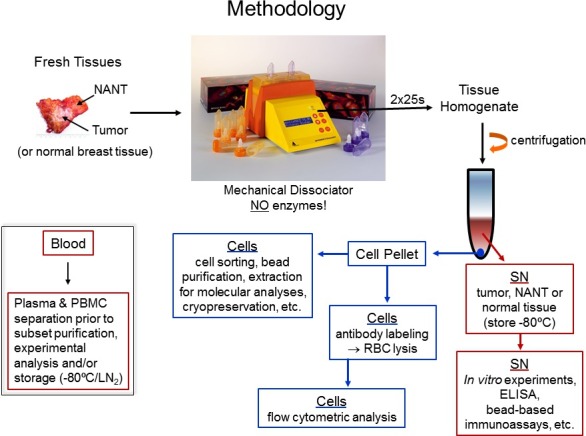 Figure 1. Protocol flow chart. Our procedure for processing fresh human tissues and some analytical approaches that can subsequently be used to assess lymphocytes infiltrating human tissues are shown.
Figure 1. Protocol flow chart. Our procedure for processing fresh human tissues and some analytical approaches that can subsequently be used to assess lymphocytes infiltrating human tissues are shown.
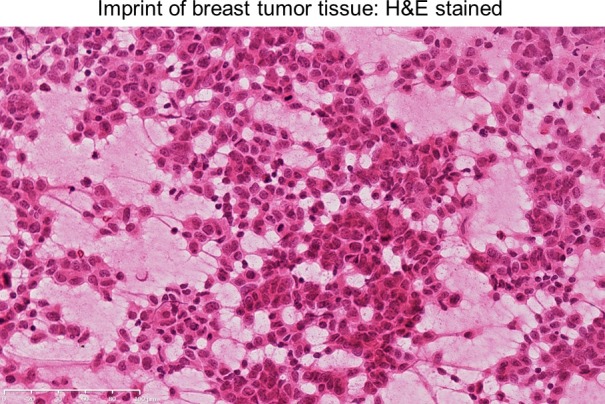 Figure 2. H&E stained image of a breast tumor tissue imprint. This imprint, taken from a fresh breast tumor tissue fragment, shows the presence of tumor cells with the open spaces reflecting areas that were not transferred. Please click here to view a larger version of this figure.
Figure 2. H&E stained image of a breast tumor tissue imprint. This imprint, taken from a fresh breast tumor tissue fragment, shows the presence of tumor cells with the open spaces reflecting areas that were not transferred. Please click here to view a larger version of this figure.
 Figure 3. Optimization of breast tissue dissociation for lymphocyte analysis. Flow cytometric analysis of: (A) CD4 surface expression on lymphocytes from undigested and Collagenase I/II digested tumor tissue; (B) CD4 expression on lymphocytes from lymph node and tumor tissue digested with Collagenase IV and compared to undigested blood and bone marrow cells from the same patient; and (C) analysis of total leukocytes (CD45+) and epithelial cells (EpCAM+) from tumor tissue rapidly mechanically dissociated using the protocol described here. The viability of epithelial cells and CD45+ cells using our protocol is assessed via the incorporation of fixable viability dye. Please click here to view a larger version of this figure.
Figure 3. Optimization of breast tissue dissociation for lymphocyte analysis. Flow cytometric analysis of: (A) CD4 surface expression on lymphocytes from undigested and Collagenase I/II digested tumor tissue; (B) CD4 expression on lymphocytes from lymph node and tumor tissue digested with Collagenase IV and compared to undigested blood and bone marrow cells from the same patient; and (C) analysis of total leukocytes (CD45+) and epithelial cells (EpCAM+) from tumor tissue rapidly mechanically dissociated using the protocol described here. The viability of epithelial cells and CD45+ cells using our protocol is assessed via the incorporation of fixable viability dye. Please click here to view a larger version of this figure.
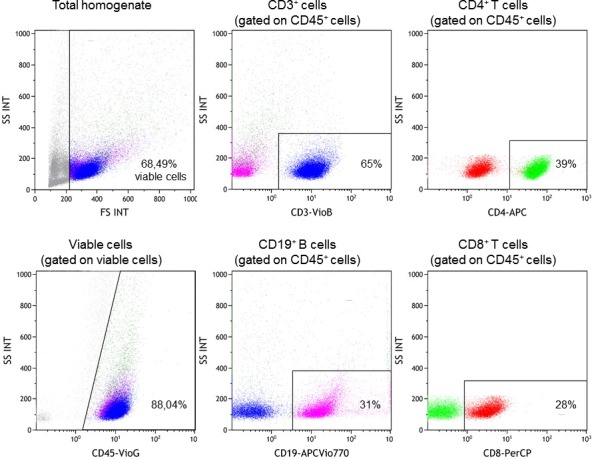 Figure 4. Tumor infiltrating lymphocyte subsets in the homogenate. Multicolor flow cytometric analysis was performed on the day of surgery after mechanical dissociation using the protocol described here. The viable cells and major lymphocyte subpopulations are shown: CD3 is a pan T cell marker, CD4 and CD8 are major T cell subset markers and CD19 is a pan B cell marker. Please click here to view a larger version of this figure.
Figure 4. Tumor infiltrating lymphocyte subsets in the homogenate. Multicolor flow cytometric analysis was performed on the day of surgery after mechanical dissociation using the protocol described here. The viable cells and major lymphocyte subpopulations are shown: CD3 is a pan T cell marker, CD4 and CD8 are major T cell subset markers and CD19 is a pan B cell marker. Please click here to view a larger version of this figure.
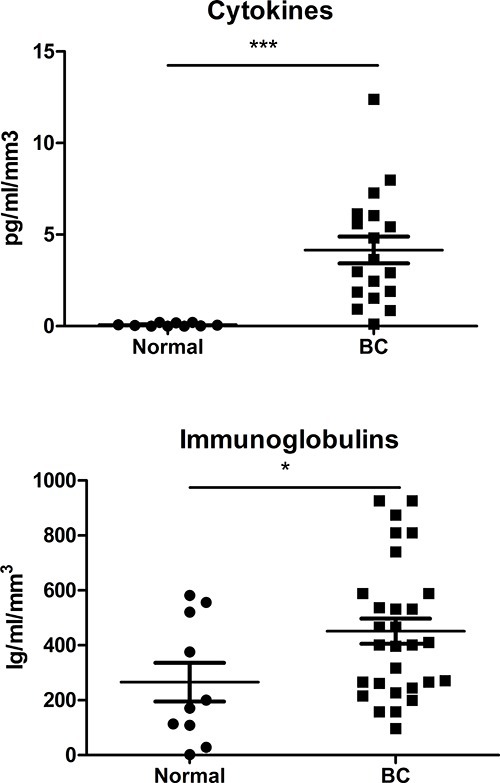 Figure 5. Soluble mediators in the supernatant. A panel of cytokines (Th1/Th2/Th7/Th9/Th17) and immunoglobulins (IgA, IgE, IgG, IgM) were assessed using bead-based immunoassays to analyze SN derived from normal and BC tissues using the protocol described here.
Figure 5. Soluble mediators in the supernatant. A panel of cytokines (Th1/Th2/Th7/Th9/Th17) and immunoglobulins (IgA, IgE, IgG, IgM) were assessed using bead-based immunoassays to analyze SN derived from normal and BC tissues using the protocol described here.
Discussion
This study describes an optimized method for the rapid preparation of normal and malignant breast tissue homogenates without enzymatic digestion for subsequent cell sorting, extraction, cryopreservation and/or phenotypic analysis of CD45+ subpopulations. The goal of this experimental approach is to produce images of the TIL that closely reflect their in vivo state and compare them to normal tissues with minimal manipulation of the tissues fresh from the operating room. To date, our laboratory has used this protocol to analyze >250 fresh BC tissues (tumor and NANT), >35 normal breast tissues from mammary reductions. In principal, this protocol should be directly applicable to other tumor or tissue types; however, some optimization may be necessary (i.e., the mechanical dissociation program, number of cycles, media volume, etc.). For tissues with low lymphocyte infiltration, using a larger tissue fragment may be necessary, which is what we do for normal tissues where the lymphocyte count is low. Alternatively, a density gradient centrifugation step could be added to enrich the leukocyte suspension. This analysis of human T and B cell subpopulations infiltrating breast tumors includes an assessment of >100 markers by flow cytometry (flow cytometry data for 74 markers is available in the supplementary data of Gu-Trantien, et al.9) and purification of specific lymphocyte subpopulations for subsequent immunological and molecular analyses. Further, we have evaluated cytokines, chemokines, immunoglobulins and tumor antigens in the primary tumor SN (compared to NANT and normal tissue SN)10,11 as a reflection of these molecular mediators in the tumor microenvironment. We have also tested the effect of treating peripheral blood lymphocytes from healthy donors with tumor SN and shown that many of the gene expression changes detected in the TIL can be specifically reproduced9.
In their microenvironment, the tumor and stromal cells are more tightly held in the tissue matrix than the immune cells, which are characteristically migratory. The method described here provides a simple and rapid means for isolating and analyzing TIL without enzymatic digestion. Our numerous attempts to join this mechanical homogenization approach with a short enzymatic digestion to produce viable and receptor positive lymphoid and epithelial cells from the same tumor fragment have been unsuccessful. Tissue homogenization efficiently releases lymphocytes but results in a significant loss of tumor cell viability. A short enzymatic digestion of the tissue liberates viable tumor cells (the total number increases as a function of digestion time) but has the consequence of a parallel decrease in the expression of many surface receptors, particularly apparent on lymphocytes. Thus, for the comparative analysis of tumor cells and TIL from the same tumor it is still necessary to separately process two sequential tumor fragments.
Currently, the majority of studies designed to characterize TIL have employed enzymatic digestion (hours to O/N) frequently coupled with some form of mechanical dissection4-8. Expansion of TIL for further analysis or therapy generally involves the subsequent ex vivo cultivation of TIL or tumor tissue fragments with stimulatory agents (days to weeks)13-15. The rapid, non-enzymatic method for tissue dissociation described here provides a simple and reproducible means for extracting intact CD45+ cells from normal and abnormal tissue fragments prior to their expansion or analysis. This speedy acquisition of CD45+ cells from patients undergoing tumorectomy could possibly be developed for use in a prognostic biomarker assay. In addition, it may yield TIL with greater potential for ex vivo expansion prior to adoptive immunotherapy.
Disclosures
The authors declare that no conflict of interest exists.
Acknowledgments
This work was supported by grants fromthe Belgian Fund for Scientific Research (FNRS), Les Amis de l’Institut Bordet, FNRS-Opération Télévie, Plan Cancer of Belgium, Fonds Lambeau-Marteaux, Fonds J.C. Heuson and Fonds Barsy.
References
- Chen DS, Mellman I. Oncology meets immunology: the cancer-immunity cycle. Immunity. 2013;39(1):1–10. doi: 10.1016/j.immuni.2013.07.012. [DOI] [PubMed] [Google Scholar]
- Boudreau A, van't Veer JL, Bissell MJ. An 'elite hacker': breast tumors exploit the normal microenvironment program to instruct their progression and biological diversity. Cell Adh Migr. 2012;6(3):236–248. doi: 10.4161/cam.20880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gajewski TF, Schreiber H, Fu YX. Innate and adaptive immune cells in the tumor microenvironment. Nat Immunol. 2013;14(10):1014–1022. doi: 10.1038/ni.2703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quezada SA, et al. Limited tumor infiltration by activated T effector cells restricts the therapeutic activity of regulatory T cell depletion against established melanoma. J Exp Med. 2008;205(9):2125–2138. doi: 10.1084/jem.20080099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grange C, et al. Phenotypic characterization and functional analysis of human tumor immune infiltration after mechanical and enzymatic disaggregation. J Immunol Methods. 2011;372(1-2):119–126. doi: 10.1016/j.jim.2011.07.002. [DOI] [PubMed] [Google Scholar]
- McCauley HA, Guasch G. Serial orthotopic transplantation of epithelial tumors in single-cell suspension. Methods Mol Biol. 2013;1035:231–245. doi: 10.1007/978-1-62703-508-8_20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gros A, et al. Myeloid cells obtained from the blood but not from the tumor can suppress T-cell proliferation in patients with melanoma. Clin Cancer Res. 2012;18(19):5212–5223. doi: 10.1158/1078-0432.CCR-12-1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zirakzadeh AA, Marits P, Sherif A, Winqvist O. Multiplex B cell characterization in blood, lymph nodes, and tumors from patients with malignancies. J Immunol. 2013;190(11):5847–5855. doi: 10.4049/jimmunol.1203279. [DOI] [PubMed] [Google Scholar]
- Gu-Trantien C, et al. CD4(+) follicular helper T cell infiltration predicts breast cancer survival. J Clin Invest. 2013;123(7):2873–2892. doi: 10.1172/JCI67428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buisseret L, et al. Lymphocytes Infiltrating Breast Cancer : Density, Composition And Organization. Annals of Oncology. 2014;25(1):17. [Google Scholar]
- Garaud S, et al. Characterization of B Cells Infiltrating Human Breast Cancer. Annals of Oncology. 2014;25(1):18. [Google Scholar]
- Gu-Trantien C, et al. Cxcl13-Producing Follicular Helper T Cells In Human Breast Cancer. Annals of Oncology. 2014;25(1):17. [Google Scholar]
- Yee C. The use of endogenous T cells for adoptive transfer. Immunol Rev. 2014;257(1):250–263. doi: 10.1111/imr.12134. [DOI] [PubMed] [Google Scholar]
- Butler MO, et al. Ex vivo expansion of human CD8+ T cells using autologous CD4+ T cell help. PLoS One. 2012;7(1):30229. doi: 10.1371/journal.pone.0030229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye Q, et al. Engineered artificial antigen presenting cells facilitate direct and efficient expansion of tumor infiltrating lymphocytes. J Transl Med. 2011;9:131. doi: 10.1186/1479-5876-9-131. [DOI] [PMC free article] [PubMed] [Google Scholar]